
Abstract
In order to enrich our taxonomic and systematic comprehension of Ovulidae within the evolutionary framework of Littorinimorpha. we present a comprehensive analysis of the mitochondrial genome (mitogenome) sequence of Volva habei using next-generation sequencing technology (GenBank accession number OR492307). The mitogenome spans a total length of 16,519 bp, encompassing a complete set of 37 genes, including 13 protein-coding genes (PCGs), 22 tRNAs and two rRNAs, demonstrating a distinct AT bias. Notably, trnS2 lacks a dihydrouracil (DHU) arm, thus preventing the formation of a typical secondary structure. In contrast, the remaining tRNAs exhibit a characteristic cloverleaf-like secondary structure. Comparative analysis with ancestral gastropods reveals substantial differences in three gene clusters (or genes), incorporating fifteen tRNAs and eight PCGs. Of particular significance are the observed inversions and translocations, representing the predominant types of rearrangements in V. habei. Phylogenetic analysis strongly supports the monophyletic grouping of all Littorinimorpha species, with V. habei forming a distinct Ovulidae clade. It is noteworthy that V. habei forms a sister group with Cypraeidae, collectively belonging to the Cypraeoidea. In summary, this study not only advances our comprehension of the entire mitochondrial dataset for Calyptraeoidea but also provides novel insights into the phylogenetic relationships within Littorinimorpha.
Similar content being viewed by others


Introduction
Initially documented by Oyama1, Volva habei is a species affiliated with the Ovulidae family. Its key features encompass a smaller shell size compared to preceding species, relatively short anterior and posterior siphonal grooves, pronounced inflation in the central region, and a shell surface exhibiting a light pink or white hue. The siphonal grooves terminate without pigmentation, and the shell size ranges between 48 and 110 mm. This species predominantly resides in China, spanning from Zhejiang to Guangdong, and can also be found internationally, including regions such as Japan. It commonly inhabits shallow sandy seabeds within the subtidal zone.
Ovulidae (Fleming, 1822), commonly known as egg cowries, constitutes a widely distributed family of specialized carnivorous mollusks with a proclivity for grazing on the polyps and tissues of Anthozoa, particularly soft corals and sea fans. Predominantly found in tropical regions, with only a limited number of species inhabiting temperate waters, Ovulidae2 traces its origins back to the early Eocene3,4 and underwent a significant increase in diversity during the Pleistocene. This diversification was especially notable in the warm seas of the central Indo-West Pacific Region, currently boasting the highest documented specific diversity within the family. Despite a scarcity of detailed anatomical studies5, a close affinity with Cypraeidae, the ‘true’ cowries, has consistently been inferred, as indicated by prior works6. In the molecular systematics of this taxonomic family, Ovulidae is acknowledged as the group closest to Cypraeidae7,8. The taxonomic classification of Ovulidae has undergone multiple revisions9,10. Historically, based on shell morphology, the family has been dichotomized into two subfamilies (Schilder 1932): (1) Ovulinae, represented by genera such as Ovula, Primovula, Diminovula, Margovula, Calpurnus and Procalpurnus, exhibit pyriform, ovate and inflated shells. These shells typically have short extremities, a posterior canal turning leftward and well-developed teeth on the outer lip. (2) Volvinae, including genera like Volva, Simnia, Phenacovolva, Cyphoma and Neosimnia, showcase narrow fusiform shells with prominent extremities, and may lack or have poorly developed teeth.
The mitochondrial genome stands as a paradigm for genome evolution and serves as a powerful tool in elucidating evolutionary relationships11. Recognized as a valuable resource, the complete mitogenome has been pivotal in resolving phylogenetic relationships across diverse mollusk groups12,13,14,15,16. Owing to maternal inheritance, the absence of recombination and a high rate of base substitution, mitogenomes have found extensive application in phylogenetic analyses17,18,19. In contrast to gene fragments, mitogenomes encapsulate a wealth of information, demonstrating utility in recovering internal nodes with robust statistical support. This attribute has been widely applied in reconstructing phylogenetic relationships within various gastropod groups. A common characteristic of metazoan mitogenomes is the presence of a covalently circular molecule that encodes a total of 37 genes. These include 13 protein-coding genes (PCGs), two ribosomal RNA (rRNA) genes and 22 transfer RNA (tRNA) genes. Despite exceptions reported20,21, the mitogenome, when compared to single gene fragments, yields a substantial amount of variant information. This information has played a crucial role in species identification13,22 and phylogenetic reconstruction14,23 within Mollusca. Recent technological advancements, such as rolling circle amplification24,25 and next-generation sequencing technologies26, coupled with decreasing costs of DNA sequencing, have standardized the amplification and sequencing of entire mt genomes.
This investigation seeks to enhance taxonomic representation within the Ovulidae family by sequencing the complete mitogenome of V. habei. Moreover, a comprehensive examination of the mitogenome of V. habei was conducted, exploring intricate genomic details and evolutionary dynamics. This investigation encompassed aspects such as gene order, nucleotide composition, codon utilization, and the secondary structure of tRNA. The precise positioning of V. habei within the phylogenetic framework was discerned through a thoroughgoing phylogenetic analysis of the Littorinimorpha. In order to attain a more profound comprehension of the evolutionary interconnections within the Littorinimorpha, the gene arrangements of mitogenomes across the entire Littorinimorpha family were seamlessly integrated. Moreover, the recently acquired mitogenome was assimilated into the Ovulidae family dataset, aimed at resolving contentious phylogenetic relationships associated with the family. The phylogenetic tree of V. habei carries noteworthy implications for comprehensive genomic and systematic inquiries at elevated taxonomic strata, particularly given the prevailing uncertainties surrounding deep-seated relationships within the Ovulidae family.
