
Abstract
Platelet-cancer cell interactions play a significant role in metastasis. Indeed, they interact via a plethora of receptors, including integrins (e.g. ⍺IIbβ3 and ⍺vβ3), and calcium is essential for both their stability and function. Additionally, calcium plays a significant role in the coagulation cascade, and the implication of calcium level changes on metastatic dissemination and cancer-associated thrombosis are not fully understood. A significant proportion of cancer patients suffer from hypercalcemia and have a worse prognosis. We hypothesized that calcium levels are important for platelet-cancer cell interactions that are mediated via integrins, thus this can be leveraged to disrupt platelet support to the metastatic process. In this study, we assessed the detection of integrins ⍺IIbβ3 and ⍺vβ3 on platelets and cancer cells, platelet function, and the respective receptors implicated in platelet function, while modulating calcium levels. The effect of calcium levels on platelet-cancer cell interactions and cancer cell invasion in vitro was also assessed. Our data demonstrates that calcium levels affect surface integrins, and receptors involved in platelet-cancer cell interactions. In addition, calcium levels significantly affect platelet activation and aggregation. In our experimental scenarios, calcium depletion modulates platelet-cancer cell interaction with MDA-MB-231 breast cancer cells, while hypercalcemic environments did not affect interaction. Meanwhile, hypercalcemia leads to enhanced cancer cell invasion for both MDA-MB-231 and A549 cells in the presence of platelets. Thus, this study provides a greater understanding of the dynamics associated with the effects of calcium and platelet-cancer cell interactions mediated by integrins.
Similar content being viewed by others


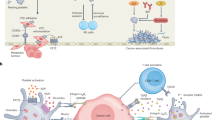
Introduction
Most cancer related deaths from solid tumors are the result of metastatic dissemination. Specifically, 66.7% of registered cancer deaths from solid tumors identify metastases as a contributing factor1. Although their primary role is to maintain hemostasis, platelets are implicated in cancer progression by interacting with circulating tumor cells (CTCs)2. Upon interacting with tumor cells that have entered the bloodstream, platelets support CTCs by protecting them from shear stress, promoting immune cell invasion (e.g., NK cells), and promoting their arrest within the vasculature3,4. In addition, platelet depletion in murine models leads to a significant decrease in metastatic dissemination5. Therefore, platelet interaction with CTCs is necessary for successful dissemination and metastatic development.
Platelets and cancer cells can interact via integrins present on their surface and bridged via an integrin binding protein such as fibrinogen or via direct receptor-receptor interaction (e.g. CLEC-2 on platelets and podoplanin on cancer cells)2. ⍺IIbβ3 is the most abundant integrin on platelets and is the mediator for platelet aggregation6. Integrin ⍺vβ3 is also expressed on platelets and is an important integrin for cancer-associated angiogenesis7. Similar to platelets, both ⍺IIbβ38,9 and ⍺vβ3 are expressed on some cancer cells10,11. ⍺IIbβ3 and ⍺vβ3 share the same β subunit and their ⍺ subunit are 36% identical12. Like platelet-platelet interaction, the interaction of platelets and cancer cells via these two specific integrins is mediated via bridging proteins (e.g., fibrinogen, vWF, fibronectin). Divalent cations (e.g., calcium) are essential to the structural stability and function of integrins13. Divalent cation removal via chelators (e.g. sodium citrate, ethylenediaminetetraacetic acid (EDTA)) leads to disassociation of the integrin heterodimer complex14,15.
Calcium is also essential in the coagulation cascade which is implicated in metastatic progression16. We hypothesized that divalent cation levels, critically calcium, have an essential role in the function of the key integrin proteins responsible for platelet-cancer cell interaction and this could be leveraged to disrupt platelet support to the metastatic process. In addition, 20–30% of advanced cancer patients suffer from hypercalcemia, and they typically have a worse prognosis17,18,19.
In this study, we aimed to better understand how calcium level can impact platelet-cancer cell interactions, as well as cancer cell invasion in vitro.
Materials and methods
Materials
APC mouse anti-human CD41a (cat# 559777), APC mouse IgG, ĸ isotype control (cat# 555751), Alexa Fluor® 647 mouse anti-human platelet GPVI (cat# 564701), Alexa Fluor® 647 mouse IgG1 κ isotype control (cat# 557714), FITC mouse anti-human CD42b (cat# 555472), FITC mouse IgG1, κ isotype control (cat# 555748), FITC mouse anti-human PAC-1 (cat# 340507), FITC mouse IgM, κ isotype control (cat# 551448) were purchased from BD Biosciences. FITC mouse anti-human CD51 (cat# 327908), FITC mouse IgG2a, κ isotype control (cat# 400209), APC mouse anti-human CD61 (cat# 336412), and APC mouse IgG1, κ isotype control (cat# 400121) were purchased from BioLegend. The adenosine diphosphate (ADP) (cat# 384) and fibrillar type I collagen (cat# 385) were obtained from Chrono-Log. Thrombin receptor agonist peptide (TRAP) (cat# 1185) and GW 4869 (cat# 6741) were obtained from Tocris. The lactate dehydrogenase (LDH)-Glo cytotoxicity assay (cat# J2380) was purchased from Promega. CellMask deep red plasma membrane stain (cat# C10046) and Hoechst 33342 (cat# H3570) were obtained from Invitrogen. Bovine fibrinogen (cat# J63276-03) was obtained from Thermo Scientific Chemicals. Bovine plasma fibronectin (cat# 8248) was obtained from ScienCell Research Laboratories. Cultrex UltiMatrix Reduced Growth Factor (RGF) Basement Membrane Extract (BME) (cat# 3445-005-01) was purchased from Bio-techne. EDTA (10% w/v) (cat# 2670-4) and sodium citrate (10% w/v) (cat# 723016) were obtained from Ricca Chemical Company. Calcium chloride (CaCl2) (cat# C1016), crystal violet (cat# V5265), acetic acid (cat# A628), and Bovine Serum Albumin (cat# 7030) were obtained from Sigma Aldrich. Triton™ X-100, 98% (cat# AC327371000) was purchased from Acros Organics.
