
Abstract
The Anopheles maculipennis complex consists of several mosquito species, including some primary malaria vectors. Therefore, the presence of a species in a particular area significantly affects public health. In this study, 1252 mosquitoes were collected in northern Italy, representing four identified species of the Anopheles maculipennis complex (Anopheles daciae sp. inq., Anopheles maculipennis s. s., Anopheles atroparvus and Anopheles melanoon). The sequences of two DNA markers, mitochondrial cytochrome c oxidase I (COI) and nuclear internal transcribed spacer 2 (ITS2), were generated. DNA-based species delimitation analyses were performed, incorporating public sequences, with distance-based and coalescent tree-based methods to confirm the actual species boundaries within the complex. While some morphospecies were unequivocally delimited by all methods and markers, COI analysis splitted An. maculipennis s. s. into two well-supported groups. However, molecular delimitation failed in recognizing An. daciae sp. inq. and An. messeae as two separate evolutionary entities. Species delimitation was further tested with a morphometric approach, which clearly differentiated species collected in the survey area. These findings underscore how challenging the characterization of the taxonomy of the complex is, providing evidence of potential introgression events in An. maculipennis s. s. and suggesting the need for robust evidence to support An. daciae sp. inq. and An. messeae as distinct species.
Similar content being viewed by others

Introduction
The Anopheles genus includes more than 480 species, 70 of which are known to transmit malaria1. The genus enumerates several complex species characterized by different vectorial competence, ecology and/or behavior1. Several of the most dangerous vectors of malaria in the Holarctic region belong to the Anopheles maculipennis complex2. The identification of the species within this complex is hampered by their morphological similarity; indeed, the species were first defined based on the morphological features of their eggs3. Subsequently, species were also defined on cytogenetic and molecular bases4,5. Moreover, wing landmarks were identified as useful for discriminating among the species of the complex6. However, the taxonomy of the complex continues to be debated due to the intrinsic difficulty of defining clear diagnostic characters for delimiting species. Over the years, new species were described, and others were synonymized, such as Anopheles subalpinus, which was synonymized with An. melanoon7,8. No consensus has been reached within the scientific community on the taxonomic status of Anopheles daciae Linton, Nicolescu & Harbach9,10,11. Therefore, in this article, we will refer to it as species inquirenda (sp. inq.), namely a species of doubtful identity that requires further investigation.
In recent decades, with the development of biotechnologies, molecular approaches have played a central role in modern entomology, especially as a support for classical morphological approaches for species identification but also for the delimitation of taxa12,13. Molecular species delimitation methods and approaches are particularly useful for delimiting sibling insect taxa or species within species complexes14,15,16; thus, they often provide answers to taxonomic questions that are debated17,18,19. These methods were also used to define the taxonomic status of the taxa within complexes of the genus Anopheles20,21,22,23. The molecular discrimination of Anopheles species and the phylogenetic relationships among taxa of the genus have often been determined by using two markers: the mitochondrial gene cytochrome c oxidase subunit I (COI), usually the 5′ region, and the nuclear internal transcribed spacer 2 (ITS2) region between the ribosomal 5.8S gene and the 26S rRNA22,23,24. Although COI is recommended as a universal marker for insect molecular identification in DNA barcoding and metabarcoding25, it can lead to misidentification26. In fact, due to maternal inheritance, variability in COI can be influenced by mitochondrial introgression due to hybridization, potentially leading to misidentifications24. On the other hand, the biparentally inherited ITS2 region has proven to be particularly suitable for the identification of species of the genus Anopheles because it is often conserved within species and variable between species21,24. The low levels of intraspecific variation of this marker is probably driven by mechanisms of concerted evolution, which promote the homogenization of repeated sequences within species. At the same time, this non-coding region can accumulate mutations between species24,27. In summary, using both COI and ITS2 markers together facilitates cross-verification of species identifications, especially in instances where hybridization may complicate interpretations.
The main aim of this study is to explore the species boundaries within the Anopheles maculipennis complex using an integrated taxonomy approach, combining DNA-based species delimitation methods and wing morphometry. Moreover, the study evaluates the congruence between DNA marker signals and their consistency with morphological information to identify the most effective approach for delimiting species within the complex. We analyzed mosquito data gathered from more than one hundred sites across the Po Plain in Italy, along with data from public repositories.
Results
COI and ITS2 features of the collected species
The ITS2 gene was successfully amplified, and high-quality sequences were obtained for 1226 individuals out of the 1276 processed, obtaining sequences from 376 to 391 bases depending on the species; the COI marker was amplified from a subsample of 827 mosquitoes, and high-quality sequences of 659 bases were obtained for 480 specimens. DNA sequences of both markers were obtained for 454 individuals.
