
Abstract
The aim of this study was to develop a quantitative feature-based model from histopathologic images to assess the prognosis of patients with gastric cancer. Whole slide image (WSI) images of H&E-stained histologic specimens of gastric cancer patients from The Cancer Genome Atlas were included and randomly assigned to training and test groups in a 7:3 ratio. A systematic preprocessing approach was employed as well as a non-overlapping segmentation method that combined patch-level prediction with a multi-instance learning approach to integrate features across the slide images. Subjects were categorized into high- or low-risk groups based on the median risk score derived from the model, and the significance of this stratification was assessed using a log-rank test. In addition, combining transcriptomic data from patients and data from other large cohort studies, we further searched for genes associated with pathological features and their prognostic value. A total of 165 gastric cancer patients were included for model training, and a total of 26 features were integrated through multi-instance learning, with each process generating 11 probabilistic features and 2 predictive labeling features. We applied a 10-fold Lasso-Cox regression model to achieve dimensionality reduction of these features. The predictive accuracy of the model was verified using Kaplan-Meyer (KM) curves for stratification with a consistency index of 0.741 for the training set and 0.585 for the test set. Deep learning-based resultant supervised pathohistological features have the potential for superior prognostic stratification of gastric cancer patients, transforming image pixels into an effective and labor-saving tool to optimize the clinical management of gastric cancer patients. Also, SLITRK4 was identified as a prognostic marker for gastric cancer.
Similar content being viewed by others

Introduction
Gastric cancer is a major public health challenge and one of the leading causes of death globally, and the treatment of gastric cancer patients remains a great challenge1,2,3. In recent years, a combination of therapeutic approaches such as immunotherapy, surgery, and chemotherapy has made great strides in adequately addressing the needs of gastric cancer patients, but there is still a need for the emergence of a promising new avenue of cancer treatment4,5,6. Therefore, the search for valuable diagnostic and prognostic biomarkers is becoming increasingly important in enhancing the care of cancer patients.
Histopathological examination of tumor specimens is considered the gold standard for tumor diagnosis and is widely used in the diagnosis of gastric cancer. As a result, pathology examinations have resulted in a wealth of valuable and reliable information, which has set the stage for the proliferation of pathomics7,8,9,10. Pathomics is an emerging image analysis technology that focuses on extracting features (including quantitative features such as morphology, texture, and biology) to characterize the various tissue types captured in digital pathology images11,12,13. Pathology embodies a variety of data captured from digital pathology image analysis, which is then analyzed to determine a diagnosis or predict survival outcomes14,15. Therefore, we sought to analyze automated digital pathology features extracted from H&E-stained slides that could predict prognosis and survival benefits for patients with gastric cancer.
With the popularization of second-generation sequencing, the popularity of transcriptomic data has brought us gene expression data from patients, and the information contained in these data is enormous16,17,18. In addition, research teams from all over the world publicize their sequencing data as well as matched patient information19,20. These large cohorts help us to unearth more potential biomarkers, thus setting the stage for the development of new therapeutic targets.
Previous studies have shown that SLITRK4 is strongly associated with uterine smooth muscle sarcoma, brain tumors, hepatocellular carcinoma and neuropsychiatric disorders21,22,23. Mechanistic studies indicated that decreased levels of miR-139-5p enhance tumor cell invasion and proliferation by increasing SLITRK4 expression, while increased levels of miR-139-5p have the opposite effect24. Furthermore, in vivo and in vitro experiments showed that SLITRK4 was involved in gastric cancer progression, invasion and metastasis25.
In this paper, we constructed a pathology model using data from the Cancer Genome Atlas and combined it with machine learning techniques to assess the prognostic level of gastric cancer patients. We also identified SLITRK4 as a prognostic marker for gastric cancer by combining transcriptomic data.
Methods
Patient cohort and data resources
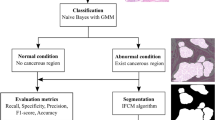
All data in this paper are from the Cancer Genome Atlas (TCGA), a public database containing whole slide image (WSI) and transcriptome data from patients with gastric cancer. The following patients were excluded: patients with other malignant tumors; patients with incomplete clinical data or lack of follow-up; patients with missing or poor-quality pathology sections and no RNA-seq data. A total of 165 patients were included, and eligible patients were randomly divided into the training group and the experimental group in the ratio of 7:3. The research flow chart is shown in Fig. 1.

The flow chart of this study.
Image processing
We selected the open-source software QuPath version 0.3.2 for annotation. Two experienced pathologists manually annotated the tumor regions in the WSI. WSIs were segmented into non-overlapping 512 × 512 patches at 0.5 μm/patch resolution. H&E-stained tissue was separated from the background by Otsu’s method26. Macenko’s method was applied for color normalization27. Z-score normalization standardized the RGB intensity distribution, which served as model input. During the training phase, we applied online data enhancement techniques, including random cropping, horizontal flipping, and vertical flipping. For the test patches, only normalization was performed.