Human blood processing and platelet isolation
Healthy human whole blood was purchased from BioIVT (Westbury, NY) following approval from the Institutional Biosafety Committee at the George Washington University. Whole blood was collected with sodium heparin and shipped overnight before being processed for our experiments the following day. Whole blood was centrifuged at 150 g for 20 min to pellet red blood cells and the platelet rich plasma (PRP) was collected. To obtain platelet poor plasma (PPP), the remaining blood fraction after PRP removal was centrifuged at 2,000 g for 20 min. Platelet concentrate (PC) was obtained by centrifuging PRP at 1,000 g for 20 min. 140 nM prostaglandin E1 (PGE1) was incubated 10 min with the PRP prior to the centrifugation at 1,000 g. After removal of the supernatant, the resulting platelet pellet was resuspended in tyrode buffer (136 mM NaCl, 12 mM NaHCO3, 2.9 mM KCl, 0.34 mM Na2HPO4, 1 mM MgCl2, and 10 mM HEPES buffer, pH 7.4). Platelet concentration was determined using the Novocyte flow cytometer (Agilent).
Cell culture
MDA-MB-231 and A549 cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA). Both cell lines were maintained in Dulbecco’s Modified Eagle Medium (DMEM) with high glucose and pyruvate (Gibco, cat# 11995065) supplemented with 10% FBS (fetal bovine serum) and 1% penicillin–streptomycin (Gibco, cat# 15140122). Both cell lines were maintained at 37 °C with 90% humidity and 5% CO2.
Receptor characterization by flow cytometry
To characterize receptor expression, platelets, MDA-MB-231 cells, and A549 cells were analyzed via flow cytometry. Platelets and MDA-MB-231 cells were assessed for GPIIb (⍺IIb) and GPIIIa (β3) detection using an APC mouse anti-human CD41a and an APC mouse anti-human CD61 antibody, respectively. Platelets were also assessed for GPVI and GPIb⍺ detection using an Alexa Fluor® 647 mouse anti-human GPVI antibody and a FITC mouse anti-human CD42b antibody, respectively. A549 cells were assessed for ⍺v and β3 detection using a FITC mouse anti-human CD51 antibody and an APC mouse anti-human CD61 antibody, respectively. Platelets, MDA-MB-231 cells, and A549 cells were incubated with treatment (sodium citrate, EDTA, or calcium chloride) for 1 h at 37 °C. 1 × 106 platelets in Tyrode buffer were incubated with treatment conditions followed by the addition of 2% BSA in Tyrode. To characterize ⍺IIb and β3, platelets were fixed with 2% paraformaldehyde following the 1 h incubation with treatment conditions. Following a 10 min incubation, platelets were incubated with 2% BSA in Tyrode for 10 min prior to a 20 min incubation with the antibody. Regarding the activated form of ⍺IIbβ3, platelets were treated 10 min with 20 uM ADP following the 1 h incubation with treatment conditions. Platelets were then incubated with FITC-conjugated mouse anti-human PAC-1 for 20 min following a blocking step using 2% BSA in Tyrode for 10 min. The samples were then fixed with 2% paraformaldehyde and analyzed with the Novocyte flow cytometer. Characterization of GPIb⍺ and GPVI were performed similarly. To access receptor detection on MDA-MB-231 and A549 cells, 1 × 105 cells resuspended in calcium-deprived DMEM (Gibco, cat# 21068-028) incubated with treatment conditions followed by a centrifugation at 1,200 rpm for 5 min. Each cell pellet was then resuspended in 2% BSA in PBS and incubated for 20 min on ice with the antibody. The samples were then centrifuged at 1,200 rpm for 5 min, resuspended in calcium-deprived DMEM and analyzed with the Novocyte flow cytometer. To block exosome biogenesis, MDA-MB-231 cells incubated with 0.5 uM GW 4869 for 1 h at 37 °C before being incubated with treatment conditions20,21. Samples remained at the appropriate treatment concentration at each step during the experiment. Heparin anti-coagulated PRP was assumed to have a normal calcium level of 9 mg/dL22. APC-conjugated mouse IgG, ĸ isotype, APC-conjugated mouse IgG1, ĸ isotype, FITC-conjugated mouse IgM, κ isotype, FITC-conjugated mouse IgG1, κ isotype, Alexa Fluor® 647 conjugated mouse IgG1 κ isotype, and FITC-conjugated mouse IgG2a, κ isotype were used as controls for non-specific binding.
Platelet aggregation by light transmission aggregometry (LTA)
Platelet aggregation was assessed using a Model 490 4 + 4 aggregometer from Chrono-Log (Havertown, PA). Platelet aggregation was measured for 15 min under constant stirring at 1,200 rpm at 37 °C at a platelet concentration of 2 × 105 platelets/uL. ADP and TRAP at concentrations of 10 uM and 5 uM were used as agonists. Aggregation was measured immediately after adding treatment (3.8% Na citrate, 0.18% EDTA, 11 mg/dL Ca2+, 13 mg/dL Ca2+, 16 mg/dL Ca2+, 18 mg/dL Ca2+) at 37 °C. 3.8% Na citrate and 0.18% EDTA are common vacutainer concentrations used for blood sample collection.
Platelet adhesion assay
A protocol using a LDH assay was designed to assess the adhesion of platelets on collagen, fibrinogen, and fibronectin following a 1 h incubation with treatment (sodium citrate, EDTA, or CaCl2) at 37 °C. 1 mg/mL fibrillar collagen solution was diluted 1:10 with phosphate buffered saline (PBS). Fibrinogen and fibronectin were diluted to 100 ug/mL with PBS. 100 uL of either collagen, fibrinogen, or fibronectin were added per well in a 96-well plate and incubated for 1 h at room temperature. The wells were then gently rinsed one time with PBS after removing the substrate solution. 100 uL of the treated PRP mixtures at 2 × 105 platelets/uL were added to the collagen-coated wells. 1.5 × 105 platelets/uL were added to fibrinogen- and fibronectin-coated wells. Untreated PRP, 2 × 105 platelets/uL or 1.5 × 105 platelets/uL, and uncoated wells (blank) were used as controls for the assay. Following the 2 h incubation, the PRP suspension was removed, and the wells gently rinsed three times with PBS. 50 uL of 10% triton X-100 was added to each well and incubated for 20 min to permeabilize the adhered platelets. 50 uL of the LDH detection solution from the Promega LDH-Glo cytotoxicity assay kit was added to each well and incubated for 30–60 min. The luminescence was measured using a SpectraMax iD5 plate reader (Molecular Devices). The concentration of platelets in each well was calculated using a standard curve created from known concentrations of lysed platelets.