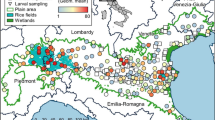
The sequences generated were ascribable to four different species An. daciae sp. inq. (1025, 81.9%), An. maculipennis s. s. (202, 16.1%), An. atroparvus (18, 1.4%) and An. melanoon (7, 0.6%) (Fig. 1). Moreover, we obtained eggs from 22 of the collected mosquitoes; all the deposed eggs belonged to the An. messeae/An. daciae morphotype. We obtained the ITS2 sequence from all but one of these specimens and the COI sequence from 12 specimens.

Position of collection sites, with reference to identified species (a) and location of the monitored area on the map of Italy (b). Haplotype median joining network of the ITS2 (c) and COI (d) sequences obtained in this study. Mutations are represented by one-step edges; the diameter of network circles is proportional to the number of sequences (empty circles represent 10 sequences). Map circles: blue, An. daciae sp. inq. (An. da.); yellow, An. maculipennis s. s. (An. ma.); green, An. da. and An. ma.; red, An. atroparvus, An.da. and An. ma.; light blue, An. melanoon (An. me.), An. da. and An. ma.; purple, An. me. and An. da.
In the haplotype network obtained from all the available ITS2 sequences, four clearly separated groups were present (Fig. 1d). For this marker, ambiguous bases were recorded in sequences of the same individuals as a result of intraindividual polymorphisms. COI produced a more dispersed network, which included subspecific groups, due to the highest variability of the marker (Fig. 1c). Conversely, the translated amino acid sequences were identical for the majority of conspecifics, and nonsynonymous mutations were detected within the same species in 26 sequences (14 from An. daciae sp. inq., 11 from An. maculipennis s. s., 1 from An. atroparvus).
Species delimitation
A subset of COI and ITS2 sequences generated in this study, along with orthologous sequences obtained from previous works, were used to construct datasets encompassing 118 individuals from the following species: An. atroparvus, An. beklemishevi, An. daciae sp. inq., An. maculipennis s. s., An. messeae, An. melanoon and An. sacharovi plus An. plumbeus (serving as an outgroup of the Maculipennis species complex).
Automatic Barcode Gap Discovery (ABGD) analyses highlighted the absence of a clear barcoding gap in the frequency distributions of pairwise nucleotide distances for both the COI and ITS2 datasets. In the case of the COI dataset, ABGD delimited from 2 to 97 hypothetical species. The highest congruence between the analyzed morphospecies and the retrieved hypothetical species was found in partitions ranging from a prior nucleotide distance of 0.34% to 0.43%, where nine groups were identified. Among these groups, a perfect match with the morphospecies was obtained only for An. atroparvus, An. beklemishevi, An. labranchiae and An. plumbeus. Within this range of nucleotide distances (0.34% to 0.43%), all the sequences of An. messeae and An. daciae sp. inq. were merged together in a single group, and those of An. melanoon clustered with a group of An. maculipennis s. s.; the latter species was split into two separate groups. In addition, An. sacharovi were split into two groups (Fig. 2b). In the case of ABGD analysis of the ITS2 dataset, perfect matches between the initial and recursive partitions were observed for 0.46% to 10% of the prior nucleotide distances tested. The number of groups in which ABGDs were identified ranged from nine (with prior nucleotide distances of 0.10–0.28%) to three (with prior nucleotide distances of 10%). In the partition with eight groups, a perfect match between the identified hypothetical species and the morphological species was observed for seven out of nine morphospecies included in the dataset. These seven morphospecies are An. atroparvus, An. beklemishevi, An. labranchiae, An. maculipennis s. s., An. melanoon and An. sacharovi. The two presumptive species—An. messeae and An. daciae sp. inq.—were lumped together (Fig. 2b).

Species delimitation of the Anopheles maculipennis species complex. (a) Bayesian ultrametric tree inferred from the COI dataset. (b) Bayesian ultrametric tree inferred from the ITS2 dataset. Morphological assignment of the individuals (an asterisk indicates individuals identified through egg morphology) and results of species delimitation analyses (ABGD, GMYC, mPTP) are reported using vertical bars, with different colors according to the different species delimitation analyses performed and different line textures indicating taxa delimited in the same unit but not adjacent to the tree. GenBank accession numbers are reported near to the species name. Support of the nodes is shown when > 0.7, omitted for minor lineages. The scale bar indicates the distance in substitutions per site. Sequences of An. messeae deposited in public repositories were renamed according to the presence of ITS2 polymorphisms diagnostic for An. daciae sp. inq.