Temporal-related labels
We found in our research that deep learning models with classification-based objectives perform significantly better than those that use regression methods. As such, we focused on the analysis of survival data, specifically the time-dependent 1-year survival risk, applying this categorization consistently throughout all patches linked to a single patient. We were able to determine time-dependent risk evaluations for 145 of the studied samples (20 truncated data were excluded). After that, a patch-by-patch training of the classification model was applied to these chosen samples.
Deep learning training
Our deep learning framework employs a two-layer prediction strategy that combines patch-level prediction with a multi-instance learning approach to integrate features across the WSI. For patch-level prediction, we used six models, DenseNet121, ResNet18, ResNet50, ResNet101, VGG19 and Inception_v3, which have been pre-trained on ImageNet respectively. Our goal was to evaluate the likelihood of each patch being assigned the corresponding WSI label. When constructing the 1-year timestamp-based pathology model, we found that these 20 patients belonged to the truncated data, i.e., the event of interest in our study (death) did not occur during the 1-year observation period. Therefore, these data were temporarily excluded from the model training phase. However, these truncated-tailed data still have important research value in survival analysis, and we characterized these patients by extracting them and including them in the subsequent Kaplan-Meier survival analysis. We utilize a pre-trained model to predict the outcome of these samples in order to obtain the predicted probability and label for each patch.
The Patch Likelihood Histogram (PLH) pipeline and the Bag of Words (BoW) pipeline are used in the WSI-level processing. PLH utilizes histograms to describe the distribution of patches across the WSI. Meanwhile, BoW utilizes Term Frequency-Inverse Document Frequency (TF-IDF) to map each patch, which produces a TF-IDF feature vector that generalizes the WSI features.
Constructing pathological signatures
To simplify this feature set, we used a correlation-based selection method that retained only one feature in each pair with a Pearson correlation coefficient greater than 0.9. To identify features that predicted stability, we further refined the features using lasso regression. A multivariate Cox proportional risk model was developed using this feature set to accomplish the prediction of patient overall survival (OS). We used a consistency index (C-index) to measure the performance of the model and further analyzed the pathological features. We also categorized subjects into high- and low-risk groups based on the median risk scores derived from the model and assessed the significance of this stratification using the log-rank test.
Model construction and performance evaluation
Covariates linked to prognosis were found. Clinicopathologic data identified as potential predictors included age, sex, T stage, N stage, M stage, Lauren classification, and Location. Multifactorial cox regression identified covariates associated with survival. p < 0.05 factors were used to construct a clinical model. Subsequently, we further developed an integrated model with pathologic and clinicopathologic features.
Exploring pathways and functional mechanisms
The z score parameter in the R package GSVA was implemented for the 14 functional state gene sets to obtain the COMBINED z-score scores, and the Pearson correlation of SLITRK4 with each gene set score was calculated28. Patients with the highest 30% expression of the SLITRK4 gene were defined as the high-expression group, and patients with the lowest 30% expression were defined as the low-expression group. Differential analysis of the two groups was performed using the Limma software package, and gene set enrichment analysis was performed based on the marker gene set versus the KEGG metabolic gene set29. Bubble plots were used for visualization.
Immune-related analysis content
Based on gene expression profiling data, patients were categorized into high/low expression groups based on the median SLITRK4 expression. A nonparametric Wilcoxon rank sum test was used to compare the differences in immune cell content assessed by different calculation methods between the two groups30,31,32. The heatmap was constructed based on the immune cell content data, with samples from left to right arranged according to low to high gene expression.
Statistical analysis
Shapiro-Wilks test was used to assess the normality of the distribution of clinical characteristics in these cohorts. The Student’s t-test or Mann-Whitney U test was used for continuous variables and the Chi-square or Fisher’s exact test for categorical variables. Statistics were conducted using two-sided tests, and statistical significance was defined as P < 0.05. A variety of software tools, including ITK-SNAP version 3.8.0, were used in this thorough examination, coupled with specially written Python version 3.7.12 scripts. The following Python libraries were used in this analysis: Matplotlib version 3.4.2, SciPy version 1.7.3, scikit-learn version 1.0.2, PyRadiomics version 3.0, Onekey version 2.2.3, OpenSlide version 1.2.0, Seaborn version 0.11.1, NumPy version 1.20.2, PyTorch version 1.8.0, and Lifelines version 0.27.0.
Results
Patients characteristics
Patient characteristics of the training and test groups are shown in Table 1. The training cohort included 70 males and 45 females, and the test cohort included 31 males and 19 females (p = 1.000, Chi-square test). The age in training cohort ranged from 41 to 86 years (mean age: 66.64 years) and in test cohort from 44 to 84 years (mean age: 66.63 years) (p = 0.078, Mann-Whitney U). From our findings, it was observed that the samples within each group did not exhibit p-values less than 0.05, indicating that our random allocation process was relatively uniform.