Cancer cell-platelet interaction by flow cytometry
The effect of calcium chelation and hypercalcemia on cancer cell-platelet interaction was assessed using flow cytometry. In brief, 500 uL PC (2 to 5 × 106 platelets/uL) was stained with CellMask deep red plasma membrane stain at a 1:1,000 dilution and incubated for 1 h under agitation at 37 °C. The APC fluorescence signal of the stained platelets was assessed on the flow cytometer. The stained PC was then washed three times in tyrode buffer by centrifuging at 1,000g for 20 min. MDA-MB-231 and A549 cells were stained with Hoechst 33342 at a 1:10,000 dilution for 5 min at 37 °C. The cells were then washed twice and resuspended in calcium-deprived DMEM. For conditions with 1% sodium citrate, 3.8% sodium citrate and 0.18% EDTA, cancer cells and platelets incubated at a ratio of 1:500 for 1 h at 37 °C under gentle agitation. Samples were washed twice via centrifugation at 1,200 rpm for 10 min to remove excess platelets. For the hypercalcemic condition (18 mg/dL Ca2+), cancer cells and platelets were mixed at ratios of 1:1, 1:2.5, 1:5 and immediately centrifuged twice at 1,200 rpm for 10 min. Samples were analyzed on the flow cytometer. The level of cancer cell-platelet interaction was determined by percentage of the population that was positive for APC (CellMask deep red) and Pacific Blue (Hoechst 33342). Samples remained at the appropriate treatment concentration at each step during the experiment. Cancer cells and platelets not treated with sodium citrate, EDTA, or calcium were used as controls at the appropriate ratios. Controls for unspecific staining were prepared for each ratio by first incubating the same number of CellMask deep red stained platelets in DMEM at the appropriate time. The samples were then centrifuged at 2,000 g for 20 min in a 2mL Amicon Ultra Centrifugal filter (Millipore, cat# UFC210024). The filtrate was then collected and allowed to incubate at the appropriate time with the same number of cancer cells. The cancer cells were washed twice via centrifugation at 1,200 rpm for 10 min similarly to the other conditions.
Cancer cell invasion
The effect of calcium chelation and hypercalcemia on cancer cell invasion, with and without platelets (PC), was assessed using a transwell assay. Invasion assays were performed in 24-well polyethylene (PET) tissue culture treated membrane cell inserts (8 µm pore size) (Nest Biotechnology, cat#725321). The inserts were coated with 50 uL of Cultrex UltiMatrix RGF BME (BioTechne, cat# BME001-05). 150,000 MDA-MB-231 or 100,000 A549 cells were plated in the inserts after being treated with (1) 1% sodium citrate, (2) platelets (1:300 cancer cell to platelet ratio) and 1% sodium citrate, (3) 0.18% EDTA, (4) platelets (1:300 ratio) and 0.18% EDTA, (5) 18 mg/dL Ca2+, or (6) platelets (1:300 ratio) and 18 mg/dL Ca2+. Untreated cancer cells were used as a control. Cancer cells with or without platelets were incubated with treatment for 15 min at 37 °C under gentle agitation. The bottom chamber contained a mixture of calcium- and FBS-deprived DMEM, PRP, and treatment (chelation or CaCl2). After an invasion time of 6 h, the upper chambers were cleaned with a cotton swab moistened with PBS, and then rinsed with PBS. The inserts were fixed with 4% paraformaldehyde for 15 min and washed with PBS. The inserts were then stained with 0.25% crystal violet for 15 min and washed with PBS. They were placed in 33% acetic acid for 30 min with agitation to lyse and release the crystal violet from the invaded cells adhered to the bottom of the inserts. The outer surface of the bottom of the inserts was gently scraped with a mini spatula to ensure that all invaded cells were effectively removed/lysed. 200 uL of the acetic acid solution from each well was transferred to a 96-well plate. The absorbance at 590 nm was measured with the SpectraMax iD5 plate reader. The number of invaded cells was calculated using a standard curve generated from known cell densities.
Statistical analysis
All graphical representations and statistical analysis were completed with GraphPad Prism software. A one-way ANOVA test followed by Dunnett’s or Šidák’s post hoc multiple comparisons test was used to determine statistical significance (*P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, and **** P ≤ 0.0001). All figures display the mean ± SEM for each group.
Results
Detection of key receptors on platelets
⍺IIbβ3, GPIb⍺, and GPVI are key receptors implicated in platelet function, platelet aggregation, and platelet-cancer cell interactions. To assess the effect of decreased or increased calcium levels on these receptors, their detection was characterized after treating platelets with chelating agents or CaCl2 for 1 h. Platelets ⍺IIb (CD41a) surface detection was significantly decreased after treatment with chelating agents, sodium citrate and EDTA, regardless of concentration (Fig. 1A, S-1A, P < 0.0001). ⍺IIb surface detection significantly increased for hypercalcemic conditions (Fig. 1A, S-1A, P < 0.0214). EDTA addition increased platelet β3 surface detection, with significance being noted at the higher concentration of 0.18% (Fig. 1B, S-1B, P < 0.0001). Hypercalcemic treatment conditions did not modulate β3 surface detection on platelets.

Characterization of the level of antibody binding on the platelet surface. Quantification of detection level following incubation with sodium citrate, EDTA, and Ca2+. Representative histograms are provided in supplementary Fig. 1. (A) CD41a (⍺IIb) [2 donors, n = 4], (B) CD61 (β3) [2 donors, n = 6], (C) PAC-1 (activated CD41a or ⍺IIb) [1 donor, n = 3], (D) CD42b (GPIb⍺) [2 donors, n ≥ 6], (E) GPVI [2 donors, n ≥ 6].
Following platelet activation with 20 uM ADP, platelets exposed to a high concentration of sodium citrate had a significant decrease in detection of active conformation of ⍺IIbβ3 (PAC-1 detection) compared to the ADP control, while calcium addition significantly increased platelet activation, regardless of calcium concentration (Fig. 1C, S-1C, P < 0.0001).