Result
Genome structure and composition
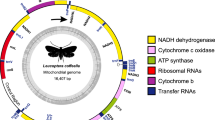
The complete mitogenome sequence of V. habei represents a typical closed-loop molecule with a length of 16,519 bp (GenBank accession number OR492307) (Fig. 1). This genome encompasses a total of 37 genes, comprising 13 PCGs, 22 tRNAs, two rRNAs (16 S rRNA and 12 S rRNA) and three concise non-coding regions. All these genes have been identified and are delineated in Fig. 1; Table 1. Among the 37 genes, the majority are located on the heavy strand, with only eight tRNAs (trnM, trnY, trnC, trnW, trnQ, trnG, trnE and trnT) situated on the light strand. Within the 13 PCGs, nad5 is the longest (1695 bp), while atp8 is the shortest (159 bp). The complete mitogenome of V. habei harbors eight overlapping regions, with two regions exceeding or equal to 10 bp in length, specifically cox2 and 16 S rRNA. Regions with fewer than 10 bp include trnW, trnG, 12 S rRNA, trnV, trnL and trnS. In these overlapping regions, two pairs of PCGs overlap (atp8 and atp6; nad4L and nad4). Additionally, the mitogenome of V. habei includes 27 intergenic regions spanning 952 bp, with lengths ranging from 1 to 232 bp (Table 1). Regarding nucleotide composition, the mitogenome of V. habei comprises A (27.97%), T (40.66%), G (17.82%) and C (13.54%), demonstrating a notable AT bias. The cumulative A + T (%) content of the mitogenome is 68.63%. In the selected complete mitogenome, the mitogenome of V. habei shows a negative AT skewness (-0.185) and a positive GC skewness (0.137), suggesting a prevalence of T and C compared to A and G (Table 2).

Maps of the mitochondrial genomes of Volva habei.
Transfer RNAs, ribosomal RNAs
Similar to widespread patterns observed in many invertebrate species27,28, the mitogenome of V. habei comprises 22 tRNA genes, with 14 encoded by the heavy strand and eight by the light strand. Throughout the entire mitogenome, the size of tRNA molecules ranges from 65 to 71 bp, resulting in a total length of 1478 bp for the 22 tRNA genes. Notably, both trnS and trnL exist in duplicate copies. In the mitogenome sequence of V. habei, the lengths of the two rRNA genes, 16 S rRNA and 12 S rRNA, are 1396 and 882 bp, respectively. They are positioned between trnV and two additional tRNA genes, trnL1 and trnE, with mutual separation facilitated by trnV (Fig. 1; Table 1). Furthermore, the tRNAs exhibit a distinct AT bias (67.12%). The values for AT skewness and GC skewness are recorded as 0.014 and 0.193, respectively, indicating a slight preference for adenine and a pronounced preference for guanine (Table 2). After predicting the secondary structure of the 22 tRNA genes of V. habei, it was observed that trnS2 cannot form the typical cloverleaf structure due to the absence of dihydrouracil (DHU) arms. This structural variation in trnS2 is consistent with similar observations in mitogenomes of other invertebrates29,30,31. In contrast, other tRNAs have the capability to fold into the conventional cloverleaf structure. Additionally, all tRNAs exhibit G-C mismatches. On the Acceptor stem, U-U mismatches are observed in trnI due to C-T transitions, and C-U mismatches are observed in trnC due to A-C transitions (Fig. 2).

Secondary structure of the tRNA genes in the mitogenome of Volva habei.
PCGs and codon usage
The results of the start and stop codons for all PCGs in V. habei are presented in Table 2. The mitogenome of V. habei comprises a total of 13 PCGs, including one cytochrome b (cob), two ATPases (atp6 and atp8), three cytochrome oxidases (cox1-3), and seven NADH dehydrogenases (nad1–6 and nad4l). This configuration aligns with established structural patterns observed in the Cypraeoidea27. The total length of these 13 PCGs is 11,025 bp. Within this set, individual PCGs vary in length from 159 to 1695 bp. Notably, the average A + T content is 67.05%, with a range from 63.84% (cox3) to 72.95% (atp8). The AT skewness and GC skewness values are calculated as -0.260 and 0.129, respectively (Table 2). Most PCGs initiate with the start codon ATG, except for nad2, nad5 and nad6, which use ATT, and nad1, which uses ATA as their start codon. Additionally, the predominant stop codons are TAG or TAA (Table 1). In V. habei, Phe is the most frequently occurring amino acid, while Gln is the least common (Fig. 3). The relative synonymous codon usage (RSCU) values are detailed in Table 4; Fig. 4. Among these, UUA (Leu) is the most prevalent codon, while CUG (Leu) is the least common (Fig. 4; Table 3).

Amino acid composition of Volva habei mitochondrial genomes.

Relative synonymous codon usage (RSCU) in the mitogenomes of Volva habei.
Gene re-arrangement
Mitogenome rearrangements in gene order provide an independent dataset for resolving evolutionary relationships. Patterns of mitochondrial gene order rearrangements shared among different taxonomic groups may indicate shared ancestral relationships rather than products of shared evolution32,33. In comparison to the ancestral gastropod gene arrangement, there are notable rearrangements in the mitogenome of V. habei. As illustrated in Fig. 5, at least three gene clusters (or genes) differ markedly from the conventional arrangement, including 15 tRNA genes (M, Y, C, W, Q, G, E, V, L, L, P, S, H, F and T) and eight PCGs (16 S rRNA, 12 S rRNA, nad1, nad6, cob, nad4l, nad4 and nad5). The rearrangements of these three gene clusters (or genes) are detailed below (Fig. 5): (1) The M-Y-C-W-Q-G-E cluster has been relocated downstream of atp6; (2) The T cluster has shifted downstream of F; (3) A rearrangement and translocation have occurred in the F-nad5-H-nad4-nad4l-S-cob-nad6-P-nad1-L-16 S-V-12 S cluster.