Species delimitation analyses implementing the coalescent tree-based methods led to somewhat similar results. In the case of COI, the Generalized Mixed Yule Coalescent (GMYC) model resulted in a greater log likelihood than the null model, identifying a statistically significant transition from the Yule model to the coalescent model (logLGMYC = 801.8, logLNULL = 777, PLR test < 0.001), indicating a threshold between the inter- and intraspecific levels at − 0.031 relative time units (root time − 1, tip time 0). This threshold identifies 11 maximum likelihood entities (95% CI 11–17 units), six of which perfectly match the morphospecies An. atroparvus, An. beklemishevi, An. labranchiae, An. melanoon, An. sacharovi and An. plumbeus (Fig. 2a). The individuals morphologically identified as An. maculipennis s. s. were paraphyletic and were split into two well-supported clades (Bayesian posterior probability (BPP) ≥ 0.99) recognized by GMYC as different Evolutionary Significant Units (ESUs). In the COI tree, individuals of An. messeae and An. daciae sp. inq. morphospecies clustered together into a well-supported clade (BBP = 1). Within the last clade, GMYC identified three separate ESUs: the first including individuals of An. daciae sp. inq. plus the individual of An. messeae KZ3 AY258182 (clade 1, C1; Fig. 2a); the second consisted of the individuals of An. daciae BG2 AY258172 only (clade 2, C2; Fig. 2a); the third group was composed of individuals of An. daciae sp. inq. plus one individual whose identification is uncertain (An. messeae/daciae A22) and An. messeae KO3 AY258178 (clade 3, C3; Fig. 2a). Interestingly, the delimitation achieved using the multi-rate Poisson Tree Processes (mPTP) method on the COI dataset (Fig. 2a) was mainly congruent with that achieved using the GMYC method; however, the mPTP recognized all the An. messeae and An. daciae sp. inq. individuals as belonging to a single ESU. For ITS2 (Fig. 2b), the GMYC model exhibited a greater log likelihood than did the null model (logLGMYC = 1019.3, logLNULL = 984.2, PLR test < 0.001), and the Yule coalescent threshold identified by the model (threshold relative time − 0.005) split the nine Anopheles morphospecies into 9 ESUs (confidence intervals [8–9]). Almost perfect congruence with the existing morphospecies was observed. The only exceptions were An. daciae sp. inq. and An. messeae, where two ESUs were identified (Fig. 2b). These ESUs are not consistent between the two morphospecies since some sequences of An. daciae sp. inq. clustered with the two present An. messeae in a single clade (not corresponding to any COI clade), while the majority of sequences of An. daciae sp. inq. was recognized within a separate clade (Fig. 2b). Delimitation using the mPTP identified eight ESUs, seven of which were consistent with the morphospecies An. atroparvus, An. beklemishevi, An. labranchiae, An. maculipennis s. s., An. melanoon, An. plumbeus and An. sacharovi, while sequences of An. messeae and An. daciae sp. inq. were grouped into a single clade (Fig. 2b).
Morphometry results
The first three CDA eigenvalues explained a cumulative percentage of 99.3 of the total variance (CDA1 = 66.3%, CDA2 = 22.4% and CDA3 = 10.6%), suggesting a difference in wing shape among the sibling species (Fig. 3). Moreover, the permutation test on the mean CDA scores showed that the pairwise comparisons of wing shape distance were all statistically significant (Fig. 3).

Wing landmarks and canonical discriminant analysis (CDA) reported in 3D for the four sibling species: An. atroparvus, An. maculipennis s. s., An. melanoon, An. daciae sp. inq. Tanglegrams between the UPGMAs of the molecular markers and the wing shape.
The first PC dimension (explained variance: PCA1 = 66%) tested against the COI Unweighted Pair-Group Method with Arithmetic mean (UPGMA) dendrogram was not statistically significant considering all the metrics (Table S1), while the ITS2 UPGMA dendrogram was significant for the metrics Cmean and lambda (p value < 0.05, Table S1). The wing shape UPGMA showed a Baker’s gamma index of − 0.23 and a cophenetic correlation of − 0.25 with COI UPGMA and a Baker’s gamma index of 0.85 and a cophenetic correlation of 0.89 with ITS UPGMA, indicating a greater association between wing shape and ITS2 than with COI UPGMA (Fig. 3).
At the intraspecific level, the first two PCs of the morphospace of An. maculipennis s. s. and An. daciae sp. inq. showed a random ordination of the groups identified by species delimitation analysis performed on the COI (Fig. S1). For each species, we tested the associations between COI groups and wing shape. The LDA model achieved poor performance considering all the performance metrics computed on the testing set and failed to record differences between the two within An. maculipennis s. s. and An. daciae sp. inq. obtained from COI species delimitation analyses (Fig. S2).