0.5% and 2% Na citrate treated platelets led to a significantly increase in GPIb⍺ (CD42b) surface detection (Fig. 1D, S-1D, P = 0.0060 and P = 0.0084, respectively). Contrary, platelets treated with 0.18% and 0.5% EDTA had significantly less GPIb⍺ (CD42b) surface detection (Fig. 1D, S-1D, P < 0.0001). Hypercalcemic conditions also led to significantly less GPIb⍺ (CD42b) surface detection (Fig. 1D, S-1D, P < 0.0468).
Platelet GPVI surface detection was significantly increased following all chelation treatments (Fig. 1E, S1-E, P < 0.0343), while calcium addition did not modulate GPVI surface detection.
Detection of integrins on MDA-MB-231 human breast cancer cell line
Integrin ⍺IIbβ3 expressed on some cancer cells also mediates platelet-cancer cell interactions, thus playing a key role in platelet promoted tumor progression and metastasis. To further assess the effect of calcium on sub integrins ⍺IIb and β3, their detection was characterized after MDA-MB-231 was incubated with chelating agents, or CaCl2. Calcium depletion with both sodium citrate and EDTA significantly decreased ⍺IIb (CD41a) surface detection on MDA-MB-231 cells (Fig. 2A, S-2A, 0.5% sodium citrate: P = 0.0004; 2% sodium citrate: P < 0.0001; 0.18% EDTA: P < 0.0001, 0.5% EDTA: P < 0.0001). Hypercalcemic conditions did not modulate ⍺IIb (CD41a) surface detection. MDA-MB-231 preincubation with exosome biogenesis inhibitor, GW 4869, resulted in a significant increase of ⍺IIb (CD41a) surface detection for 2% sodium citrate treated samples (Fig. 2A, S-2A, P < 0.0001 vs. 2% sodium citrate without GW 4869). β3 (CD61) surface detection on MDA-MB-231 was decreased with calcium depletion (Fig. 2B, S-2B, 0.5% Na citrate: P = 0.0383; 0.18% EDTA: P = 0.0442). In addition, hypercalcemic conditions resulted in a significant decrease in β3 (CD61) surface detection (Fig. 2B, S-2B, P < 0.0161).

Characterization of the level of integrin antibody binding on MDA-MB-231. Quantification of integrin detection following incubation with sodium citrate, EDTA, and Ca2+, with and without GW 4869, an inhibitor of exosome biogenesis. Representative histograms are provided in supplementary Fig. 2. (A) CD41a (⍺IIb) [n ≥ 5], (B) CD61 (β3) [n ≥ 5].
Detection of integrins on A549 human lung cancer cell line
Similarly, integrin αvβ3 that is expressed on some cancer cells mediates platelet-cancer cell interactions, thus contributing to the metastatic process. The detection of integrin subunits CD51 (αv) and CD61 (β3) was characterized after treating A549 with chelating agents or CaCl2. A549 CD51 (αv) surface detection significantly decreased following treatment with all chelation conditions, except 2% Na citrate (Fig. 3A, S-3A, P < 0.0001). Hypercalcemic conditions significantly increased CD51 (αv) surface detection on A549 (Fig. 3A, S-3A, P < 0.0399). β3 (CD61) detection was not significantly modulated regardless of treatment (Fig. 3B, S-3B).

Characterization of the level of integrin antibody binding on A549. Quantification of detection level following incubation with sodium citrate, EDTA, and Ca2+. Representative histograms are provided in supplementary Fig. 3. (A) CD51 (⍺v) [n ≥ 5], (B) CD61 (β3) [n ≥ 5].
Aggregation of platelets with calcium chelation and hypercalcemia
Platelet aggregation is an important platelet function that also contributes to key aspects of the metastatic process (e.g. protecting CTCs as they travel through the bloodstream, promoting CTC arrest in the vasculature and extravasation). The effect of calcium depletion and hypercalcemia on platelet aggregation was assessed using LTA immediately after adding chelating agents or CaCl2. 10 uM ADP and TRAP were used as platelet agonists for calcium chelating conditions. However, lower concentrations (5 uM ADP and TRAP) were used for hypercalcemic conditions to better observe gradual increases in platelet aggregation with increments of calcium concentrations. A complete inhibitory effect on platelet aggregation was observed with both, 3.8% Na citrate (P < 0.0001) and 0.18% EDTA (P < 0.0001) (Fig. 4A,B) with 10 uM ADP. Platelet aggregation was significantly increased for severe hypercalcemia, 18 mg/dL Ca2+ (P = 0.0012) (Fig. 4C,D) with 5 uM ADP. Also, an increasing trend in platelet aggregation was observed at 13 mg/dL Ca2+ and 16 mg/dL Ca2+, though not statistically significant (Fig. 4C,D). A similar complete inhibitory effect was observed with 10 uM TRAP for both chelating agents (P < 0.0001) (Fig. 4E,F). Platelet aggregation was significantly increased immediately after adding treatment for 13, 16, and 18 mg/dL Ca2+ (P < 0.0005) with 5 uM TRAP.

Aggregation of platelets in the presence of 3.8% sodium citrate, 0.18% EDTA, and 11 to 18 mg/dL Ca2+. Quantification (left) and representative LTA curves (right) for the maximum aggregation of platelets using a normal platelet concentration of 200,000 platelets/uL. (A, B) 10uM ADP [4 donors, n ≥ 4], (C, D) 5uM ADP [4 donors, n = 7], (E, F) 10uM TRAP [6 donors, n ≥ 16], (G, H) 5uM TRAP [2 donors, n ≥ 5].