Development and performance evaluation of pathomics-based model
At the patch level, six CNN-based models were compared and evaluated using ROC curves. resnet-50 performed the best, with an AUC value of 0.979 (95% CI: 0.977–0.981) for the training cohort and 0.717 (95% CI: 0.703–0.731) for the test cohort.
A total of 26 features were integrated through multi-instance learning, with each process producing 11 probabilistic features and 2 predictive labeling features. Feature selection was based on the Pearson correlation coefficient, with only one of a pair of highly correlated features retained if the correlation coefficient exceeded 0.9. These features underwent further selection and optimization using a lasso regression model, culminating in the retention of a pair of highly correlated features. culminating in the retention of 8 features, including 7 TF-IDF probabilistic features “prob01”, “prob02”, “prob03” “prob04”, “prob05”, “prob06”, " prob07”, and a predictive feature “pred1” with the highest feature weight. The 8 features were retained for subsequent analysis.
Integration with clinicopathological characteristics
Table 2 shows that both T-stage and N-stage were significant (P < 0.05) in cox regression analysis, and a clinical prediction model was constructed based on this. The predictive accuracy of the model was verified by stratification using Kaplan-Meyer (KM) curves with a consistency index (c-index) of 0.741 for the training set and 0.585 for the test set.
Analysis of changes over time
We also performed a ROC analysis of these models for time-correlation analysis (Fig. 2). These results indicate that the model has good efficacy. Overall, the combined model provided reliable predictions across all groups and time scales. For short-term prediction, the pathology model showed better accuracy in both the training and validation sets, with AUCs of 0.914 and 0.818, respectively. For long-term prediction, the combined model (combining clinical and pathological information) had an AUC of 0.844 in the training set and 0.7 in the testing set, showing the stability of the model across longer time scales. Supplementary Table 1 reveals more details about the model.

(A–F) ROC curves for the training and test cohorts for time-related analysis. The subject operating characteristic (ROC) curves for the clinical, pathology, and combined models are illustrated. These curves assess the performance of the models in predicting survival outcomes over time.
SLITRK4 is a prognostic biomarker for gastric cancer
In order to further explore the model-related gastric cancer biomarkers, we calculated the correlation between genes and features by analyzing transcriptomics using pearson correlation analysis, and ultimately found that SLITRK4 had high correlation with all of the screened features. Therefore, we analyzed SLITRK4 in depth.
To further confirm the value of SLITRK4 as a prognostic marker for gastric cancer, we analyzed the predictive value of SLITRK4 for three types of survival (Overall survival, Progression-free interval, and Disease-specific survival) in gastric cancer patients in the TCGA dataset (Fig. 3A-C). Specifically, gastric cancer patients in the TCGA database were categorized into high and low expression groups based on SLITRK4 expression. The difference in survival time between patients in the high and low expression groups was compared using three survival periods, respectively, all of which indicated that patients with low SLITRK4 expression had a better level of prognosis. This was also demonstrated in two large gastric cancer datasets (GSE84433,GSE62254) that publicize patient prognostic information (Fig. 3D,E). This therefore suggests that SLITRK4 has potential as a prognostic marker for gastric cancer. In addition, the results of further unifactorial and multifactorial cox analyses also indicated that SLITRK4 was an independent prognostic factor for gastric cancer patients (Fig. 3F,G). Accordingly, we developed a nomogram model (Fig. 3H). The model had good predictive ability, and the calibration curve further confirmed the accuracy of the model (Fig. 3I).
We also found that SLITRK4 also showed a better ability to predict prognosis in subgroups stratified by various clinical characteristics, including T-stage, age, gender, pathologic stage, and N-stage (Fig. 4).

(A–C) Prognostic value of SLITRK4 in the TCGA- stomach adenocarcinoma dataset. (D,E) Prognostic value of SLITRK4 in two external validation datasets. (F,G) Results of univariate and multivariate regression analyses. (H) Nomogram created based on SLITRK4 expression. (I) Calibration curves demonstrating accuracy. The survival package was used to test the proportional risk hypothesis and perform fitted survival regression.

(A–J) SLITRK4 has good prognostic value in multiple subgroups of the population.
SLITRK4 and immunotherapy and immune correlation analysis
Our different immune infiltration algorithms calculated the difference in immune levels between populations with high SLITRK4 expression and populations with low SLITRK4 expression (Fig. 5A). To further validate our conclusions, we calculated immune cell infiltration as well as immune scores using the ssgsea algorithm and the estimate algorithm (Fig. 5B,C). The results showed that the level of immune infiltration was significantly higher in patients with high SLITRK4 expression than in patients with low SLITRK4 expression. Correlation analysis also showed that SLITRK4 showed a positive correlation with the immune score as well as immune-related genes (Fig. 5D,E).