Comparison of mitochondrial gene rearrangements of the Volva habei. The blue squares represent PCGs, the green squares represent tRNAs, and the orange squares represent rRNAs.
Phylogenetic relationships
The comparative analysis of individual genes or gene segments provides a more comprehensive understanding of genomic-level features, including genetic resources, molecular evolution, genome evolution, and phylogenetic relationships. In this study, we conducted a phylogenetic analysis using the sequences of 13 PCGs to gain insights into the relationships within the Littorinimorpha clade, with a particular focus on V. habei. Additionally, we included 66 other Littorinimorpha species in the analysis, using A. heterodon and A. varicosa as outgroups. Maximum Likelihood (ML) and Bayesian Inference (BI) methods were employed to construct two phylogenetic trees with identical topologies but different support values. The topology based on the support values from the maximum likelihood tree was selected for presentation and display (Fig. 6).

The phylogenetic tree was inferred from the nucleotide sequences of 13 mitogenome PCGs using BI and ML methods. Numbers on branches indicate posterior probability (BI) and bootstrap support (ML).
Within the phylogenetic tree encompassing the 20 families, each family formed a monophyletic group, supported by high confidence values. Phylogenetic analysis revealed relationships within nine superfamilies in Littorinimorpha as follows: (((Truncatelloidea + Rissooidea) + (Littorinoidea + Naticoidea)) + ((Stromboidea + Xenophoroidea) + (Tonnoidea + Cypraeoidea)) + Vermetoidea). All nine were identified as monophyletic groups, and some previous studies have supported this notion34,35. The phylogenetic tree indicates that Cypraeidae forms a clade, the Ovulidae family, where V. habei belongs, constitutes another clade, and the families Ovulidae and Cypraeidae are sister groups, collectively forming the Cypraeoidea.
Discussion
In comparison to previously reported mitogenomes of other Cypraeoidea members36,37, the sequenced mitogenome in this study exhibits highly similar patterns in terms of genome size, nucleotide composition, and AT content. Investigation into the nucleotide composition of the V. habei mitogenome reveals a distinct AT bias within its genomic structure. This feature is reminiscent of the mitochondrial composition observed in the majority of bivalve mollusks and is a shared characteristic across various animal taxa, including fish and crustaceans38,39. V. habei also exhibits overlapping adjacent genes, a phenomenon commonly observed in molluscan mitogenomes40,41,42. All thirteen protein-coding genes in the V. habei mitogenome are encoded on the heavy strand, aligning with a pattern observed in certain gastropod mollusks, such as Nassariids43, which also encode proteins on the heavy strand. V. habei possesses a total of thirteen intact PCGs, consistent with the quantity and types of PCGs found in the majority of gastropod mollusks. This distinguishes it from some bivalve mollusks, such as true oysters, scallops, and other bivalves, which only possess twelve PCGs44.
In the context of gene rearrangement patterns, three main types are typically identified45: (1) Shuffle, where genes move from their original positions to adjacent positions on the same strand, often without crossing PCGs; (2) Transposition, where genes cross multiple genes, often including protein-coding genes, moving from their original positions to new locations; (3) Inversion, involving genes switching from one strand to the other. Based on the features of mitochondrial sequences, our analysis suggests that inversion and transposition are the primary rearrangement types observed in V. habei. However, mitochondrial gene rearrangements are often associated with an elevated rate of evolution46. Previous studies have found a significant positive correlation between the rate of gene order rearrangement in mitogenomes and an accelerated rate of evolution47. Interestingly, in contrast to the extensive gene rearrangements observed in Lottiidae, the differences in gene order within Littorinimorpha are relatively minor48, excluding Vermetoidea, Truncatelloidea and Stromboidea. We speculate that this could be attributed to the relatively small variations in genome size among Littorinimorpha species, ranging from 15,078 bp to 17,504 bp (Table 1), compared to the mitogenome size in Lottiidae, which spans from 16,319 bp to 26,835 bp. Further research is needed to explore this correlation in a broader range of gastropod taxa. Additionally, we compared the gene order in V. habei with other superfamilies within Littorinimorpha. Except for Vermetoidea, Truncatelloidea, and Stromboidea, the gene order in other superfamilies remains relatively consistent. Notably, in Truncatelloidea, the species Helicostoa sinensis and in Stromboidea, the species Ministrombus variabilis exhibit the absence of trnF in their gene orders. In Vermetoidea, deviations from the conserved order primarily involve tRNAs. Furthermore, the M-Y-C-W-Q-G-E cluster in the mitogenome of Vermetoidea undergoes inversion, a phenomenon observed in other gastropod mitogenomes12, resulting in the disruption and rearrangement of the cluster. Interestingly, a closely related set of genes experienced rearrangement in the common ancestor of Caenogastropoda, although the integrity of the M-Y-C-W-Q-G-E cluster was maintained40,49,50. These findings align with conclusions drawn from phylogenetic analyses based on gene order, emphasizing the value of comparing mitochondrial gene rearrangements as a tool in systematic studies.