Adhesion of platelets with calcium chelation and hypercalcemia
To further understand the effect of calcium depletion and hypercalcemia on platelet functions, platelet adhesion to three thrombogenic surfaces was examined. Platelet GPVI plays a key role in collagen-induced platelet activation. Following platelet activation and adhesion to collagen, further platelet activation leads to aggregation and formation of the platelet plug. Platelet tethering on collagen is mediated via GPIb-IX-V, while firm adhesion involves the recruitment of GPVI, GPIIb-IIIa, and ⍺2β112. GPIIb-IIIa, ⍺vβ3, and ⍺5β1 are able to bind to both fibrinogen and fibronectin23,24,25,26. Platelet GPIIb-IIIa plays a key role in adhesion of activated platelets to fibrinogen which mediates platelet aggregation27. A LDH fluorescence assay was used to assess platelet adhesion to collagen, fibrinogen, and fibronectin following a 1 h incubation with Na citrate, EDTA, and calcium at various concentrations. There was no statistical difference in platelet adhesion to collagen between the conditions with calcium depletion or hypercalcemia in comparison to the control (Fig. 5A). In contrast, 2% Na citrate, 0.18% EDTA, and 0.5% EDTA significantly decreased platelet adhesion to fibrinogen (Fig. 5B, P < 0.0001, P < 0.0001, P = 0.0013, respectively). 0.5% EDTA significantly decreased platelet adhesion to fibronectin (Fig. 5C, P = 0.0291). A decreasing trend was observed for 2% Na citrate and 0.18% EDTA, though not significant compared to the control (Fig. 5C). Hypercalcemia conditions did not significantly modulate platelet adhesion to either fibrinogen or fibronectin (Fig. 5B,C).

Adhesion of platelets on thrombogenic surfaces. Normalized quantification of adhesion of platelets following incubation with sodium citrate, EDTA, and Ca2+ at various concentrations. (A) Collagen surface [6 donors, n ≥ 3], (B) Fibrinogen surface [3 donors, n ≥ 4], (C) Fibronectin surface [3 donors, n ≥ 4].
Cancer cell interaction with platelets
Cancer cell-platelet interaction was assessed to corroborate the results observed for the effects of calcium levels on platelet function and the key receptors that mediate platelet-cancer cell interaction. A flow cytometry assay was used to study the direct effect of calcium chelation and hypercalcemia on the interaction of cancer cells and platelets.
Calcium depletion via 3.8% Na citrate significantly decreased MDA-MB-231 and platelet interaction (P < 0.0001), while 0.18% EDTA did not (Fig. 6A, S-4A, S-5A-C). However, a downward trend was observed with 0.18% EDTA. Calcium depletion via 1% Na citrate did not modulate this interaction. Interestingly, calcium depletion did not modulate A549 and platelet interaction (Fig. 6B, S-4B, S-5A-C).

Platelet – cancer cell interaction assessed via flow cytometry. Quantification for the interaction of platelets and cancer cells in the presence of 1% sodium citrate, 3.8% sodium citrate, 0.18% EDTA, and 18 mg/dL Ca2+. Representative flow cytometry plots are provided in supplementary Fig. 4. (A) Platelets + MDA-MB-231 using a 1:500 MDA-MB-231 to platelet ratio [12 donors, n ≥ 5], (B) Platelets + A549 using a 1:500 A549 to platelet ratio [4 donors, n ≥ 9], (C) Platelets + MDA-MB-231 using a 1:1, 1:2.5, and 1:5 MDA-MB-231 to platelet ratio [3 donors, n ≥ 5], (D) Platelets + A549 using a 1:1, 1:2.5, and 1:5 A549 to platelet ratio [4 donors, n ≥ 3].
Adding more platelets (from 1:1 to 1:5 cancer cell-to-platelet ratio) led to a significant increase in MDA-MB-231 and platelet interaction (Fig. 6C, S-4C, S-5A-C, P = 0.0085). However, a hypercalcemic environment did not significantly affect MDA-MB-231 and platelet interaction (Fig. 6C, S-4C, S-5A-C). Similar trends were observed with A549 and hypercalcemic conditions did not alter their interaction with platelets (Fig. 6D, S-4D, S-5A-C).
Assessment of cancer cell invasion with calcium modulation
To further understand the effect of calcium on platelet-cancer cell interactions and the pivotal step in the metastatic cascade, cancer cell invasion was assessed using a transwell assay. Treating MDA-MB-231 with 1% Na citrate, 0.18% EDTA, and 18 mg/dL Ca2+ did not significantly modulate MDA-MB-231 invasion, but a downward trend was observed for the chelation conditions while a positive trend was observed with calcium addition (Fig. 7A). However, MDA-MB-231 invasion with platelets was significantly increased in the presence of 18 mg/dL of calcium (Fig. 7A , P= 0.0140 vs. platelets). The hypercalcemic condition was significantly enhancing invasion in the presence of platelets (Fig. 7A, P= 0.0163 vs. calcium). Similar trends were observed with A549 invasion (Fig. 7B).

Cancer cell invasion without and with platelets in chelation or hypercalcemic conditions. (A) MDA-MB-231 invasion (3 donors and 3 independent experiments, n = 9), (B) A549 invasion (3 donors and 3 independent experiments, n = 9).
Discussion
The objective of this study was to further understand how calcium affects platelet-cancer cell interaction and tumor cell invasion. The first phase of this study was the detection of the key receptors of interest, ⍺IIbβ3 and αvβ3, available on the surface of platelets and cancer cells following treatment with chelating agents or calcium at various concentrations. The resulting data shows that the surface detection of the most abundant platelet sub-integrin, ⍺IIb (CD41a), which is essential for platelet-platelet aggregation and tumor cell-induced platelet aggregation28, is significantly decreased when there is a lack of calcium or divalent cations (Fig. 1A, S-1A). This is in agreement with literature as past observations have shown that under calcium chelation, the alpha subunit of platelet ⍺IIbβ3 structurally collapses leading to the separation of the alpha and beta subunits, further inhibiting platelet aggregation29. Interestingly, platelet β3 (CD61) detection was unaffected with calcium/divalent cation depletion via sodium citrate, while it was increased with depletion via EDTA (Fig. 1B). The increase in β3 detection with EDTA treated platelets agrees with previous studies that have shown that the binding of anti-CD61 is increased in the presence of EDTA, even following dissociation of the ⍺IIbβ3 complex with EDTA30,31,32,33. Our results have shown that effect of calcium/divalent cation levels is more significant on the alpha subunit, compared to the beta subunit, of the ⍺IIbβ3 integrin on platelets.
Interestingly, neither calcium/divalent cation chelation, nor calcium addition, activates platelet ⍺IIbβ3 integrin at baseline as suggested by our PAC-1 data (Fig. 1C).