Discussion
The morphological identification of species of the Anopheles maculipennis complex is challenging, which is why DNA-based molecular identification is widely used to discriminate between these species. In this study, we generated DNA sequences for COI and ITS markers, which are widely used in the identification of Anopheles species, and used these sequences coupled with orthologous sequences available in online databases to investigate the species boundaries within the complex. We used sequences generated from individuals identified with alternative methods to DNA-based methods, such as analyses of egg morphology or chromosome inversions, to integrate morphological and molecular evidence.
The two molecular markers analyzed in this study showed different levels of nucleotide variability. ITS2 showed limited intraspecific variability compared to the mitochondrial marker COI, which was more variable within species. This finding is consistent with the expected low intraspecific variability of ITS2, likely due to concerted evolution, while its higher interspecific variability may be attributed to the ability of this non-coding region to accumulate variations between species24. Additionally, the results confirm that COI is less suited for discriminate among recently diverged taxa24,36. The morphospecies An. atroparvus, An. beklemishevi, An. sacharovi and An. labranchiae were clearly delimited in separate evolutionary units (ESUs) by the molecular species delimitation methods adopted, both with COI and ITS2, while nonunivocal results were obtained for An. melanoon, An. maculipennis s. s., An. daciae sp. inq. and An. messeae. Considering the results obtained with the two markers independently, the ITS2 gene tree showed well-supported clades, and all the delimitation methods used largely corroborated the classical division into morphospecies of the complex. The only exceptions were An. messeae and An. daciae sp. inq., which were lumped in a single ESU or, in the case of GMYC, in two ESUs. Indeed, GMYC split An. messeae and An. daciae sp. inq. into two groups whose structure had no apparent congruence with the taxonomy or with the geography or climatic conditions of the collection sites of the individuals.
On the other hand, the COI tree exhibited a more complex branching pattern than the ITS2 tree. The species delimitation obtained from the COI marker identified meaningful evolutionary units that do not always reflect the current species boundaries, such as in the case of An. maculipennis s. s. and An. messeae-An. daciae sp. inq. Commonly, the high intraspecific nucleotide variability that this marker possesses in some groups of insects allows the detection of cryptic species28,29 and phylogeographic patterns30,31. These COI features, on the other hand, can also lead to an overestimation of species number. The most relevant split observed involved An. maculipennis s. s., where the individuals of this morphospecies were clustered into two separate but well-supported monophyletic clades in the ultrametric tree inferred from COI sequences. Based on this marker, An. maculipennis s. s. morphospecies was polyphyletic, and the two groups were separated by monophyletic An. labranchiae and An. melanoon. All the species delimitation methods adopted on COI—including ABGD, which does not rely on a gene tree as input—were congruent in identifying two ESUs within the An. maculipennis morphospecies, contrary to ITS2, which supported the monophyletic status of all An. maculipennis s. s. morphospecies. The polyphyly observed in An. maculipennis s. s. based on mitochondrial gene analysis could be attributed to at least four potential explanations: i. introgression events between a lineage of An. maculipennis and closely related species; ii. vertical transmission of bacteria, such as Wolbachia and Cardinium, influencing host reproduction and spread within populations of ancestral or introgressed haplotypes; iii. artifactual tree topology resulting from high nucleotide variation in COI sequences; iv. the presence of cryptic diversity within the morphospecies An. maculipennis s. s.
In detail, introgression, defined as the genetic exchange between species through the backcrossing of interspecific hybrids, is a common phenomenon in mosquitoes32. This phenomenon has previously been proposed as the most plausible explanation for the topology observed in the COI phylogenetic tree of Anopheles species from the southwest Pacific26, which, similar to what was observed in this study, contrasted with that obtained with the ITS gene. Moreover, introgression was recently proposed as a fundamental phenomenon driving speciation in Anopheles complexes33. Based on the results obtained in this study with the ABGD method, which merged in one taxonomic entity An. melanoon and one group of An. maculipennis s. s., we can suppose that introgression among the progenitors of these groups occurred. However, we cannot disregard the possibility that the observed results may be influenced by vertically transmitted bacteria, such as Wolbachia and Cardinium, which can induce reproductive segregation and incompatibility. Although this phenomenon has already been documented in Anopheles mosquitoes34, the topology of the COI tree of this study, notably the clustering of one of the two groups of An. maculipennis s. s. with An. melanoon and An. labranchiae, diminishes the likelihood of this hypothesis.
The artifactual topology of the COI tree can indeed be linked to the high nucleotide variability of the COI gene, which is particularly notable at third codon positions. This variability can impede the accurate resolution of relationships, even among closely related taxa35. Consequently, the observed tree topology may be a result of inherent COI features intertwined with the evolutionary history of An. maculipennis s. s. This interplay might have generated a tree in which two An. maculipennis s. s. clusters are separated by a basal split, and the artifact is represented by the An. labranchiae and An. melanoon, which clustered with one of them. Moreover, we cannot dismiss incomplete lineage sorting as an alternative explanation, given its challenging differentiation from other phenomena based solely on phylogenetic evidence36.