The results of the phylogenetic analysis indicate that Naticoidea and Littorinoidea are the closest sister groups, consistent with findings by Santos51. Vermetoidea is positioned at the base of the monophyletic Littorinimorpha, aligning with previous research50. This placement may also be linked to results of gene rearrangement, as the gene sequences of Vermetoidea significantly differ from those of other species, leading to its basal position in the phylogenetic tree.
Stromboidea, encompassing the highest number of families, constitutes a highly diverse group. Currently recognized to include six extant families: Aporrhaidae, Rostellariidae, Seraphsidae, Strombidae, Struthiolariidae and Xenophoridae. At the family level, each family’s species forms a distinct clade. Strombidae, Seraphsidae, and Rostellariidae exhibit close phylogenetic relationships, while Aporrhaidae, Struthiolariidae and Xenophoridae form another closely related group. This is consistent with the findings of Irwin52, who suggested that in mitochondrial gene analysis, combined with nuclear gene analysis, Xenophoridae resolves as the sister group with the highest support to Struthiolariidae + Aporrhaidae. The positions of Rostellariidae and Seraphsidae vary in different analyses. Prokaryotic analyses often recover Rostellariidae as the sister group to Strombidae, while Bayesian inference analysis places it as the sister group to Seraphsidae. Only nuclear gene analysis recovers Seraphsidae as the sister group to Strombidae, with varying support levels52.
Previous studies have established Tonnoidea as a monophyletic group, identifying Ficoidea as its sister group. Our study not only reaffirms the monophyly of Tonnoidea but also uncovers that Tonnoidea is the sister group to Cypraeoidea. Although this study does not include species from the superfamily Ficoidea, we hypothesize, based on existing data, that Ficoidea species may also share a sister group relationship with Cypraeoidea, similar to Tonnoidea53,54,55. In 1973, Cate classified the Ovulidae within the Cypraeoidea taxonomy, a classification that is further substantiated by the phylogenetic analysis results obtained in this study10. Due to the faster evolutionary rate of Ovulidae compared to other gastropod groups, they exhibit high phenotypic plasticity56,57,58,59, making classification based solely on morphology more challenging. Recent studies have utilized cox1 and 16 S rRNA to investigate the phylogeny of Ovulidae, revealing the ability of both markers to distinguish specimens at the species level. This led to the discovery of synonymy among Ovulidae species58,59,60,61 employed barcode technology to analyze sequences of over 2000 individuals from 263 taxonomic groups within Cypraeidae, achieving a 98% accuracy in identifying Evolutionarily Significant Units (ESUs) using neighbor-joining methods. While some have discussed the limitations of mitogenomics for inferring deep-sea mollusk relationships62,63, the presented phylogenetic tree offers a robust framework, enhancing our understanding of Caenogastropoda relationships.
Conclusion
In this investigation, we employed next-generation sequencing technology to meticulously sequence the mitogenome of V. habei, with the goal of obtaining pertinent mitochondrial data for Ovulidae. An extensive analysis of the mitogenome of V. habei unveiled considerable similarity with other members of Littorinimorpha, marked by distinctive features such as AT bias and specific preferences in codon usage. Through comparative analysis with ancestral gastropods, notable rearrangements in the gene order of the mitogenome of V. habei were identified. Meanwhile, Littorinimorpha exhibited five distinct rearrangement patterns. The consistent similarity in these rearrangements underscores their phylogenetic relationships. Our phylogenetic tree, when compared to previous studies, demonstrates both consistency and some variations. Phylogenetic analysis indicates that V. habei forms a distinct Calyptraeoidea clade, while (((Truncatelloidea + Rissooidea) + (Littorinoidea + Naticoidea)) + ((Stromboidea + Xenophoroidea) + (Tonnoidea + Cypraeoidea)) + Vermetoidea) constitutes a separate clade. Despite the limitation in the number of species, our phylogenetic tree garners statistical support, providing a rational framework for future phylogenetic studies in the Calyptraeoidea field. These findings not only contribute insights into the gene arrangement characteristics in the mitogenome of Littorinimorpha but also establish the foundation for a more profound exploration of the phylogeny within Littorinimorpha. The study sheds light on features of mollusk mitogenomes, advancing our understanding of the phylogeny within the gastropod order, crucial for further species identification research and the implementation of effective management and control strategies.
Materials and methods
Sampling and DNA extraction
The specimens were meticulously collected in Zhejiang, adhering to strict sampling protocols. After collection, they were immersed in ethanol and preserved at -20 °C. Expert taxonomists at the Marine Biological Museum of Zhejiang Ocean University conducted a preliminary morphological assessment to authenticate their taxonomic classification. To achieve taxonomic precision, complete genomic DNA was carefully extracted from muscle tissues using the salt-extraction method and stored at -20 °C64, laying the foundation for subsequent in-depth analyses in mitochondrial whole-genome research.
Genome sequencing, assembly and annotation
The mitogenomes of V. habei were meticulously sequenced by Origin Gene Co. Ltd. in Shanghai, China, leveraging the advanced Illumina HiSeq X Ten sequencing platform. Following a protocol akin to that employed for V. habei, HiSeq X Ten libraries were meticulously crafted, featuring insert sizes spanning 300 to 500 bp, sourced from genomic DNA samples, resulting in an impressive 10 gb of raw data. Stringent preprocessing procedures were implemented, encompassing the exclusion of low-quality reads, adapters, sequences containing a notable proportion of ambiguous bases (“N” bases), and those with a length below 25 bp.The assembly process was executed with the aid of the sophisticated NOVOPlasty software65, accessible at https://github.com/ndierckx/NOVOPlasty. Annotation and manual refinement of the assembly were conducted with meticulous attention to established mitogenome datasets. De novo assembled mitogenomes were generated using the powerful MITOS tools66, accessed through the MITOS Web Server at uni-leipzig.de. Sequence accuracy was systematically validated through alignment with mitochondrial genes from other Muricidae species, further confirmed by employing the COI barcode sequence and conducting NCBI BLAST searches67.