Our results also indicate that calcium addition enables the recruitment of ⍺IIbβ3 at the platelet surface (Fig. 1A, S-1A, P < 0.0214), which remain in inactive conformation at baseline (Fig. 1C, red plain symbols), however, which enables an exacerbated ⍺IIbβ3 activation in response to platelet agonists such as ADP (Fig. 1C, red open symbols). This also translates in a significantly enhanced response of platelet aggregation in calcium addition conditions (Fig. 4C,D, 4G,H). Together these results highlight for the first time that a hypercalcemic microenvironment primes platelets for deploying a significantly enhanced response in pro-thrombotic conditions.
In comparison to platelets, both the detection of the alpha and beta subunits of integrin ⍺IIbβ3 in MDA-MB-231 cells tend to decrease with both calcium chelation and calcium addition (Fig. 2A-B, S-2A-B). This trend became significant for the ⍺IIb subunit with chelation, and for the β3 subunit with both chelation or in the presence of calcium addition (Fig. 2A-B, S-2A-B). We hypothesized that the observed reduced detections correlate with the production of MDA-MB-231-derived ⍺IIb/β3 positive exosomes. Indeed, CaCl2 incubation has been shown to increase intracellular calcium in MDA-MB-231 cells with a subsequent increase in the production of cancer cell-derived exosomes34. A study also highlights that exosome production by ovarian cancer cells is promoted in calcium chelated environments35. This was confirmed for the 2% sodium citrate treated samples, where ⍺IIb (CD41a) expression shifted from significantly decreased (Fig. 2A, green plain symbols, P < 0.0001 vs. control), to significantly increased when the sample was preincubated with the exosome biogenesis inhibitor GW 4869 (Fig. 2A, green open symbols, P < 0.0001 vs. Na citrate). In contrast, the same concentration of GW 4869 failed to modulate the expression of ⍺IIb (CD41a) in EDTA treated samples (Fig. 2A).
Platelet adhesion was assessed to understand the effect of calcium on another key aspect of platelet function. Contrary to the effect of calcium levels on platelet activation and aggregation, our results demonstrated that neither increasing or depleting calcium modulates platelet adhesion to collagen. This was corroborated by similar or even increased levels of GPVI across calcium addition and chelation, respectively (Fig. 1E). GPVI is involved in platelet activation via collagen and is also involved in the consolidation of platelet adhesion on collagen, along with ⍺IIbβ3 and ⍺2β1. However, it has been reported that GPVI, not ⍺2β1 is essential for platelet interaction with collagen36. Because GPVI is not directly involved in platelet tethering37, the observed significant increase in GPVI detection for some conditions does not correlate with increased adhesion, but could rather indicate a stronger/firmer consolidation of platelet adhesion. Our results demonstrate that GPVI is still present on the platelet surface, thus facilitating platelet interaction with collagen and consequential platelet activation and adhesion. Meanwhile, GPIb⍺ detection significantly decreased in the presence of the treatment with EDTA and calcium (Fig. 1D). GPIb is the subunit of GPIb-IX-V that binds to vWF during platelet tethering on exposed collagen38. Despite the decrease in GPIb⍺ detection, the lack of effect on adhesion would indicate that this drop is not functionally significant in altering platelet activity and that sufficient levels of GPIb⍺ are still present for platelet tethering to collagen via vWF. In contrast to the results observed for platelet adhesion to collagen, calcium depletion reduced platelet adhesion to both, fibrinogen (Fig. 5B) and fibronectin (Fig. 5C) with a dose dependent effect. Compared to the control, calcium depletion via EDTA presented a more significant downward trend of platelet adhesion to fibrinogen and fibronectin compared to the trend observed with sodium citrate. Indeed, EDTA has a higher cation binding constant and is a stronger chelator than sodium citrate39,40. The similar trend observed for the effect of calcium depletion on platelet adhesion to fibrinogen and fibronectin, could be explained by the fact that platelet ⍺IIbβ3, αvβ3, and α5β1 are responsible for platelet binding to these substrates via the fibrinogen binding domain on ⍺IIbβ323 and the RGD affinity of both αvβ3 and α5β1 integrins41. Because ⍺IIbβ3 is the most abundant receptor on platelets, the decrease in platelet adhesion to fibrinogen and fibronectin with calcium depletion corroborates with our results observed for the effect of calcium chelation on platelet ⍺IIb (Fig. 1A-B). Direct platelet-cancer cell interaction was then assessed via flow cytometry. The results demonstrated that calcium depletion via sodium citrate at a concentration of 3.8% significantly decreases platelet-MDA-MB-231 interaction (Fig. 6A). This could potentially be explained by the structural or conformational change of the integrin in response to calcium depletion as suggested by the receptor assessments for both platelets and cancer cells, as well as literature14,15. Disrupting the integrin complexes on both cells would indeed disrupt platelet-MDA-MB-231 interaction. Meanwhile, although hypercalcemic conditions increased ⍺IIb on platelets (Fig. 1A), this does not promote increased platelet-MDA-MB-231 interaction in our experimental conditions (Fig. 6C). Interestingly, neither calcium depletion nor severe hypercalcemia modulates platelet-A549 interactions (Fig. 6B,D). Although detection of the ⍺v subunit of ⍺vβ3 was significantly decreased with EDTA and a downward trend of the β3 subunit was observed in calcium chelating environments, this decreased detection does not impact platelet-A549 interaction. It could imply that the A549 ⍺vβ3 integrin is not essential in the A549-platelet interaction. In addition, our experimental conditions emulating platelet-cancer cell interactions are limited by the two following aspects: the need for at least 10,000 events to be recorded on the flow cytometer and selected platelet-cancer cell ratios that do not induce clotting in our samples, which necessitated the use of non-physiological ratios.
Tumor cell invasion is a significant step in the metastatic cascade as tumor cell ability to penetrate the basement membrane and disseminate as CTCs initiates the eventual formation of metastatic sites19,20. Our results corroborate with previous studies that platelets tend to promote tumor cell invasion. Additionally, it is described in literature that activated platelets and platelet-cancer cell interactions can enhance tumor cell invasion by increasing the expression of pro-invasion transcription factors such as NF-κB2,42,43. Meanwhile, a hypercalcemic environment tends to increase cancer cell invasion and this invasion is even more exacerbated in the presence of platelets (Fig. 7). Our results also implicate that hypercalcemia creates an environment that promotes additional platelet support to tumor cell invasion and could potentially contribute to the poor prognosis of cancer patients also suffering from hypercalcemia44. A severe hypercalcemic environment promoting the invasion of cancer cells in the presence of platelets further emphasizes both the contribution of hypercalcemia and platelet-cancer cell interactions to metastasis.
