
Abstract
Formic acid is one of the most promising candidates for the long-term storage of hydrogen in liquid form. Herein, we present a new collection of ruthenium pincer complexes of the general formula [RuHCl(POP)(PPh3)] using commercially available or easy-to-synthesize tridentate xantphos-type POP pincer ligands. We applied these complexes in the dehydrogenation of formic acid to CO2 and H2 using the ionic liquid BMIM OAc (1-butyl-3-methylimidazolium acetate) as solvent under mild, reflux-free conditions. The best performing catalyst with respect to maximum turnover frequency, the literature-known complex [RuHCl(xantphos)(PPh3)] Ru-1, produced a maximum turnover frequency of 4525 h−1 with 74% conversion after 10 min at 90 °C and complete conversion (> 98%) occurring within 3 h. On the other hand, the best overall performing catalyst, the novel complex [RuHCl(iPr-dbfphos)(PPh3)] Ru-2, facilitated full conversion within 1 h leading to an overall turnover frequency of 1009 h−1. Moreover, catalytic activity was observed at temperatures as low as 60 °C. Only CO2 and H2 are observed in the gas phase, with no CO detected. High-resolution mass spectrometry suggests the presence of N-heterocyclic carbene complexes in the reaction mixture.
Similar content being viewed by others

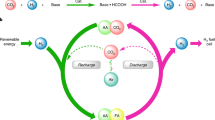
Introduction
The increasing market share of renewable energy and its intermittency leads to the need for industrial scale energy storage technologies in the electricity, heat, industry, and transport sectors1,2. Hydrogen being considered one of the most common energy carriers3, liquid organic hydrogen carriers (LOHCs) have recently come into the focus of researchers, offering the prospect of storing hydrogen in easy-to-handle form without hazards associated with pressurization or cryogenic techniques4,5,6. Due to their physical properties, significant parts of the already existing infrastructure for the transportation of gasoline and other liquid fuels can be utilized for the transport of LOHCs7,8. The physical properties of formic acid (FA) make it a promising hydrogen storage candidate with a gravimetric hydrogen load of 4.4 weight%9,10. Published catalytic systems for formic acid dehydrogenation, however, typically require the use of volatile organic solvents, water, or neat formic acid11,12,13,14, potentially leading to the need for solvent vapor separation techniques such as condensation, which would pose an additional burden for consumer applications. This issue is eliminated by using a solvent with negligible vapor pressure, such as ionic liquids. Previously, our workgroup showed the ionic liquid butyl-methyl-imidazolium acetate (BMIM OAc) to be a suitable solvent in this reaction using commercially available Ru-PNP pincer complexes of the Ru-MACHO type15. For example, we demonstrated FA dehydrogenation in a continuous flow system using BMIM OAc, achieving TONs exceeding 18,000,000 at 95 °C. Although several systems before have achieved high TONs, many relied on volatile organic solvents such as THF or DMF, or the use of additives like bases. In contrast, our work employed practically non-volatile ionic liquids (ILs) and no additive.
Hazari and Bernskoetter reported the use of an Fe-PNP catalyst in the presence of dioxane and LiBF₄ for the dehydrogenation of formic acid (FA) at 80 °C, achieving an impressive turnover number (TON) of ~ 1,000,00016. Laurenczy employed a Ru(II)-TPPTS complex catalyst in a continuous FA dehydrogenation system. This approach enabled almost complete dehydrogenation of FA with minimal CO traces detected at 80 °C17. Further advancing the field, Pidko demonstrated reversible FA dehydrogenation using a Ru-PNP pincer catalyst in DMF/DBU and DMF/NHex₃ mixtures, achieving TON values between 310,000 and 706,500 at 90 °C18. Hull, Himeda, and Fujita explored a dinuclear Ir-complex catalyst with sacrificial KHCO₃ and H₂SO₄, alternating between CO₂ hydrogenation and FA dehydrogenation. Their system achieved TONs greater than 3,500,000 and 308,000, respectively, with hydrogenation occurring at 30 °C under CO₂/H₂ (1:1) at 1 bar, and dehydrogenation occurring between 60 and 90 °C19. Sponholz, Junge, and Beller developed a Mn-PNP complex for reversible CO₂ hydrogenation and FA dehydrogenation at 90 °C20.