High-throughput sequencing reads underwent de novo assembly, and the comprehensive annotation of the mitogenomes was meticulously performed using Sequin version 16.0. The intricate landscape of the V. habei mitogenome was vividly portrayed using the web-based tool Poksee, accessible at https://proksee.ca68, enhancing our understanding of its structural organization.The tertiary structures of tRNA genes were anticipated and visually presented through the sophisticated MITOS Web Server. In-depth insights into coding sequence characteristics were garnered through the computation of relative synonymous codon usage (RSCU) values, and an evaluation of substitution saturation for the 13 PCGs was executed using DAMBE 5. Subsequent analysis and interpretation of these values were meticulously conducted using MEGA 769. Furthermore, a nuanced exploration of base compositional disparities and strand asymmetry among samples was undertaken, involving the calculation of GC-skews and AT-skews. These parameters were intricately determined using the following formulas: AT-skew=[A − T]/[A + T] and GC skew=[G − C]/[G + C]. The extent of substitution saturation for the 13 PCGs was quantified with precision using DAMBE 570. This comprehensive analytical approach provides a robust foundation for understanding the intricacies of the V. habei mitogenome.
Gene order analysis
Beyond the mitogenomes subjected to sequencing in this investigation, an additional set of 66 complete mitogenomes belonging to the Littorinimorpha was procured from GenBank (Table 4) to facilitate comparative analyses. This expansive dataset underwent meticulous scrutiny, as all 66 mitogenomes were juxtaposed against the ancestral Gastropoda gene arrangement. The aim was to pinpoint potential novel gene orders that had not been documented previously. To mitigate the risk of misinterpretation arising from potential misannotations, any mitogenomes within the Littorinimorpha exhibiting deviations from the ancestral pattern underwent a comprehensive reannotation process employing MITOS66. This rigorous approach ensures the accuracy and reliability of observed gene order differences, thereby enhancing the robustness of our comparative analyses.
Phylogenetic analysis
The study aimed to investigate the phylogenetic relationships within Littorinimorpha using 13 PCGs. A dataset of 69 mitogenome sequences was compiled from GenBank, including Alasmidonta heterodon and Alasmidonta varicosa as outgroups. Both ML and BI methods were applied71,72,73. IQ-TREE 1.6.2 with 1000 bootstrap replicates identified the optimal TIM2 + F + R10 model based on Bayesian Information Criterion (BIC). Interdisciplinary associations were established using MrMTgui74, integrating PAUP, ModelTest, and MrModelTest 2.3. Bayesian phylogenetic analyses in MrBayes 3.2 used the GTR + I + G model chosen by Akaike Information Criterion (AIC). Markov Chain Monte Carlo (MCMC) chains ran for 2,000,000 generations, sampling every 1,000 steps, with a 25% burn-in. Convergence was assessed by mean standard deviation of split frequencies (< 0.01). Resulting phylogenetic trees, revealing nuanced Littorinimorpha relationships, were visualized and refined using Figure Tree v.1.4.375. This analytical framework enhances understanding of evolutionary dynamics in the studied taxa.
Data availability
The mitochondrial genome data has been submitted to NCBI GenBank under the following Accession Numbers OR492307.
References
Oyama, K. Review of nomenclature on Japanese shells (3). Venus (Jpn. J. Malacol.) 20, 361–362. https://doi.org/10.18941/venusjjmb.20.4_361 (1961).
Cate, C. N. The ovulidae: replacement names for four ovulid homonyms. Veliger 16, 238–239 (1973).
Fehse, D. Beitrage Zur Kenntnis Der ovulidae (mollusca: Cypraeoidea) viii. Einleitung sur familie sowie katalog, taxonomie und bibliographie und bemerkungen zu verwandten gruppen. Acta Conchyliorum 5, 3–47 (2001).
Dolin, L. & Ledon, D. Nouveaux Taxons Et discussion de la systématique des genres correspondants d’ovulidae (mollusca, caenogastropoda) de l’éocène inférieur de gan (France). Geodiversitas 24, 329–347 (2002).
Wilson, B. & Superfamily Cypraeoidea Mollusca: The Southern Synthesis. Fauna of Australia 5, 780–786 (1998).
Gosliner, T. M. Aspects of the morphology of the endemic South African cypraeidae with a discussion of the evolution of the Cypraeacea and Lamellariacea. Ann. S. Afr. Mus. 96, 67–122 (1985).
Meyer, C. P. Molecular systematics of cowries (Gastropoda: Cypraeidae) and diversification patterns in the tropics. Biol. J. Linn. Soc. 79, 401–459. https://doi.org/10.1046/j.1095-8312.2003.00197.x (2003).
Meyer, C. P. Toward comprehensiveness: Increased molecular sampling within Cypraeidae and its phylogenetic implications. Malacologia-Philadelphia-. 46, 127–156 (2004).
Allan, J. Cowry Shells of World Seas (Georgian House, 1956). https://doi.org/10.5962/p.341903
Cate, C. N. A systematic revision of the recent cypraeid family Ovulidae (Mollusca: Gastropoda). Veliger 15, 1-116 (1973).
Boore, J. L. Animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780. https://doi.org/10.1093/nar/27.8.1767 (1999).