Despite the aforementioned inconsistencies in the delimitation of An. messeae and An. daciae sp. inq. across markers and methods, attributed solely to the molecular delimitation performed with GMYC, the results of the other two molecular delimitation methods are concordant. Specifically, these methods merge the two species into one ESU, aligning with morphological evidence. In fact, the discrimination between An. messeae and An. daciae sp. inq. is unattainable through adult features and the differences in their egg characteristics are negligible and lack statistical significance37,38. Initially, An. daciae sp. inq. was distinguished from An. messeae based on the presence of five single-nucleotide polymorphisms in the ITS2 sequences37. However, subsequent studies have identified intraindividual polymorphisms in this marker9, including within three of the five diagnostic sites for An. daciae sp. inq. and An. messeae39, leaving only two informative sites for discriminating between these two species. Other methodologies that could aid in determining the species status of these taxa include crossing experiments to evaluate hybrid sterility, cytogenetic markers and molecular investigations, which have yielded conclusive results for several Anopheles species complexes5. However, laboratory experiments on hybridizations between An. daciae sp. inq. and An. messeae may be challenging to conduct, and hybrids have been reported in various studies based on molecular and cytogenetic evidence9,10,40,41. The occurrence of hybridization between these species is also conceivable considering the variability observed in several ITS2 sequences from Russia and Kazakhstan, locally recorded in a large proportion of specimens, featuring polymorphic sites at the two key positions distinguishing An. messeae and An. daciae sp. inq.42.
Moreover, inversions in polytene chromosomes are widely used as markers to discriminate between species in complexes, including those within the genus Anopheles and even within the Anopheles maculipennis complex2. Specifically, regarding An. messeae, inversions in polytene chromosomes led to the early definition of chromosomal forms A and B for this species and subsequently, An. messeae A was proposed to be synonymous with An. daciae43. Differences in the frequencies of inversions have been reported in various populations of An. messeae and An. daciae sp. inq.10,41.
The morphometric analysis of mosquito wings revealed significant differences between An. maculipennis s. s., An. daciae sp. inq., An. melanoon and An. atroparvus, confirming previous results6,44. At the same time, the intraspecific groups obtained by the COI marker were not supported by morphometric data, suggesting that this marker should be used with caution in species delimitation work, at least in those concerning Anopheles species, while the ITS2 marker showed better performance in this context.
Unfortunately, we have not classified the mosquito wings as An. messeae to screen morphometric differences with specimens of the An. daciae sp. inq. However, notably, the morphometric results for other species corroborated the results obtained for ITS2, and this marker does not support subgroups of An. messeae.
The use of morphometry in this work allowed us to evaluate, with an independent approach, the species delimitation results obtained for the COI and ITS2 markers. This approach is promising and deserves wider recognition because of the improvements it can achieve by exploiting new information tecnology methods. The use of tools to capture images and wingbeat frequency, the analysis of such data by artificial intelligence and deep learning, the use of a species distribution model combined with machine learning algorithms and global sensitivity and uncertainty analysis are innovative and widespread approaches in ecology45. Convolutional neural networks have demonstrated high accuracy in image and spectrogram classification46. Advances in automated mosquito identification and machine learning could produce a critical tool for monitoring mosquito populations, surveillance and spatial and temporal prediction using environmental layers47.
Conclusions
The present study revealed that An. atroparvus, An. beklemishevi, An. sacharovi and An. labranchiae species can be accurately distinguished using multiple approaches, including the analysis of morphological features and DNA variation of both COI and ITS2 markers. However, the COI cannot be considered an adequate marker for identifying An. maculipennis s. s. due to the likely introgression events or incomplete lineage sorting phenomenon. Furthermore, all the delimitation methods and approaches adopted failed to adequately distinguish between An. daciae sp. inq. and An. messeae. In the absence of unequivocal results, caution is suggested in accepting the species status of An. daciae sp. inq., at least until more robust evidence is produced.
Although it might seem to be a very specialist issue, the definition of taxonomic boundaries inside mosquito complexes is important from a public health perspective, particularly for the Anopheles genus, which includes species vectors of malaria. The occurrence of cryptic species can lead to significant problems in surveillance and control when they differ in vector capacity due to differences in their biology, ethology and propensity to bite humans1. In addition, hybridization between evolving lineages can produce mosquitoes with intermediate behavioral and ecological characteristics, which can give rise to speciation events48. This picture is complicated by the strong evolutionary pressure to which mosquitoes are subjected, often due to anthropic actions, such as landscape variations and chemical control, which shape the drivers of speciation processes for these insects. Nevertheless, the definition of systematic relationships within the complex is a stimulating task because Anopheles complexes are an ideal context for defining the boundaries of sibling species and the mechanism of speciation.