(A) Differences in immune cell infiltration between populations with high SLITRK4 expression and populations with low SLITRK4 expression. (B,C) Two algorithms showing differences in immune levels between populations with high SLITRK4 expression and populations with low SLITRK4 expression. (D,E) Co-expression of SLITRK4 with immune-related genes and correlation. The non-parametric Wilcoxon rank sum test was used to compare the difference in immune cell content assessed by different methods between the two groups. *p < 0.05, **p < 0.01, ***p < 0.001.
Role of SLITRK4 in cancer pathways
To understand the possible role and specific function of SLITRK4 in gastric cancer, we reflected the activity of a given pathway by integrating characterized gene expression. The z score parameter in gene set variation analysis (GSVA) was implemented for 14 functional state gene sets (angiogenesis, apoptosis, cell cycle, differentiation, DNA damage, DNA repair, epithelial mesenchymal transition (EMT), hypoxia, inflammation, invasion, metastasis, proliferation, quiescence, and stemness), and the combined z-score score was obtained, and the Pearson correlation between SLITRK4 and each of the gene set scores was further calculated of Pearson correlation (Fig. 6A). The results showed that SLITRK4 was significantly and positively correlated with differentiation, epithelial mesenchymal transition, and angiogenesis. Further, we collected a dataset of 17 gastric cancers for gene set enrichment analysis (GSEA) analysis. The results indicated that SLITRK4 may be involved in a large number of tumor-related pathways and metabolic processes, such as epithelial mesenchymal transition and myogenesis (Fig. 6B).

(A) SLITRK4 expression was highly associated with 14 malignant features of gastric cancer. (B) Bubble plots illustrating the GSEA results of signature gene features between high and low SLITRK4 expression groups in the gastric cancer dataset.
Discussion
Because a variety of histological characteristics in distinct tumor cells correspond to varying degrees of disease development, clinical outcomes, and treatment responses, pathology is a novel method that has been utilized to investigate tumor heterogeneity. In order to evaluate the properties of tumor cells, pathologists do traditional pathology examinations at various magnifications. However, they are not able to consistently describe more specific information for each slide. As a result, pathohistology can be a helpful technique to enhance conventional pathologic assessment.
Previous studies have demonstrated the use of multiple imaging histology and pathohistology features to predict the prognosis of patients with gastric cancer5,33,34,35,36,37,38,39. However, the main focus of these studies was to establish associations between different image features and lacked reproducibility for reasons of patient information protection. In addition, valuable image features extracted using artificial intelligence methods have not been genetically linked. This study used the TCGA database for pathohistology, which offers several significant advantages. Specifically, the TCGA data contains imaging data from multiple medical institutions and research centers, and the data undergoes rigorous quality control and standardization to ensure data reliability and consistency. This helps researchers to compare and validate between different studies, improving the reproducibility and scientific validity of the studies. More importantly, the data in the TCGA database contains information in multiple dimensions, such as patients’ clinical information, imaging features, gene expression data, etc. This information provides us with a more comprehensive perspective for the study of gastric cancer, which helps to explore the disease mechanism and diagnosis and treatment methods in depth. As described in our results, image features identified based on a combination of artificial intelligence and machine learning algorithms are extremely valuable for prognostic prediction of gastric cancer patients. Further, we linked image features to patients’ gene expression to explore potential biological markers for gastric cancer patients.
In this study, pathologic models, with their wealth of clinical information and timely disease monitoring, offer significant advantages in short-term prediction. In addition, pathologic models provide unique insights into the mechanisms of disease progression, especially when used in conjunction with clinical information. By integrating multiple sources of information, comprehensive models provide more consistent results in long-term prediction, which is crucial for assessing disease progression and developing long-term treatment strategies. Therefore, time scales and specific disease contexts should be fully considered when applying models.
Our study indeed indicates that the CNN model, specifically ResNet-50, was trained extensively on a sufficiently large training set, leading to well-trained models as evidenced by AUC values approaching 140. This suggests that the model performs well in general. When the task in the testing set involves morphological or tissue region identification, we generally observe good accuracy41,42. However, when survival-related information is used, there is a notable decline in performance in the testing set43,44,45. This decline is likely attributed to the inherent complexity of survival-related problems, which pose additional challenges in achieving an optimal fit of the training model to these specific tasks. The survival-related information may encompass a wide range of biological and clinical factors that are not fully captured or represented in the training data, leading to the observed performance drop.
The TNM staging system is a widely used method of classifying tumors to describe the severity and extent of cancer spread46,47. These staging stages are critical for developing treatment plans and predicting patient prognosis48,49,50. Our clinical prediction model is constructed based on the significant difference between T stage and N stage, which occupy a central position in tumor staging. Therefore, incorporating these two stages into the model helps to predict the prognosis of patients more accurately.