Uribe, J. E., Kano, Y., Templado, J. & Zardoya, R. Mitogenomics of Vetigastropoda: insights into the evolution of pallial symmetry. Zool. Scr. 45, 145–159. https://doi.org/10.1111/zsc.12146 (2016).
Abalde, S. et al. Phylogenetic relationships of cone snails endemic to cabo verde based on mitochondrial genomes. BMC Evol. Biol. 17, 1–19. https://doi.org/10.1186/s12862-017-1069-x (2017).
Uribe, J. E., Irisarri, I., Templado, J. & Zardoya, R. New Patellogastropod mitogenomes help counteracting long-branch attraction in the deep phylogeny of gastropod mollusks. Mol. Phylogenet. Evol. 133, 12–23. https://doi.org/10.1016/j.ympev.2018.12.019 (2019).
Irisarri, I., Uribe, J. E., Eernisse, D. J. & Zardoya, R. A mitogenomic phylogeny of chitons (Mollusca: Polyplacophora). BMC Evol. Biol. 20, 1–15. https://doi.org/10.1186/s12862-019-1573-2 (2020).
Wang, Y. et al. Phylogeny of Veneridae (Bivalvia) based on mitochondrial genomes. Zool. Scr. 50, 58–70. https://doi.org/10.1111/zsc.12454 (2021).
Brown, W. M., George Jr, M. & Wilson, A. C. Rapid evolution of animal mitochondrial DNA. Proc. Nati. Acad. Sci.. 76, 1967–1971. https://doi.org/10.1073/pnas.76.4.1967 (1979).
Gissi, C., Iannelli, F. & Pesole, G. Evolution of the mitochondrial genome of metazoa as exemplified by comparison of congeneric species. Heredity (Edinb) 101, 301–320. https://doi.org/10.1038/hdy.2008.62 (2008).
Kurabayashi, A. et al. Phylogeny, recombination, and mechanisms of stepwise mitochondrial genome reorganization in mantellid frogs from Madagascar. Mol. Biol. Evol. 25, 874–891. https://doi.org/10.1093/molbev/msn031 (2008).
Lavrov, D. V. et al. Mitochondrial DNA of Clathrina Clathrus (Calcarea, Calcinea): six Linear chromosomes, fragmented rRNAs, tRNA editing, and a Novel Genetic Code. Mol. Biol. Evol. 30, 865–880. https://doi.org/10.1093/molbev/mss274 (2013).
Lavrov, D. V. & Pett, W. Animal mitochondrial dna as we do not know it: Mt-genome organization and evolution in nonbilaterian lineages. Genome Biol. Evol. 8, 2896–2913. https://doi.org/10.1093/gbe/evw195 (2016).
Abalde, S., Tenorio, M. J., Afonso, C. M. & Zardoya, R. Mitogenomic phylogeny of cone snails endemic to Senegal. Mol. Phylogenet. Evol. 112, 79–87. https://doi.org/10.1186/s12862-017-1069-x (2017).
Uribe, J. E. & Zardoya, R. Revisiting the phylogeny of cephalopoda using complete mitochondrial genomes. J. Molluscan Stud. 83, 133–144. https://doi.org/10.1093/mollus/eyw052 (2017).
Boore, J. L., Macey, J. R. & Medina, M. Sequencing and comparing whole mitochondrial genomes of animals. Methods Enzymol. 395, 311–348. https://doi.org/10.1016/S0076-6879(05)95019-2 (2005).
Simison, W. B., Lindberg, D. R. & Boore, J. L. Rolling circle amplification of metazoan mitochondrial genomes. Mol. Phylogenet. Evol. 39, 562–567. https://doi.org/10.1016/j.ympev.2005.11.006 (2006).
Hudson, M. E. Sequencing breakthroughs for genomic ecology and evolutionary biology. Mol. Ecol. Resour. 8, 3–17. https://doi.org/10.1111/j.1471-8286.2007.02019.x (2008).
Yu, Y., Kong, L. & Li, Q. Mitogenomic phylogeny of muricidae (Gastropoda: Neogastropoda). Zool. Scr. 3, 1892–1894. https://doi.org/10.1111/zsc.12598 (2023).
Yang, H. R. et al. The complete mitochondrial genome of the mudsnail Cipangopaludina Cathayensis (Gastropoda: Viviparidae). Mitochondr. DNA A 27, 1892–1894. https://doi.org/10.3109/19401736.2014.971274 (2016).
Yang, J. & Yang, W. The complete mitochondrial genome sequence of the hydrothermal vent galatheid crab Shinkaia Crosnieri (Crustacea: Decapoda: Anomura): a novel arrangement and incomplete TRNA suite. BMC Genom. 9, 1–13. https://doi.org/10.1186/1471-2164-9-257 (2008).
Tan, M. H. et al. Comparative mitogenomics of the decapoda reveals evolutionary heterogeneity in architecture and composition. Sci. Rep. 9, 10756. https://doi.org/10.1038/s41598-019-47145-0 (2019).
Guo, Y. et al. Characterization of the complete mitochondrial genomes of two species of penaeidae (decapoda: Dendrobranchiata) and the phylogenetic implications for penaeoidea. Genomics 113, 1054–1063. https://doi.org/10.1016/j.ygeno.2020.11.001 (2021).
Kilpert, F., Held, C. & Podsiadlowski, L. Multiple rearrangements in mitochondrial genomes of isopoda and phylogenetic implications. Mol. Phylogenet. Evol. 64, 106–117. https://doi.org/10.1016/j.ympev.2012.03.013 (2012).