Furthermore, the inability to use fresh blood samples for our platelet studies is not ideal for platelet functional testing and serves as a limitation in our study. Although platelets in our study may not fully replicate the functional characteristics of fresh platelets, stored platelets retain their biological activity as evidenced by the clinical use of platelets up to five days following apheresis collection.
Conclusion
The results of this study have demonstrated that platelet function and platelet-cancer cell interactions are calcium dependent. Upon fully understanding the effect that calcium levels have on platelet-cancer cell interaction and the key receptors involved in platelet-cancer cell interaction, this knowledge could then be leveraged to disrupt platelet-circulating tumor cell interactions that are allies in metastatic progression. Although the scope of this study mainly focused on the effect of calcium levels on integrins, chelation via sodium citrate and EDTA not only affects calcium ions, but also additional metal ions which are essential for receptor function and structural stability (i.e. Mg2+ and Mn2+)15.
Importantly, we also demonstrated for the first time that a hypercalcemic environment enables the recruitment of ⍺IIbβ3 at the platelet surface, which remain in an inactive conformation at time of recruitment. However, these receptors have an exacerbated response to platelet agonists such as ADP. In addition, hypercalcemia and platelets enhance cancer cell invasion more than either factor in isolation. These novel results demonstrate that hypercalcemia in patients can potentially promote increased platelet activation and the subsequent formation of platelet-tumor cell aggregates, which could be a factor in increased risk of cancer-associated thrombosis and increased metastatic dissemination.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Dillekås, H., Rogers, M. S. & Straume, O. Are 90% of deaths from cancer caused by metastases?. Cancer Med. 8(12), 5574–5576. https://doi.org/10.1002/cam4.2474 (2019).
Morris, K., Schnoor, B. & Papa, A.-L. Platelet cancer cell interplay as a new therapeutic target. Cancer 1877(5), 188770. https://doi.org/10.1016/j.bbcan.2022.188770 (2022).
Schlesinger, M. Role of platelets and platelet receptors in cancer metastasis. J. Hematol. Oncol. 11(1), 125. https://doi.org/10.1186/s13045-018-0669-2 (2018).
Labelle, M. & Hynes, R. O. The initial hours of metastasis: The importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov. 2(12), 1091–1099. https://doi.org/10.1158/2159-8290.CD-12-0329 (2012).
Camerer, E. et al. Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood 104(2), 397–401. https://doi.org/10.1182/blood-2004-02-0434 (2004).
Wagner, C. et al. Analysis of GPIIb/IIIa receptor number by quantification of 7E3 binding to human platelets. Blood 88(3), 907–914. https://doi.org/10.1182/blood.V88.3.907.907 (1996).
Liu, Z., Wang, F. & Chen, X. Integrin Alpha(v)Beta(3)-targeted cancer therapy. Drug Dev. Res. 69(6), 329–339. https://doi.org/10.1002/ddr.20265 (2008).
Oleksowicz, L. et al. Characterization of tumor-induced platelet aggregation: The role of immunorelated GPIb and GPIIb/IIIa expression by MCF-7 breast cancer cells. Thromb. Res. 79(3), 261–274. https://doi.org/10.1016/0049-3848(95)00113-6 (1995).
Strasenburg, W. et al. Tumor cell-induced platelet aggregation as an emerging therapeutic target for cancer therapy. Front. Oncol. 12, 909767. https://doi.org/10.3389/fonc.2022.909767 (2022).
Lavergne, M., Janus-Bell, E., Schaff, M., Gachet, C. & Mangin, P. H. Platelet integrins in tumor metastasis: Do they represent a therapeutic target?. Cancers 9(10), 133. https://doi.org/10.3390/cancers9100133 (2017).
Alonso-Escolano, D., Strongin, A. Y., Chung, A. W., Deryugina, E. I. & Radomski, M. W. Membrane type-1 matrix metalloproteinase stimulates tumour cell-induced platelet aggregation: Role of receptor glycoproteins. Br. J. Pharmacol. 141(2), 241–252. https://doi.org/10.1038/sj.bjp.0705606 (2004).
Platelets, 3rd ed.; Michelson, A. D., Ed.; Academic Press: London ; Waltham, MA, (2013).
Haas, T. A. & Plow, E. F. The cytoplasmic domain of: A ternary complex of the integrin α AND β subunits and a divalent cation. J. Biol. Chem. 271(11), 6017–6026. https://doi.org/10.1074/jbc.271.11.6017 (1996).
Gachet, C. et al. Alpha IIb Beta 3 integrin dissociation induced by EDTA results in morphological changes of the platelet surface-connected Canalicular system with differential location of the two separate subunits. J. Cell Biol. 120(4), 1021–1030. https://doi.org/10.1083/jcb.120.4.1021 (1993).
Zhang, K. & Chen, J. The regulation of integrin function by divalent cations. Cell Adh. Migr. 6(1), 20–29. https://doi.org/10.4161/cam.18702 (2012).
Hill, C. N. et al. Deciphering the role of the coagulation cascade and autophagy in cancer-related thrombosis and metastasis. Front Oncol 10, 605314. https://doi.org/10.3389/fonc.2020.605314 (2020).
Vakiti, A., Anastasopoulou, C., & Mewawalla, P. (2023) Malignancy-related hypercalcemia. In StatPearls. (StatPearls Publishing, Treasure Island).
Endres, D. B. Investigation of hypercalcemia. Clin. Biochem. 45(12), 954–963. https://doi.org/10.1016/j.clinbiochem.2012.04.025 (2012).
Almuradova, E. & Cicin, I. Cancer-related hypercalcemia and potential treatments. Front. Endocrinol. 14, 1039490. https://doi.org/10.3389/fendo.2023.1039490 (2023).
Essandoh, K. et al. Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochim. Biophys. Acta 1852(11), 2362–2371. https://doi.org/10.1016/j.bbadis.2015.08.010 (2015).
McNamee, N., Catalano, M., Mukhopadhya, A. & O’Driscoll, L. An Extensive study of potential inhibitors of extracellular vesicles release in triple-negative breast cancer. BMC Cancer 23(1), 654. https://doi.org/10.1186/s12885-023-11160-2 (2023).