In this study, we found that gastric cancer patients with low expression of SLITRK4 had better prognostic levels in multiple datasets. This finding reveals the great potential of SLITRK4 as a prognostic marker for gastric cancer, which can help physicians more accurately determine the prognosis of patients and thus develop more personalized treatment plans. For example, for gastric cancer patients with high SLITRK4 expression, more aggressive treatment strategies, such as enhanced immunotherapy or chemotherapy, may be needed to improve the therapeutic efficacy and patient survival51,52,53,54. The study also developed a prognostic nomogram model that includes SLITRK4, which has good predictive ability. The establishment of this model provides a new tool and method for prognostic assessment of gastric cancer and helps to promote the development and improvement of prognostic models for gastric cancer. In addition, these findings also provide new directions and ideas for gastric cancer-related research. For example, the role mechanism of SLITRK4 in gastric carcinogenesis and development can be further explored, and these studies will contribute to a more in-depth understanding of the biological behavior and immune microenvironment of gastric cancer, providing a more comprehensive theoretical basis and practical guidance for the prevention and treatment of gastric cancer55,56,57.
Patients with high SLITRK4 expression exhibited significantly greater immune cell infiltration compared to those with low SLITRK4 expression. This suggests a strong correlation between SLITRK4 expression levels and immune cell infiltration. Immune cell infiltration is crucial for the body’s response to pathogens and the removal of abnormal cells58. Thus, high SLITRK4 expression may enhance immune cell infiltration, strengthening the immune response. Understanding how SLITRK4 functions in the immune response could lead to new treatment strategies for diseases59. For instance, targeted therapies against SLITRK4 might modulate the immune response, improving therapeutic outcomes60,61. By regulating SLITRK4 expression levels, it may also be possible to influence immune cell infiltration and the expression of immune-related genes, thereby achieving disease treatment goals62,63.
We found a close relationship between SLITRK4 and differentiation, EMT, and angiogenic cell differentiation. Cell differentiation is the process by which cells gradually develop from a primitive state into cell types with specific morphology and functions64,65,66. The expression level of SLITRK4 may be related to the degree of differentiation of gastric cancer cells, which in turn affects their biological behaviors and the degree of malignancy. EMT and angiogenesis are closely associated with tumor growth, invasion, metastasis, and drug resistance67,68. The significant positive correlation between SLITRK4 and both suggests that it may promote the progression and deterioration of gastric cancer by regulating the EMT process and promoting angiogenesis69,70. Consequently, these results facilitate future mechanistic studies.
The interpretability of deep learning models is frequently challenged, particularly in mission-critical applications like healthcare71,72. We cannot correlate these features in a very reasonable way in relation to a specific clinical context. This does limit our ability to accurately interpret these features. However, we still wish to emphasize the potential importance of these features in the diagnosis and prognostic assessment of gastric cancer. These features were extracted from pathology image data by machine learning methods and they represent the frequency and importance of different visual features in pathology images. In future studies, we will endeavor to explore the relationship between these features and the clinical context.
Despite the extensive comparisons and analyses performed in this study in terms of model selection and performance evaluation, certain limitations remain. In particular, in terms of model selection, while we considered classical CNN models such as ResNet and DenseNet, we did not cover all the current state-of-the-art model architectures such as EfficientNetV2 and Vision Transformers73,74,75,76,77,78. These novel models have demonstrated excellent performance in image analysis tasks, but were not included in this study as it mainly focuses on model fitness to specific tasks and elimination of model selection bias. These novel models have demonstrated excellent performance in image analysis tasks, but were not included in this study because it focuses on model fitness to specific tasks and eliminating model selection bias. Future research is expected to further explore the potential of these novel models in medical image classification tasks.
Although this study employs a variety of data enhancement techniques, including random cropping, horizontal flipping, and vertical flipping, to enrich the diversity of training data, there may be a problem of limited variety of enhancement techniques76. This limited data enhancement strategy may not be sufficient to adequately simulate complex variations in the real world, especially when dealing with heterogeneous histopathology data, which may limit the enhancement of the model generalization ability. We are actively exploring a wider range of data enhancement methods, such as fuzzy techniques, with a view to further improving the performance of the model in future studies
Although our results are encouraging, it is important to recognize the limitations of this study. Its relatively small sample size may introduce bias and affect generalizability. Prospective studies with larger and more diverse cohorts are needed to further validate these models and their clinical utility. Meanwhile, the mechanism of action of SLITRK4 in gastric cancer needs to be further elucidated.
Data availability
All data generated or analyzed during this study are included in this published article. The data used to support the findings of the present study are available from the corresponding author upon request.
References
Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513(7517), 202–209 https://doi.org/10.1038/nature13480 (2014).
Lei, Z. et al. Identification of molecular subtypes of gastric cancer with different responses to PI3-kinase inhibitors and 5-fluorouracil. Gastroenterology 145, 554–565 (2013).
Cristescu, R. et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 21, 449–456 (2015).
Chia, N. Y. & Tan, P. Molecular classification of gastric cancer. Ann. Oncol. 27, 763–769 (2016).
Chen, D. et al. Prognostic and predictive value of a pathomics signature in gastric cancer. Nat. Commun. 13, 6903 (2022).