Zhang, Y. et al. Gene rearrangements in the mitochondrial genome of chiromantes eulimene (brachyura: Sesarmidae) and phylogenetic implications for brachyura. Int. J. Biol. Macromol. 162, 704–714. https://doi.org/10.1016/j.ijbiomac.2020.06.196 (2020).
Zhong, S., Huang, L., Huang, G., Liu, Y. & Wang, W. The first complete mitochondrial genome of mammilla from Mammilla Mammata (Littorinimorpha: Naticidae). Mitochondr. DNA B 5, 96–97. https://doi.org/10.1080/23802359.2019.1698350 (2020).
Choi, E. H. & Hwang, U. W. The complete mitochondrial genome of an endangered triton snail Charonia lampas (littorinimorpha: Charoniidae) from South Korea. Mitochondr. DNA B 6, 956–958. https://doi.org/10.1080/23802359.2021.1889416 (2021).
Fukumori, H., Itoh, H. & Irie, T. The mitochondrial genome of the gold-ringed cowry Monetaria Annulus (mollusca: Gastropoda: Cypraeidae) determined by whole-genome sequencing. Mitochondr. DNA B 4, 2305–2307. https://doi.org/10.1080/23802359.2019.1627946 (2019).
Pu, L. et al. Complete mitochondrial genome of tiger cowrie cypraea tigris (Linnaeus, 1758). Mitochondr. DNA B 4, 2755–2756. https://doi.org/10.1080/23802359.2019.1627933 (2019).
Yamauchi, M. M., Miya, M. U. & Nishida, M. Complete mitochondrial dna sequence of the swimming crab, Portunus Trituberculatus (Crustacea: Decapoda: Brachyura). Gene 311, 129–135. https://doi.org/10.1016/s0378-1119(03)00582-1 (2003).
Prosdocimi, F., de Carvalho, D. C., de Almeida, R. N. & Beheregaray, L. B. The complete mitochondrial genome of two recently derived species of the fish genus Nannoperca (Perciformes, Percichthyidae). Mol. Biol. Rep. 39, 2767–2772. https://doi.org/10.1007/s11033-011-1034-5 (2012).
Cunha, R. L., Grande, C. & Zardoya, R. Neogastropod phylogenetic relationships based on entire mitochondrial genomes. BMC Evol. Biol. 9, 1–16. https://doi.org/10.1186/1471-2148-9-210 (2009).
Rawlings, T. A., MacInnis, M. J., Bieler, R., Boore, J. L. & Collins, T. M. Sessile snails, dynamic genomes: Gene rearrangements within the mitochondrial genome of a family of caenogastropod molluscs. BMC Genom. 11, 1–24. https://doi.org/10.1186/1471-2164-11-440 (2010).
Wang, J., Zhang, D., Jakovlić, I. & Wang, W. Sequencing of the complete mitochondrial genomes of eight freshwater snail species exposes pervasive paraphyly within the viviparidae family (caenogastropoda). PLoS One 12, e0181699. https://doi.org/10.1371/journal.pone.0181699 (2017).
Yang, Y., Liu, H., Qi, L., Kong, L. & Li, Q. Complete mitochondrial genomes of two toxin-accumulated nassariids (neogastropoda: Nassariidae: Nassarius) and their implication for phylogeny. Int. J. Mol. Sci. 21, 3545. https://doi.org/10.3390/ijms21103545 (2020).
Xia, L. P., Xu, M. H. & Guo, B. Y. Complete mitochondrial genome sequencing and phylogeny analysis of Macridiscus Multifarius. J. Zhejiang Ocean. Univ. (Nat. Sci.) 40, 93–100. https://doi.org/10.3969/j.issn.1008-830X.2021.02.001 (2021).
Zhang, Y. Mitochondrial genome rearrangement of sesarmidae species and its phylogenetic implication. Zhejiang Ocean Univ. https://doi.org/10.27747/d.cnki.gzjhy.2022.000441 (2022).
Bernt, M. et al. A comprehensive analysis of bilaterian mitochondrial genomes and phylogeny. Mol. Phylogenet. Evol. 69, 352–364. https://doi.org/10.1016/j.ympev.2013.05.002 (2013).
Xu, W., Jameson, D., Tang, B. & Higgs, P. G. The relationship between the rate of molecular evolution and the rate of genome rearrangement in animal mitochondrial genomes. J. Mol. Evol. 63, 375–392. https://doi.org/10.1007/s00239-005-0246-5 (2006).
Xu, T., Qi, L., Kong, L. & Li, Q. Mitogenomics reveals phylogenetic relationships of patellogastropoda (mollusca, gastropoda) and dynamic gene rearrangements. Zool. Scr. 51, 147–160. https://doi.org/10.1111/zsc.12524 (2022).
Osca, D., Templado, J. & Zardoya, R. The mitochondrial genome of ifremeria nautilei and the phylogenetic position of the enigmatic deep-sea abyssochrysoidea (Mollusca: Gastropoda). Gene 547, 257–266. https://doi.org/10.1016/j.gene.2014.06.040 (2014).
Osca, D., Templado, J. & Zardoya, R. Caenogastropod mitogenomics. Mol. Phylogenet. Evol. 93, 118–128. https://doi.org/10.1016/j.ympev.2015.07.011 (2015).
Santos, C. A., Bezerra, F. O. & Andrade, S. C. L. Ittoraria Flava (Gastropoda: Littorinidae) mitogephylogeneticenetic considerations within the caenogastropoda and evidence of microscale local adaptation. Mar. Biol. 169, 121. https://doi.org/10.1007/s00227-022-04108-w (2022).