Herein, we pursued the IL approach but rather than utilizing Ru-PNP, we explored the use of a Ru-POP catalyst, which, to the best of our knowledge, has not been previously demonstrated in this context.
Due to their excellent metal ligand cooperation (MLC), amino-based PNP pincer complexes based on the Noyori-type concept with a cooperating secondary amine functionality21 such as Ru-MACHO-BH have generally become increasingly popular several small molecule manipulations, such as the hydrogenation of CO222, alkenes and carbonyls, transfer hydrogenations23, as well as acceptorless dehydrogenations of alcohols24. N-Methylating the PNP pincer ligand has been reported to completely stop catalyst activity25, which is explained by the amine acting as a proton source as a fundamental requirement during the catalytic cycle using MLC. However, Beller recently observed the opposite trend with formic acid dehydrogenation where an N-methylated Ru-PNP complex in fact showed superior catalytic formic acid dehydrogenation over the non-methylated congener26. As the former complex is unable to participate in MLC via the amino unit, it strongly indicates that MLC, and hence the amino unit, could play a less important role in some (de)hydrogenative transformations than previously accepted.
Compared to PNP pincers, POP pincer complexes of ruthenium are underexplored in this field. The POP ligands have traditionally been mainly used in hydroformylation, in which their characteristic bidentate bite angles as chelating rather than pincer ligands of approximately 120° have been exploited to optimize the selectivity of linear over branched products27,28,29. Ru-POP complexes have since been sparingly used in hydrogenation catalysis, but examples of their activity in transfer hydrogenations have previously been reported30. Here, we show that Ru-POP complexes are competent catalysts for formic acid dehydrogenation, corroborating the findings of Beller that the amino unit in classical amino-based Ru-PNP catalysts is less essential in this reaction.
Results and discussion
Our investigation commenced with the synthesis of two representative catalysts of the general formula [RuHCl(POP)(PPh3)] (Fig. 1a). To vary the steric and electronic structures, the commercially available Xantphos (9,9-Dimethyl-9H-xanthene-4,5-diyl)bis(diphenylphosphane) ligand, and the isopropyl-substituted DBFphos derivative 4,6-bis(diisopropylphosphaneyl)dibenzo[b,d]furan were chosen (Fig. 1b)31. The catalysts investigated in this study were synthesized using a general approach developed by Whittlesey32 using [RuHCl(PPh3)3]•toluene adduct33 as precursor. The metal precursor and the POP pincer ligands were stirred in THF under strictly anhydrous and anaerobic conditions. The reaction is accompanied by a prominent color change from dark purple to yellow and produces clean product after refluxing for 4 h or at 40 °C for 72 h. Washing twice in hexane or diethyl ether after removing THF in vacuo removed triphenylphosphine, yielding the products as yellow powders in high to quantitative yields.

Synthesis of complexes Ru-1 and Ru-2. a) Synthetic approach to complexes. b) Structures of synthesized complexes.