The global, regional, and national burden of stomach cancer in 195 countries, 1990–2017: A systematic analysis for the Global Burden of Disease study 2017. The Lancet Gastroenterology & Hepatology. https://www.thelancet.com/journals/langas/article/PIIS2468-1253(19)30328-0/fulltext.
Bera, K., Schalper, K. A., Rimm, D. L., Velcheti, V. & Madabhushi, A. Artificial intelligence in digital pathology - new tools for diagnosis and precision oncology. Nat. Rev. Clin. Oncol. 16, 703–715 (2019).
Song, H. et al. Development and interpretation of a multimodal predictive model for prognosis of gastrointestinal stromal tumor. npj Precis Onc. 8, 1–13 (2024).
Ehteshami Bejnordi, B. et al. Diagnostic assessment of deep learning algorithms for detection of lymph node metastases in women with breast cancer. JAMA 318, 2199–2210 (2017).
Niazi, M. K. K., Parwani, A. V. & Gurcan, M. N. Digital pathology and artificial intelligence. Lancet Oncol. 20, e253–e261 (2019).
Li, B. et al. From pixels to patient care: Deep learning-enabled pathomics signature offers precise outcome predictions for immunotherapy in esophageal squamous cell cancer. J. Transl. Med. 22, 195 (2024).
Liu, Y. et al. Prediction of TNFRSF9 expression and molecular pathological features in thyroid cancer using machine learning to construct Pathomics models. Endocrine. https://doi.org/10.1007/s12020-024-03862-9 (2024).
The Emergence of Pathomics. Current Pathobiology Reports. https://link.springer.com/article/10.1007/s40139-019-00200-x
Gilley, P. et al. Utilizing a pathomics biomarker to predict the effectiveness of bevacizumab in ovarian cancer treatment. Bioengineering 11, 678 (2024).
Yoo, S. Y. et al. Whole-slide image analysis reveals quantitative landscape of tumor-immune microenvironment in colorectal cancers. Clin. Cancer Res. 26, 870–881 (2020).
Li, W., Zhao, X., Fu, J. & Cheng, L. Identification of lysosome-related hub genes as potential biomarkers and immune infiltrations of moyamoya disease by multiple bioinformatics methods and machine-learning strategies. Heliyon 10, e34432 (2024).
Jiang, H. et al. Identification of prostate cancer bone metastasis related genes and potential therapy targets by bioinformatics and in vitro experiments. J. Cell. Mol. Med. 28, e18511 (2024).
Yang, C., Liu, Y. H. & Zheng, H. K. Identification of TFRC as a biomarker for pulmonary arterial hypertension based on bioinformatics and experimental verification. Respir. Res. 25, 296 (2024).
Cole-Skinner, B. et al. Oleate alters the immune response in non-small cell lung adenocarcinoma through regulation of HMGB1 release. Front. Cell. Dev. Biol. 12, 1348707 (2024).
Zhang, C. et al. Pan-cancer analyses reveal the molecular and clinical characteristics of TET family members and suggests that TET3 maybe a potential therapeutic target. Front. Pharmacol. 15, 1418456 (2024).
Davidson, B. et al. Gene expression signatures of primary and metastatic uterine leiomyosarcoma. Hum. Pathol. 45, 691–700 (2014).
Aruga, J., Yokota, N. & Mikoshiba, K. Human SLITRK family genes: Genomic organization and expression profiling in normal brain and brain tumor tissue. Gene 315, 87–94 (2003).
Proenca, C. C., Gao, K. P., Shmelkov, S. V., Rafii, S. & Lee, F. S. Slitrks as emerging candidate genes involved in neuropsychiatric disorders. Trends Neurosci 34, 143–153 (2011).
Wu, J., Zhang, T., Chen, Y. & Ha, S. MiR-139-5p influences hepatocellular carcinoma cell invasion and proliferation capacities via decreasing SLITRK4 expression. Biosci. Rep. 40, BSR20193295 (2020).
Zhou, Y. Q. et al. The SLITRK4-CNPY3 axis promotes liver metastasis of gastric cancer by enhancing the endocytosis and recycling of TrkB in tumour cells. Cell. Oncol. 46, 1049–1067 (2023).
Otsu, N. A threshold selection method from gray-level histograms. IEEE Trans. Syst. Man Cybern. 9, 62–66 (1979).
Macenko, M. et al. A method for normalizing histology slides for quantitative analysis. In 2009 IEEE International Symposium on Biomedical Imaging: From Nano to Macro 1107–1110 (2009). https://doi.org/10.1109/ISBI.2009.5193250
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7 (2013).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015).
Aran, D., Hu, Z. & Butte, A. J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220 (2017).
Li, T. et al. TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 77, e108–e110 (2017).
Chen, B., Khodadoust, M. S., Liu, C. L., Newman, A. M. & Alizadeh, A. A. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol. Biol. 1711, 243–259 (2018).