Methods
Sample collection and identification
The specimens were collected in the Po Plain area in the Emilia-Romagna and Lombardy regions (Northern Italy). This zone is a suitable environment for Anopheles life and reproduction since it features many potential breeding sites, such as rice fields (e.g., Lomellina area) or wetland areas near the Po River delta, which is one of the largest wetland areas in Europe (Valli di Comacchio and Po River Delta). The majority of the specimens were collected between 2017 and 2018, while a few specimens were collected between 2011 and 2016. Some specimens were retrieved during entomological surveillance of the West Nile virus in Italy at 103 sites on the Po Plain; these mosquitoes were sampled using attractive traps baited with CO249. Further samples were collected by direct aspiration of resting adults at 43 sites, including farms with a variety of animals (cattle, horses, goats and poultry), to collect engorged and host-seeking individuals and uninhabited buildings to collect overwintering mosquitoes50.
Some live engorged females were allowed to lay eggs in glass tubes equipped with a wet blotting paper strip. Laid eggs were used to identify adults to the species level following the egg identification keys4.
DNA extraction, PCR and sequencing
DNA was extracted from one leg of the sampled mosquitoes, including individuals identified to the species level through egg analysis, using the Qiagen DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The two markers—COI and ITS2—were amplified by PCR according to the conditions reported by Jalali et al.51 and Marinucci et al.52, respectively. Both strands of the obtained amplicons were sequenced using ABI technology (Applied Biosystems, Foster City, CA, USA) and marker-specific primers. The obtained electropherograms were edited, and the consensus sequences were acquired using Geneious Pro 11 (Biomatters Ltd., NZ). In the case of COI sequences, the open reading frame was checked using the tool EMBOSS Transeq (https://www.ebi.ac.uk/Tools/st/emboss_transeq/). The sequences were deposited in the Bold system53 under the project ‘Anopheles maculipennis complex in Northern Italy (ANMA)’ (BOLD IDs: ANMA001-21 to ANMA1247-21) and in GenBank database54 (COI accession numbers from PQ640095 to PQ640174, ITS2 accession numbers from PQ640180 to PQ640259).
The map of the distribution of mosquitoes in the surveyed area according to the geolocated sampling site was generated using QGIS software (https://www.qgis.org/en/site/index.html#).
Sequence dataset preparation
Orthologous COI and ITS2 sequences of selected Palearctic species from 33 individuals of the Anopheles maculipennis complex (i.e., An. atroparvus, An. beklemishevi, An. daciae sp. inq., An. labranchiae, An. maculipennis s. s., An. melanoon, An. messeae and An. sacharovi) were retrieved from previously published studies37,43,55,56. Preference was given to studies in which the specimens were identified to the species level by observing egg morphology. COI and ITS2 sequences were retrieved only when both markers were obtained from the same individual. In 76 individuals, for which the morphological identification was not possible, molecular identification was performed using Basic Local Alignment Search Tool analysis (BLAST: http://www.ncbi.nlm. nih.gov/BLAST) with the default parameters. Species-level identification was assigned only when a similarity ≥ 99% and an E-value < 1 × 10−20 were obtained from the comparison between the query and the reference sequence.
The retrieved sequences were merged with the de novo COI and ITS2 sequences obtained in this study. The COI sequences were aligned using MUSCLE with default parameters. COI haplotypes were identified using R version 3.6.2 (R Core Team, 2019), and library haplotypes were identified (https://biolsystematics.wordpress.com/r/). One representative sequence for each COI haplotype was selected. In the cases in which the same haplotype was shared between two groups (or more), the sequences were maintained for all species (the same procedure was also adopted in Bellin et al.44). The COI dataset obtained was further reduced to balance the number of individuals belonging to each species (balancing the intraspecific sample size before performing the species delimitation analysis) by randomly removing the COI haplotypes of overrepresented species according to previous studies57. In this step, the sequences developed within this study were removed. The ITS2 dataset was then pruned to retain the sequences of the individuals maintained in the COI dataset. The ITS2 sequences of An. messeae obtained were further checked and, if needed, renamed according to the presence of the diagnostic polymorphisms of the ITS2 that distinguish An. daciae sp. inq. (according to Lilja et al.39). The COI sequences developed from the same individuals were also relabeled accordingly. The ITS2 sequences were aligned using MAFFT with G-INS-i as the search strategy58.