With Ru-1 being already literature-known32, further characterization concentrated on Ru-2. The 1H-NMR spectrum of Ru-2 confirmed the cis-configuration of the hydride towards the phosphine atoms in the ligands. The dt pattern of the peak (Fig. 2a) displayed 2JP−H coupling constants of 28.6 and 22.0 Hz, which is well within the expected range reported previously32. In the hydrogen-decoupled 31P{1H} spectrum (Fig. 2b), a 2JP−P coupling constant of approximately 27.6 Hz was observed, confirming that both phosphines of the pincer ligand are cis to PPh3. Moreover, ATR-IR revealed a characteristic ruthenium-hydrogen stretching band at 2054 cm−1. For further structural elaboration, the complex Ru-2 was crystallized in sufficient quality for XRD studies by vapor diffusion at room temperature (Fig. 3, Supplementary Table 1). It crystallized in a triclinic crystal system of the space group P-1, with one unit of co-crystallized benzene per unit cell. It displayed a wide P-Ru-P bite angle of 153.94°, significantly wider than the 130 ° bite angle in bidentate DBFphos34. At 2.401 and 2.382 Å, the Ru-PPOP bond lengths were significantly longer than the bond length from Ru to PPh3 of 2.232 Å, which is likely a result of the wide bite angle of the DBFphos backbone caused by its central 5-ring. The geometry at the metal center was otherwise essentially octahedral, with an O-Ru-PPh3 angle of 179.5°. The H-Ru-Cl coordination was not completely linear, with an angle of approximately 175°, pointing away from the triphenylphosphine ligand. Atom distances and bond lengths are listed in Table 1.

NMR spectra of Ru-2. a) Hydride region of 1 H NMR spectrum showing a dt signal for Ru- H. b) 31 P{ 1 H} NMR spectrum, showing the signals for triphenylphosphino (blue) and POP ligands (green).

Structure of Ru-2. Thermal ellipsoids are shown at 70% probability. Co-crystallized benzene and hydrogen atoms on carbon were omitted for clarity.
To assess the capability of the complexes to dehydrogenate formic acid, reaction conditions were chosen under which related PNP-pincer complexes, such as Ru-MACHO-BH, are highly active15. Using 1.0 mL (5.35 mmol) of the ionic liquid (IL) BMIM OAc, 0.5 mL (13.25 mmol) formic acid were dehydrogenated using 0.1 mol% (1000 ppm, 13 µmol) of ruthenium complex Ru-1 or Ru-2 (Table 2; Fig. 4).
To get a benchmark, the reaction was first performed with the precursor [RuHCl(PPh3)3]•toluene adduct. Reactions were performed at temperatures between 60 and 90 °C. By simple eye inspection, the complex was not completely soluble in the IL even after prolonged stirring at 90 °C, but dissolution occurred upon injection of formic acid. At 90 °C, 56% (TOF = 3424 h−1) conversion was reached within the first 10 min, which increased to an almost quantitative (97%) conversion after three hours (Entry 1). Reducing the temperature to 80 °C lowers the conversion after 10 min by more than half to 24% (TOF = 1467 h−1, Entry 2), further being decreased to 18% and 6% at 70 and 60 °C, respectively (Entries 3 and 4). In all cases, no induction periods were found, showing this catalyst to likely be the active species, or that the conversion to the active species is too fast to be detected with this dataset.
Following the precursor evaluation, the Ru-POP pincer complexes Ru-1 and Ru-2 were used under identical conditions. At 90 °C, immediate high conversions were observed. Ru-1 delivered 74% conversion within the first 10 min of the experiment (TOFmax =4525 h−1, Entry 5). Ru-2 showed slightly lower but more durable activity, facilitating 60% conversion in 10 min (TOFmax = 3669 h−1) and full conversion (> 99%) within 60 min (Entry 9). Notably, at full conversion Ru-2 is significantly superior to both the metal precursor and Ru-1. Thus, whereas the metal precursor and Ru-1 have similar TOFoverall values at reaction completion of 330 h−1 and 333 h−1, respectively, Ru-2 has a TOFoverall of 1009 h−1.
Then, a temperature variation was performed with Ru-1 and Ru-2, in which the temperatures were progressively lowered in steps of 10 °C to a minimum of 60 °C (Fig. 3). While the complexes were instantly active at 90 °C, yielding near-complete conversions within one hour, the activity fell drastically at lower temperatures. At 80 °C and 70 °C, low conversions of 14% and 23%, respectively, were found after 10 min for Ru-1, increasing to 79% and 73%, respectively, after 30 min (Entries 6 and 7). Both experiments showed conversions of ≥ 90% within two hours. A similar behavior was observed with Ru-2 (Entries 10 and 11). Interestingly, Ru-1 is slightly superior at 70 °C at the end of the reaction with a TOFoverall of 315 h−1 compared to 292 h−1 for Ru-2 and 299 h−1 for the metal precursor.