Chen, H. et al. 18F-FDG PET/CT radiomics-based multimodality fusion model for preoperative individualized noninvasive prediction of peritoneal metastasis in advanced gastric cancer. Ann. Surg. Oncol. https://doi.org/10.1245/s10434-024-15631-z (2024).
Han, Y. et al. A study predicting long-term survival capacity in postoperative advanced gastric cancer patients based on MAOA and subcutaneous muscle fat characteristics. World J. Surg. Oncol. 22, 184 (2024).
Chen, H. et al. ASO Visual Abstract: 18F-FDG PET/CT radiomics-based multimodality fusion model for preoperative individualized noninvasive prediction of peritoneal metastasis in advanced gastric cancer. Ann. Surg. Oncol. https://doi.org/10.1245/s10434-024-15822-8 (2024).
Wu, A. et al. Comparative assessment of the capability of machine learning-based radiomic models for predicting omental metastasis in locally advanced gastric cancer. Sci. Rep. 14, 16208 (2024).
O’Sullivan, N. J. et al. Computed tomography (CT) derived radiomics to predict post-operative disease recurrence in gastric cancer; a systematic review and meta-analysis. Curr. Probl. Diagn. Radiol. S0363-0188(24), 00114–00112. https://doi.org/10.1067/j.cpradiol.2024.07.011 (2024).
Zhi, H. et al. Development and validation of a machine learning-based 18F-fluorodeoxyglucose PET/CT radiomics signature for predicting gastric cancer survival. Cancer Imaging 24, 99 (2024).
Wang, W. et al. LightGBM is an effective predictive model for postoperative complications in gastric Cancer: A study integrating radiomics with ensemble learning. J. Imaging Inf. Med. https://doi.org/10.1007/s10278-024-01172-0 (2024).
Cai, X., Zhang, H., Wang, Y., Zhang, J. & Li, T. Digital pathology-based artificial intelligence models for differential diagnosis and prognosis of sporadic odontogenic keratocysts. Int. J. Oral Sci. 16, 1–10 (2024).
Wei, J. W. et al. Evaluation of a Deep Neural Network for Automated Classification of Colorectal Polyps on Histopathologic Slides. JAMA Netw Open 3, e203398 (2020).
Hölscher, D. L. et al. Next-generation morphometry for pathomics-data mining in histopathology. Nat. Commun. 14, 470 (2023).
Chen, S. et al. Clinical use of machine learning-based pathomics signature for diagnosis and survival prediction of bladder cancer. Cancer Sci. 112, 2905–2914 (2021).
Lee, J. H. et al. Prediction of immunochemotherapy response for diffuse large B-cell lymphoma using artificial intelligence digital pathology. J. Pathol. Clin. Res. 10, e12370 (2024).
Yao, J., Wei, L., Hao, P., Liu, Z. & Wang, P. Application of artificial intelligence model in pathological staging and prognosis of clear cell renal cell carcinoma. Discov. Oncol. 15, 545 (2024).
Perrotta, G. et al. Accuracy of clinical staging in early-stage pancreatic ductal adenocarcinoma. JAMA 332, 1108–1110 (2024).
Ebrahimi, A. et al. Determinants of prognosis in Head and Neck cutaneous squamous cell Carcinoma with nodal metastases. JAMA Otolaryngol. Head Neck Surg. e243103 https://doi.org/10.1001/jamaoto.2024.3103 (2024).
Jj, P. et al. Ninth version of the AJCC and UICC nasopharyngeal cancer TNM staging classification. JAMA Oncol. https://doi.org/10.1001/jamaoncol.2024.4354 (2024).
Steventon, L., et al. The impact of inter-cycle treatment delays on 5-year all-cause mortality in early-stage breast cancer: A retrospective cohort study. Eur. J. Cancer (Oxford, England: 1990) 210 (2024).
von Buchwald, C., et al. TNM 8 staging system beyond p16: Double HPV/p16 status is superior to p16 alone in predicting outcome in oropharyngeal squamous cell carcinoma. Eur. J. Cancer (Oxford, England: 1990) 211 (2024).
Liu, Y. et al. Cytokine conjugation to enhance T cell therapy. Proc. Natl. Acad. Sci. U S A. 120, e2213222120 (2023).
Meric-Bernstam F, Larkin J, Tabernero J, Bonini C. Enhancing anti-tumour efficacy with immunotherapy combinations. Lancet. 397(10278), 1010–1022 (2021).
Topalian, S. L. et al. Neoadjuvant immune checkpoint blockade: A window of opportunity to advance cancer immunotherapy. Cancer Cell 41, 1551–1566 (2023).
Hirschhorn, D. et al. T cell immunotherapies engage neutrophils to eliminate tumor antigen escape variants. Cell 186, 1432–1447e17 (2023).
Lan, Y., Zhang, S., Pan, Y., Wang, M. & Chen, G. Research progress on the mechanism of anti-tumor immune response induced by TTFields. Cancers (Basel) 15, 5642 (2023).