Haplotype networks and molecular species delimitation analysis
The alignments of COI and ITS sequences obtained from the specimens collected in the Po Valley (Italy) were used for haplotype network reconstruction. Haplotype networks were obtained via the median joining network method using PopArt software59.
Different molecular species delimitation methods were adopted to delimit the evolutionarily significant units present in the datasets (ESUs)60. In detail, the following distance-based and coalescent tree-based methods were adopted: a. the automatic barcode gap discovery (ABGD) tool61, which is known to be efficient in delimiting phylogenetically closely related species as well as distantly related species13; b. the generalized mixed Yule-coalescent (GMYC) model62; and c. the multirate Poisson Tree Processes (mPTP)63. These approaches have been widely and successfully adopted in previous studies addressing similar questions on different insect groups15,16,64,65. Species delimitation analyses were performed separately for the COI and ITS2 datasets. ABGD analyses were performed through the web-based interface (http://wwwabi.snv.jussieu.fr/public/abgd/abgdweb.html) adopting the Kimura two parameter as a model of nucleotide evolution (transition/transversion = 2); prior divergence of intraspecific diversity ranging from 0.001 to 0.1; relative gap width of 0.1 and 0.5 for the COI and ITS2 datasets, respectively; and number of bins = 20; moreover, 20 steps were set in the case of COI, and the remaining parameters were left at the default setting.
Bayesian single-locus ultrametric phylogenetic trees for each of the two markers considered in this study were inferred using BEAST 2.666 and used as input for the species delimitation analyses. The best nucleotide substitution models and partition scheme were estimated for each dataset using PartitionFinder267 and selected according to the corrected Akaike Information Criteria. The COI dataset was partitioned according to codon positions: the 1st codon position with the GTR model of nucleotide substitution + gamma distribution (\(\Gamma\)) and nucleotide frequencies estimated with maximum likelihood from the data; the 2nd codon position with the Tamura-Nei model + \(\Gamma\) and the estimated nucleotide frequencies; and the 3rd codon position with the HKY model and estimated nucleotide frequencies. Regarding the ITS2 dataset, the Tamura-Nei model + \(\Gamma\) and estimated nucleotide frequencies was the best model of nucleotide substitution.
Two independent BEAST runs were performed for each dataset using the following parameters: Markov chain length of 150 × 106 generations for the COI and ITS2 datasets; sampling of trees and parameters every 1000 generations; models of nucleotide evolution as previously mentioned; birth–death process as the tree prior, suitable for trees describing the relationships between individuals from different species, with a uniform prior; and the other priors were set to their default values. The convergence of the two BEAST runs was visualized and examined using TRACER68. The runs were then pooled after removal of the tree burn-in fraction, and the maximum clade credibility tree was obtained using TreeAnnotator67.
Species delimitation analyses using single-threshold GMYC were performed through the R package Splits (species limits by threshold statistics), available at http://r-forge.r-project.org/projects/splits/, using the maximum clade credibility ultrametric trees as input after outgroup removal. Tree manipulation was performed using the R package ape69. The tree-based mPTP method was applied to the same trees used as inputs for GMYC. mPTP analyses were performed through the web interface available at https://mptp.h-its.org/ adopting maximum likelihood delimitation.
Geometric morphometrics
The wings of the selected specimens were removed mechanically, brushed gently with a thin-tipped brush to remove the scales and mounted on a glass slide in Hoyer’s medium. The landmarks were then fixed manually using the Clic package70 for 356 mosquitoes of the four species An. maculipennis s. s. (n = 67), An. daciae sp. inq. (n = 165), An. atroparvus (n = 10) and An. melanoon (n = 4) and were then digitized6,44. The landmark coordinates were aligned in a common reference system using generalized Procrustes analysis (GPA). Canonical discriminant analysis (CDA) was used to fit a linear combination of the GPA coordinates to estimate the maximum separation among wing shapes and to visualize differences between species in a reduced space. A permutation test (n = 1000) was used to determine differences in the CDA scores among each pair of species. The p value of each test was adjusted using Bonferroni correction. Linear discriminant analysis (LDA) was applied to morphometric data at the intraspecific COI group level recorded in An. maculipennis s. s. and An. daciae sp. inq.
GPA was performed using the R package geomorph71. The CDA was fitted with the R package MorphoTools272 and visualized with ggplot273. The permutation test was performed using the R package Morpho74.