A further temperature reduction to 60 °C led to no conversion being observed in the first 30 min of the experiment. This apparent inactivity of Ru-1 at the lowest temperature at the beginning of the experiment, followed by an increase in activity, points towards the requirement of an activation period, in which the pre-catalyst Ru-1 is transformed to the catalytically active species. While likely present at all temperatures, the timeframe of 10 min in the beginning of the experiment is not sufficient to probe the activation period at higher temperatures. A similar behavior was found for Ru-2. At 70 and 60 °C, no conversion was observed in the first 10 min of the experiment. Importantly, in neither experiment formation of carbon monoxide was observed within the detectable limit of our instrumentation (< 300 ppm), with H2 and CO2 being the only observed products.
Comparing this to previously achieved results of formic acid dehydrogenation in this workgroup, representing the state of the art and using a Ru-PNP pincer complex, shows similar activity of the newly synthesized Ru-POP pincers compared to their PNP counterparts15. While the PNP pincers reached turn-over frequencies of 500–1260 h−1 in batch experiments, the new POP pincers reached similar TOFoverall values of 326 h−1 and observed TOFmax values of 1988 h−1 for Ru-1 and 1590 h−1 for Ru-2 at 80 °C, as well as 4525 h−1 for Ru-1 and 3669 h−1 for Ru-2 at 90 °C, respectively.

Temperature screening of formic acid dehydrogenation using catalysts Ru-1 and Ru-2. Conditions: 13 µmol catalyst, 0.5 mL (13.25 mmol) formic acid, 1.0 mL (5.35 mmol) BMIM OAc.
NMR was used to gather insight into the reaction mechanism. Due to the very pronounced differences in 2JH−P between the hydrides and the phosphine ligands, the hydride peaks were the primary focus of this investigation. In the case of Ru-1, the typical dt pattern was found for the hydrido unit during the first 60 min of the dehydrogenation. While shifted significantly downfield from − 16.29 to -13.35 ppm, it showed coupling constants to phosphine of 27.2 and 18.4 Hz, respectively (Fig. 5, peak A). This is compatible with all three phosphines being in a cis-configuration to the hydrido ligand and shows that the ligand configuration is somewhat stable in the IL over the course of approximately one hour at the optimized reaction conditions. The strong downfield shift might be explained by the abstraction of the chlorido ligand and the formation of the corresponding acetato and formato complexes, the in-situ formation of the d3-MeCN complex in the NMR tube, or the formation of the corresponding N-heterocyclic carbene (NHC) complex. Over the course of the dehydrogenation reaction, the intensity of this signal continuously decreased, and after 180 min, it was no longer observed. Instead, two new signals were detected. The first shows an apparent dd pattern and occurs at -6.4 ppm (peak B). The doublet has a big coupling constant of approximately 130.4 Hz, suggesting that one of the phosphine units has moved to a trans-position relative to the hydrido. This could mean the rearrangement of the POP pincer into a κ2-P, P-configuration. The appearance of this complex late in the catalysis might point to this species leading to a deactivation route over time forming a catalyst sink. Alternatively, the low chemical shift suggests that this might be a dihydrido species15. The second new peak is found at -17.5 ppm. While its multiplicity is not clear, we suggest it to be a triplet with a small coupling constant of 17.3 Hz, indicating a coupling of the hydrido ligand solely with the phosphine ligands of the POP pincer, pointing towards the liberation of triphenylphosphine (peak C) and its possible replacement by another ligand such as acetato or an NHC formed in situ from the ionic liquid. Dissociation of PPh3 was further indicated by a strong singlet at -5.9 ppm in the 31P{1H} spectrum of Ru-1 at 90 °C after 180 min (see supplementary information).