Zhang, C., Xu, C., Gao, X. & Yao, Q. Platinum-based drugs for cancer therapy and anti-tumor strategies. Theranostics 12, 2115–2132 (2022).
Liu, J. et al. Selective autophagy in cancer: Mechanisms, therapeutic implications, and future perspectives. Mol. Cancer 23, 22 (2024).
Mantilla-Rojas, C., et al. Enhanced T-cell priming and improved anti-tumor immunity through lymphatic delivery of checkpoint blockade immunotherapy. Cancers 14 (2022).
Amouzegar, A., Chelvanambi, M., Filderman, J. N., Storkus, W. J. & Luke, J. J. STING agonists as cancer therapeutics. Cancers (Basel) 13, 2695 (2021).
Sellars, M. C., Wu, C. J. & Fritsch, E. F. Cancer vaccines: Building a bridge over troubled waters. Cell 185, 2770–2788 (2022).
Ramirez-Valdez, R. A. et al. Intravenous heterologous prime-boost vaccination activates innate and adaptive immunity to promote tumor regression. Cell Rep 42, 112599 (2023).
Pulendran, B. & Davis, M. M. The science and medicine of human immunology. Science 369, eaay4014 (2020).
Tong, W. et al. Vaccination with tumor-ganglioside glycomimetics activates a selective immunity that affords cancer therapy. Cell. Chem. Biol. 26, 1013-1026e4 (2019).
Zhang, Y. et al. Bibliometric analysis of global research on tumor dormancy. Cancers 15, 3230 (2023).
Pouliquen, D. L., & Núñez González, C. Biomarkers of tumor metastasis and invasiveness. Cancers 15 (2023).
Hoeppner, J. & Bronsert, P. Metastasis and tumor cell migration of solid tumors. Cancers (Basel) 13, 5576 (2021).
San Juan, B. P., Garcia-Leon, M. J., Rangel, L., Goetz, J. G. & Chaffer, C. L. The complexities of metastasis. Cancers (Basel) 11, 1575 (2019).
Steeg, P. S. Tumor metastasis: Mechanistic insights and clinical challenges. Nat. Med. 12, 895–904 (2006).
Valastyan, S. & Weinberg, R. A. Tumor metastasis: molecular insights and evolving paradigms. Cell 147, 275–292 (2011).
Lin, Y., Xu, J. & Lan, H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol 12, 76 (2019).
Yao, J., Wei, L., Hao, P., Liu, Z. & Wang, P. Application of artificial intelligence model in pathological staging and prognosis of clear cell renal cell carcinoma. Discov. Onc. 15, 545 (2024).
Cao, R. et al. Development and interpretation of a pathomics-based model for the prediction of microsatellite instability in colorectal cancer. Theranostics 10, 11080 (2020).
Ma, Y. et al. A study of machine learning models for rapid intraoperative diagnosis of thyroid nodules for clinical practice in China. Cancer Med. 13, e6854 (2024).
Aldakhil, L. A., Alhasson, H. F. & Alharbi, S. S. Attention-based deep learning approach for breast cancer histopathological image multi-classification. Diagnostics (Basel) 14, 1402 (2024).
Tran, M. H. et al. Detection and margin assessment of thyroid carcinoma with microscopic hyperspectral imaging using transformer networks. J. Biomed. Opt. 29, 093505 (2024).
Prezja, F. et al. Improving performance in colorectal cancer histology decomposition using deep and ensemble machine learning. Heliyon 10, e37561 (2024).
An J, Wang Y, Cai Q, Zhao G, Dooper S, Litjens G, Gao Z. Transformer-Based Weakly Supervised Learning for Whole Slide Lung Cancer Image Classification. IEEE J Biomed Health Inform. 2024 Jul 9;PP.
Hu Z, Wang J, Gao Q, Wu Z, Xu H, Guo Z, Quan J, Zhong L, Du M, Tong T, Chen G. Weakly Supervised Classification for Nasopharyngeal Carcinoma with Transformer in Whole Slide Images. IEEE J Biomed Health Inform. 2024 Jul 3;PP.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82073244).
Author information
Authors and Affiliations
Contributions
Y.Z.(Ye Zhang) and Y.G. designed the research; Y.Z.(Yuzhe Zhang) performed the research; Y.X. and Y.Z.(Yuzhe Zhang) contributed new reagents or analytic tools; Y.X. and Y.Z.(Yuzhe Zhang) analyzed the data; Y.X., Y.G. and Y.Z.(Yuzhe Zhang) wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Our study used publicly available database that had been approved by the Ethics Committee and no new data were collected, so no additional ethical approval was required.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Y., Xue, Y., Gao, Y. et al. Prognostic and predictive value of pathohistological features in gastric cancer and identification of SLITRK4 as a potential biomarker for gastric cancer. Sci Rep 14, 29241 (2024). https://doi.org/10.1038/s41598-024-80292-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-80292-7