Irwin, A. R., Strong, E. E., Kano, Y., Harper, E. M. & Williams, S. T. Eight new mitogenomes clarify the phylogenetic relationships of stromboidea within the caenogastropod phylogenetic framework. Mol. Phylogenet. Evol. 158, 107081. https://doi.org/10.1016/j.ympev.2021.107081 (2021).
Lemarcis, T. et al. Neogastropod (Mollusca, Gastropoda) phylogeny: a step forward with mitogenomes. Zool. Scr. 51, 550–561. https://doi.org/10.1111/zsc.12552 (2022).
Strong, E. E. et al. Frogs and tuns and tritons-A molecular phylogeny and revised family classification of the predatory gastropod superfamily Tonnoidea (Caenogastropoda). Mol. Phylogenet. Evol. 130, 18–34. https://doi.org/10.1016/j.ympev.2018.09.016 (2019).
Wang, Q. et al. Characterization of the complete mitochondrial genome of Ficus Variegata (Littorinimorpha: Ficidae) and molecular phylogeny of Caenogastropoda. Mitochondr. DNA B 6, 1126–1128. https://doi.org/10.1080/23802359.2021.1901628 (2021).
Reijnen, B. T. & van der Meij, S. E. Coat of many colours—dna reveals polymorphism of mantle patterns and colouration in caribbean cyphoma röding, 1798 (Gastropoda, Ovulidae). PeerJ 5, e3018. https://doi.org/10.7717/peerj.3018 (2017).
Lorenz, F. The black morph cyphoma from the Netherlands antilles (Gastropoda: Ovulidae). Acta Conchyliorum 19, 69–75 (2020).
Sánchez, J. A. et al. The masquerade game: Marine mimicry adaptation between egg-cowries and octocorals. PeerJ 4, e2051. https://doi.org/10.7717/peerj.2051 (2016).
Reijnen, B. T. & van der Meij, S. E. Systematics of the subfamily aclyvolvinae (Caenogastropoda: Ovulidae) based on molecular and morphometric analyses. J. Molluscan Stud. 85, 336–347. https://doi.org/10.1093/mollus/eyz020 (2019).
Schiaparelli, S., Barucca, M., Olmo, E., Boyer, M. & Canapa, A. Phylogenetic relationships within ovulidae (gastropoda: Cypraeoidea) based on molecular data from the 16s rRNA gene. Mar. Biol. 147, 411–420. https://doi.org/10.1007/s00227-005-1566-0 (2005).
Meyer, C. P. & Paulay, G. DNA barcoding: error rates based on comprehensive sampling. PLoS Biol. 3, e422. https://doi.org/10.1371/journal.pbio.0030422 (2005).
Stöger, I. & Schrödl, M. Mitogenomics does not resolve deep molluscan relationships (yet?). Mol. Phylogenet. Evol. 69, 376–392. https://doi.org/10.1016/j.ympev.2012.11.017 (2013).
Zapata, F. et al. Phylogenomic analyses of deep gastropod relationships reject Orthogastropoda. Proc. R. Soc. B Biol. Sci. 281, 20141739. https://doi.org/10.1098/rspb.2014.1739 (2014).
Aljanabi, S. M. & Martinez, I. Universal and rapid salt-extraction of high quality genomic dna for pcr-based techniques. Nucleic Acids Res. 25, 4692–4693. https://doi.org/10.1093/nar/25.22.4692 (1997).
Dierckxsens, N., Mardulyn, P. & Smits, G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45, e18. https://doi.org/10.1093/nar/gkw955 (2017).
Bernt, M. et al. Mitos: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319. https://doi.org/10.1016/j.ympev.2012.08.023 (2013).
Altschul, S. F. et al. Gapped blast and psi-blast: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. https://doi.org/10.1093/nar/25.17.3389 (1997).
Grant, J. R. & Stothard, P. The cgview server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 36, W181–W184. https://doi.org/10.1093/nar/gkn179 (2008).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. https://doi.org/10.1093/molbev/msw054 (2016).
Xia, X. & Xie, Z. DAMBE: software package for data analysis in molecular biology and evolution. J. Hered. 92, 371–373. https://doi.org/10.1093/jhered/92.4.371 (2001).
Perna, N. T. & Kocher, T. D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358. https://doi.org/10.1007/bf00186547 (1995).
Huelsenbeck, J. P. & Ronquist, F. MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. https://doi.org/10.1093/bioinformatics/17.8.754 (2001).
Nguyen, L., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. https://doi.org/10.1093/molbev/msu300 (2015).
Ronquist, F. et al. MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. https://doi.org/10.1093/sysbio/sys029 (2012).
Rambaut, A. Figtree, a graphical viewer of phylogenetic trees (2009).
Acknowledgements
The present manuscript was financially supported by the Project of Bureau of Science and Technology of Zhoushan (No. 2021C21017) and the National Key R&D Program of China (2019YFD0901204).
Author information
Authors and Affiliations
Contributions
X.J.L. conceived and designed the experiments, performed the experiments, analyzed the data, prepared figures and/or tables, and approved the final draft. J.J.L analyzed the data, prepared figures and/or tables, and approved the final draft. Y.Y.Y. conceived and designed the experiments, authored or reviewed drafts of the article, and approved the final draft.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liang, X., Li, J. & Ye, Y. A comprehensive study on the mitochondrial genome of Volva habei and exploring phylogenetic relationships in Littorinimorpha. Sci Rep 14, 29212 (2024). https://doi.org/10.1038/s41598-024-80695-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-80695-6