Hydride region of the 1H NMR spectrum of Ru-1 during formic acid dehydrogenation. Reaction conditions: 0.5 mL formic acid, 1.0 mL BMIM OAc, 13.0 µmol catalyst, 90 °C. NMR taken in MeCN-d 3 , 500 µL deuterated solvent, ~ 10µL reaction mixture.
To further corroborate the active species in the catalytic system, high-resolution mass spectrometry (HRMS) was performed on Ru-1 10 min after injection of the formic acid at 90 °C. This showed the presence of a species lacking the chlorido ligand of the pre-catalyst, as well as two NHC complexes in the reaction mixture, whose proposed structures are listed in Fig. 6. The corresponding HRMS spectrum can be found in Supplementary Fig. 7.

Species present in the reaction mixture of Ru-1 in BMIM OAc and formic acid shown via HRMS.
Based on this data, we suggest an inner-sphere reaction mechanism similar to the one proposed by Beller for an N-methylated PNP pincer catalyzing the same reaction26. An additional experiment excluding ionic liquid showed no activity, hence its direct involvement seems necessary. We propose that activation of Ru-1 and Ru-2 occurs through dissociation of chloride, potentially followed by addition of the NHC and dissociation of triphenylphosphine (Scheme 1a). All species of this activation have previously been observed by HRMS. The IL acetate is a stronger Brønsted base than formic acid, rendering a high degree to full deprotonation of the latter likely35.We propose that during the catalytic cycle (Scheme 1b), active species A, carrying either NHC or PPh3, is coordinated by formate, leading to species B. Reconfiguration of this complex to C eventually leads to CO2 liberation and a trans-dihydrido complex D. The following protonation towards a dihydrogen complex with the previously formed acetic acid leading to dihydrogen hydrido complex E is analogous to what Beller proposed to be the key step using the N-methylated PNP pincer congener. Moreover, the L = PPh3 analog of complex E has previously been synthesized in stoichiometric reactions using Ru-1 under a hydrogen atmosphere after chloride abstraction by a sodium salt36. Elimination of hydrogen gas and coordination of formate provided A and completes the cycle.

Proposed inner-sphere reaction mechanism for the dehydrogenation of formic acid with a Ru-POP pincer complex Ru-1.
Conclusions
A new complex, [RuHCl(iPr-dbfphos)(PPh3)], was synthesized. The complex was characterized by NMR, ATRIR, EA, and single-crystal XRD. We also report the first successful application of Ru-POP pincer complexes in the dehydrogenation of formic acid to CO2 and H2. The complexes reached maximum turnover frequencies of up to 4525 h−1 at 90 °C albeit with the metal precursor reaching a similar activity (up to 3424 h−1). Furthermore, at 90 °C the novel complex [RuHCl(iPr-dbfphos)(PPh3)] reached an overall TOF at completion of formic dehydrogenation (1009 h−1) that significantly exceeded that of both the metal precursor (330 h−1) and the previously reported complex [RuHCl(xantphos)(PPh3)] (333 h−1). Catalytic performance is comparable to Ru-PNP pincer complexes under similar conditions. HRMS data suggests the presence of carbene complexes in the reaction mixture albeit in minor amounts. We are currently addressing the role of the carbene complexes for catalysis.
Data availability
All data generated or analysed during this study are included in this published article [and its supplementary information files].
References
Azarpour, A., Suhaimi, S., Zahedi, G. & Bahadori, A. A review on the drawbacks of renewable energy as a promising energy source of the future. Arab. J. Sci. Eng. 38, 317–328 (2013).
Moriarty, P. & Honnery, D. What is the global potential for renewable energy? Renew. Sustain. Energy Rev. 16, 244–252 (2012).
Turner, J. A. A. & Realizable Renewable Energy Future Sci. 285, 687–689 (1999).
Rao, P. C. & Yoon, M. Potential liquid-organic hydrogen carrier (Lohc) systems: a review on recent progress. Energies 13, 6040 (2020).
Niermann, M., Beckendorff, A., Kaltschmitt, M. & Bonhoff, K. Liquid Organic hydrogen carrier (LOHC) – Assessment based on chemical and economic properties. Int. J. Hydrogen Energy. 44, 6631–6654 (2019).