Goldstein, D. A. Serum calcium. In Clinical Methods: The History, Physical, and Laboratory Examinations (eds Walker, H. K. et al.) (Butterworths, 1990).
Parise, L. V. & Phillips, D. R. Fibronectin-binding properties of the purified platelet glycoprotein IIb-IIIa complex. J. Biol. Chem. 261(30), 14011–14017 (1986).
Danen, E. H. J., Sonneveld, P., Brakebusch, C., Fässler, R. & Sonnenberg, A. The fibronectin-binding integrins Α5β1 and Αvβ3 differentially modulate RhoA–GTP loading, organization of cell matrix adhesions, and fibronectin fibrillogenesis. J. Cell Biol. 159(6), 1071–1086. https://doi.org/10.1083/jcb.200205014 (2002).
Felding-Habermann, B., Ruggeri, Z. M. & Cheresh, D. A. Distinct biological consequences of integrin Alpha v Beta 3-mediated melanoma cell adhesion to fibrinogen and its plasmic fragments. J. Biol. Chem. 267(8), 5070–5077 (1992).
Suehiro, K., Gailit, J. & Plow, E. F. Fibrinogen is a ligand for integrin Α5β1 on endothelial cells. J. Biol. Chem. 272(8), 5360–5366. https://doi.org/10.1074/jbc.272.8.5360 (1997).
Madan, M., Berkowitz, S. D. & Tcheng, J. E. Glycoprotein IIb/IIIa integrin blockade. Circulation 98(23), 2629–2635. https://doi.org/10.1161/01.CIR.98.23.2629 (1998).
Bambace, N. M. & Holmes, C. E. The platelet contribution to cancer progression. J. Thromb. Haemost. 9(2), 237–249. https://doi.org/10.1111/j.1538-7836.2010.04131.x (2011).
Phillips, D. R., Charo, I. F., Parise, L. V. & Fitzgerald, L. A. The platelet membrane glycoprotein IIb-IIIa complex. Blood 71(4), 831–843 (1988).
Nomura, S. et al. Differences between platelet and microparticle glycoprotein IIb/IIIa. Cytometry 13(6), 621–629. https://doi.org/10.1002/cyto.990130610 (1992).
Scavone, M., Bossi, E., Podda, G. M. & Cattaneo, M. MgSO 4 anticoagulant prevents pseudothrombocytopenia by preserving the integrity of the platelet GPIIb-IIIa complex. Br. J. Haematol. https://doi.org/10.1111/bjh.17298 (2021).
Fitzgerald, L. A. & Phillips, D. R. Calcium regulation of the platelet membrane glycoprotein IIb-IIIa complex. J. Biol. Chem. 260(20), 11366–11374 (1985).
Pidard, D., Didry, D., Kunicki, T. & Nurden, A. Temperature-dependent effects of EDTA on the membrane glycoprotein IIb- IIIa complex and platelet aggregability. Blood 67(3), 604–611. https://doi.org/10.1182/blood.V67.3.604.604 (1986).
Pan, S. et al. STIM1 promotes angiogenesis by reducing exosomal miR-145 in breast cancer MDA-MB-231 cells. Cell Death Dis. 12(1), 38. https://doi.org/10.1038/s41419-020-03304-0 (2021).
Lee, A. H., Ghosh, D., Quach, N., Schroeder, D. & Dawson, M. R. Ovarian cancer exosomes trigger differential biophysical response in tumor-derived fibroblasts. Sci. Rep. 10(1), 8686. https://doi.org/10.1038/s41598-020-65628-3 (2020).
Nieswandt, B. et al. Glycoprotein VI but not Alpha2beta1 integrin is essential for platelet interaction with collagen. EMBO J. 20(9), 2120–2130. https://doi.org/10.1093/emboj/20.9.2120 (2001).
Varga-Szabo, D., Pleines, I. & Nieswandt, B. Cell adhesion mechanisms in platelets. ATVB 28(3), 403–412. https://doi.org/10.1161/ATVBAHA.107.150474 (2008).
Constantinescu-Bercu, A. et al. The GPIbα intracellular tail—Role in transducing VWF- and collagen/GPVI-mediated signaling. Haematol 107(4), 933–946. https://doi.org/10.3324/haematol.2020.278242 (2021).
Keowmaneechai, E. & McClements, D. J. Influence of EDTA and citrate on physicochemical properties of whey protein-stabilized oil-in-water emulsions containing CaCl2. J. Agric. Food Chem. 50(24), 7145–7153. https://doi.org/10.1021/jf020489a (2002).
Bournazos, S., Rennie, J., Hart, S. P. & Dransfield, I. Choice of anticoagulant critically affects measurement of circulating platelet-leukocyte complexes. ATVB https://doi.org/10.1161/ATVBAHA.107.153387 (2008).
Hynes, R. O. Integrins: Bidirectional, allosteric signaling machines. Cell 110(6), 673–687. https://doi.org/10.1016/s0092-8674(02)00971-6 (2002).
Liao, K. et al. The role of platelets in the regulation of tumor growth and metastasis: The mechanisms and targeted therapy. MedComm 4(5), e350. https://doi.org/10.1002/mco2.350 (2023).
Xia, Y., Shen, S. & Verma, I. M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2(9), 823–830. https://doi.org/10.1158/2326-6066.CIR-14-0112 (2014).
Goldner, W. Cancer-related hypercalcemia. JOP 12(5), 426–432. https://doi.org/10.1200/JOP.2016.011155 (2016).
Acknowledgments
This work has been supported by the George Washington University (GWU) and the Department of Defense (DoD) Breast Cancer Research Program Expansion Award (W81XWH-19-1-0667) to ALP. KM has been supported by the GW Provost Graduate Fellowship Program.
Author information
Authors and Affiliations
Contributions
Conceptualization: KM, ALP, Experimental design: KM, BS, ALP, Data collection: KM (all but invasion data), SM (invasion data), BS (adhesion data), Data analysis: KM, ALP, Writing and editing: KM, ALP.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Morris, K., Masri, S., Schnoor, B. et al. Calcium levels modulate platelet function, platelet-cancer cell interaction, and cancer cell invasion. Sci Rep 15, 7750 (2025). https://doi.org/10.1038/s41598-024-79280-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-79280-8