Teichmann, D., Arlt, W., Wasserscheid, P. & Freymann, R. A future energy supply based on Liquid Organic Hydrogen Carriers (LOHC). Energy Environ. Sci. 4, 2767–2773 (2011).
Niermann, M., Timmerberg, S., Drünert, S. & Kaltschmitt, M. Liquid Organic Hydrogen Carriers and alternatives for international transport of renewable hydrogen. Renew. Sustain. Energy Rev. 135, 110171 (2021).
Rong, Y. et al. Techno-economic analysis of hydrogen storage and transportation from hydrogen plant to terminal refueling station. Int. J. Hydrogen Energy. 1–12 https://doi.org/10.1016/j.ijhydene.2023.01.187 (2023).
Guo, J. et al. Formic acid as a potential On-Board Hydrogen Storage Method: development of homogeneous Noble Metal catalysts for dehydrogenation reactions. ChemSusChem. 14, 2655–2681 (2021).
Müller, K., Brooks, K. & Autrey, T. Hydrogen Storage in formic acid: a comparison of process options. Energy Fuels. 31, 12603–12611 (2017).
Wang, Z., Lu, S. M., Li, J., Wang, J. & Li, C. Unprecedentedly high formic acid dehydrogenation activity on an Iridium Complex with an N,N′-Diimine Ligand in Water. Chem. - Eur. J. 21, 12592–12595 (2015).
Hong, D. et al. Cooperative effects of heterodinuclear IrIII–MII complexes on catalytic H2 evolution from formic acid dehydrogenation in water. Inorg. Chem. 59, 11976–11985 (2020).
Fink, C., Laurenczy, G. A. & Precious Catalyst Rhodium-catalyzed formic acid dehydrogenation in Water. Eur. J. Inorg. Chem. 2381–2387 (2019).
Celaje, J. J. A. et al. A prolific catalyst for dehydrogenation of neat formic acid. Nat. Commun. 7, 11308 (2016).
Piccirilli, L. et al. Versatile CO2 hydrogenation-dehydrogenation catalysis with a Ru-PNP/Ionic Liquid System. J. Am. Chem. Soc. 145, 5655–5663 (2023).
Bielinski, E. A. et al. Lewis Acid-assisted formic acid dehydrogenation using a Pincer-supported Iron Catalyst. J. Am. Chem. Soc. 136, 10234–10237 (2014).
Henricks, V., Yuranov, I., Autissier, N. & Laurenczy, G. Dehydrogenation of formic acid over a homogeneous Ru-TPPTS Catalyst: unwanted CO production and its successful removal by PROX. Catalysts. 7, 348 (2017).
Filonenko, G. A. et al. Efficient reversible hydrogenation of Carbon Dioxide to Formates using a ruthenium PNP-Pincer Catalyst. ChemCatChem. 6, 1526–1530 (2014).
Hull, J. et al. Reversible hydrogen Storage using CO2 and a Proton-Switchable Iridium Catalyst in aqueous media under mild temperatures and pressures. Nat. Chem. 4, 383–388 (2012).
Wei, D. et al. Reversible hydrogenation of Carbon Dioxide to Formic Acid using a Mn-Pincer Complex in the Presence of Lysine. Nat. Energy. 7, 438–447 (2022).
Piccirilli, L., Pinheiro, D. L. J. & Nielsen, M. Recent progress with Pincer Transition Metal Catalysts for sustainability. Catalysts. 10, 773 (2020).
Wei, D., Junge, H. & Beller, M. An amino acid based system for CO2 capture and catalytic utilization to produce formates. Chem. Sci. 12, 6020–6024 (2021).
Subaramanian, M. et al. General and selective homogeneous Ru-catalyzed transfer hydrogenation, deuteration, and methylation of functional compounds using methanol. J. Catal. 425, 386–405 (2023).
Ni, Z., Padilla, R., Pramanick, R., Jørgensen, M. S. B. & Nielsen, M. Base-free, acceptorless dehydrogenative coupling of ethanol to ethyl acetate with PNP complexes. Dalton Trans. 52, 8193–8197 (2023).
Fu, S., Shao, Z., Wang, Y. & Liu, Q. Manganese-catalyzed upgrading of ethanol into 1-Butanol. J. Am. Chem. Soc. 139, 11941–11948 (2017).
Agapova, A., Alberico, E., Kammer, A., Junge, H. & Beller, M. Catalytic Dehydrogenation of Formic Acid with Ruthenium-PNP-Pincer complexes: comparing N-Methylated and NH-Ligands. ChemCatChem. 11, 1910–1914 (2019).
Kranenburg, M. et al. New Diphosphine Ligands based on Heterocyclic Aromatics Inducing Very High Regioselectivity in rhodium-catalyzed hydroformylation: Effect of the Bite Angle. Organometallics. 14, 3081–3089 (1995).
Hillebrand, S., Bruckmann, J., Kriiger, C. & Haenel, M. W. Bidentate Phosphines of heteroarenes: 9,9-Dimethyl-4,5-bis(diphenylphosphino)xanthene). Tetrahedron Lett. 36, 75–78 (1995).
Van Der Veen, L. A. et al. Electronic effect on rhodium diphosphine catalyzed hydroformylation: the bite angle effect reconsidered. J. Am. Chem. Soc. 120, 11616–11626 (1998).
Alós, J. et al. POP-pincer ruthenium complexes: d6 counterparts of osmium d4 species. Inorg. Chem. 53, 1195–1209 (2014).
Asensio, G. et al. Osmium(III) complexes with POP Pincer ligands: Preparation from commercially available OsCl3·3H2O and their X-ray structures. Inorg. Chem. 49, 8665–8667 (2010).
Ledger, A. E. W. et al. Pincer Phosphine Complexes of Ruthenium: formation of Ru(P-O-P)(PPh3) HCl(P-O-P = xantphos, DPEphos, (Ph2PCH2CH2)2O) and Ru(dppf)(PPh3)HCl and characterization of Cationic Dioxygen, Dihydrogen, Dinitrogen, and Arene Coordinated Phosphine products. Inorg. Chem. 49, 7244–7256 (2010).
Schunn, R. A., Wonchoba, E. R. & Wilkinson, G. Chlorohydridotris(triphenylphosphine)ruthenium(II). Inorg. Synth. 13, 131–134 (1972).
Van Leeuwen, P. W. N. M. & Kamer, P. C. J. Featuring Xantphos. Catal. Sci. Technol. 8, 26–113 (2018).
Calder, G. V. & Barton, T. J. Actual effects controlling the acidity of carboxylic acids. J. Chem. Educ. 48, 338–340 (1971).
Ledger, A. E. W. Chelating phosphine complexes of ruthenium for the coordination and activation of small molecules (University of Bath, 2011).
Acknowledgements
The authors thank the VILLUM FONDEN (19049), Carlsberg Foundation (CF20-0365), Novo Nordisk Foundation (NNF20OC0064560), and Independent Research Fund Denmark (1127-00172B) for generous research funding. We thank Mikroanalytisches Laboratorium Kolbe for carrying out Elemental Analysis.
Author information
Authors and Affiliations
Contributions
The project was conceived and designed by M.N and A.T.N. All experimental work was performed by A.T.N and B.R. A.T.N performed all synthetic work of catalysts, and A.T.N and B.R. shared the catalytic screening work. R.W.L conducted the IR measurement. M.S.B.J. conducted the HRMS measurements. M.N supervised and planned the project. A.T.N wrote the first draft of the manuscript and all authors contributed to writing the manuscript and have given consent to the publication of the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nikol, A.T., Rabell, B., Jørgensen, M.S.B. et al. Formic acid dehydrogenation using Ruthenium-POP pincer complexes in ionic liquids. Sci Rep 14, 26209 (2024). https://doi.org/10.1038/s41598-024-76782-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-76782-3