Moreover, at the intraspecific level, we also assessed the statistical relationships among the wing shape and molecular marker groups (ITS2 and COI) identified throughout phylogenesis. We identified two main represented species in the dataset: An. maculipennis s. s. and An. daciae sp. inq. At the intraspecific level, the GPA coordinates were used to fit a principal component analysis (PCA) morphospace, and the COI group identities were superimposed to improve data visualization. We used a statistical learning framework to test the phylogenetic signal. For each species, linear discriminant analysis (LDA) was performed considering the PCAs of the morphospace (selected using the elbow plot method) as predictors and the COI groups as dependent variables. The first 10 PCs of the morphospaces were used as predictor variables in the LDA models according to the elbow plot. The individuals were subsampled considering the proportion of the COI groups toward the minority class to balance the dataset. A training set was used to train the LDA model using 70% of the data, while the remaining 30% was used as a testing set to further assess the model’s performance. The training set was further split into 5 folds, and the cross validation of the LDA model was computed. For both the fivefold cross-validation and testing sets, five different performance metrics were computed: accuracy, F1 score, kappa, sensitivity and specificity. The LDA model and the performance metrics were computed using the R package caret75.
Assessing the phylogenetic signal between wing shape and gene trees
The statistical dependence between the resemblance of the right-wing landmark coordinates and the molecular marker (ITS2 and COI) gene tree was quantified according to the definition of phylogenetic signals as the “tendency for related species to resemble each other more than they resemble species drawn at random from the tree”76. The mean wing shape of each species was computed using the GPA coordinates and projected into the PCA morphospace. The Euclidean distances between pairs of species were computed. The distance matrix between pairs of species was used to fit a UPGMA-generated wing shape tree. The COI and ITS2 sequences of the specimens analyzed with geometric morphometrics were considered. For each species, both molecular marker sequences were aligned using the ClustalW algorithm, and representative species consensus sequences were obtained. A further multiple sequence alignment among the species consensus sequences was performed using the DECIPHER algorithm77, and a UPGMA phylogenetic tree was fitted.
To assess the statistical dependence between wing shape and phylogeny, we considered two different levels of representation of the species’ mean shapes: PCA morphospace and UPGMA wing shape tree. To evaluate the phylogenetic signal, the first dimension of the PCA morphospace was tested against the UPGMA phylogenetic tree considering five different statistics: Cmean, Moran’s I, K-statistic, K* and Pagel’s λ using the R package phylosignal78. The statistical correlation and similarity between the UPGMA trees of the wing shapes and the UPGMA of the molecular markers were estimated using Baker’s Gamma Index and the cophenetic correlation was estimated using the R package dendextend79.
Data availability
The sequences obtained in this work, as well as pictures of the wings, have been deposited in the Bold system under the project “Anopheles maculipennis complex in Northern Italy (ANMA)”. The project includes 1247 records available on the Bold system database (https://www.boldsystems.org/index.php) with de IDs: ANMA001-21-ANMA1247-21. Sequences used for species delimitation were deposed also in GenBank (https://www.ncbi.nlm.nih.gov/genbank/) with the accession numbers PQ640095-PQ640174 for COI and PQ640180-PQ640259 for ITS2.
References
Manguin, S., Carnevale, P. & Mouchet, J. Biodiversity of Malaria in the World (John Libbey Eurotext, 2008).
White, G. B. Systematic reappraisal of the Anopheles maculipennis complex. Mosq. Syst. 10, 13–44 (1978).
Falleroni, D. Fauna anofelica italiana e suo ‘habitat’ (paludi, risaie, canali). Metodi di lotta contro la malaria. Riv. Malariol. 5, 553–559 1926
Becker, N., Petrić, D., Zgomba, M., Boase, C., Madon, M., Dahl, C. & Kaiser, A. Mosquitoes: Identification, Ecology and Control 3rd edn (Springer Nature, 2020).
Choochote, W. & Saeung A. Systematic techniques for the recognition of Anopheles species complexes. In Anopheles mosquitoes—New Insights into Malaria Vectors (ed Manguin, S.) (IntechOpen, 2013). https://doi.org/10.5772/54853
Bellin, N. et al. Geometric morphometrics and machine learning as tools for the identification of sibling mosquito species of the Maculipennis complex (Anopheles). Infect. Genet. Evol. 95, 105034. https://doi.org/10.1016/j.meegid.2021.105034 (2021).
Linton, Y. M., Smith, L. & Harbach, R. E. Observations on the taxonomic status of Anopheles subalpinus Hackett & Lewis and An. melanoon Hackett. Eur. Mosq. Bull. 13, 1–7 (2002).
Boccolini, D., Di Luca, M., Marinucci, M. & Romi, R. Further molecular and morphological support for the formal synonymy of Anopheles subalpinus Hackett & Lewis with An. melanoon Hackett. Eur. Mosq. Bull. 16, 1–5 (2003).
Bezzhonova, O. V. & Goryacheva, I. I. Intragenomic heterogeneity of rDNA internal transcribed spacer 2 in Anopheles messeae (Diptera: Culicidae). J. Med. Entomol. 45(3), 337–341. https://doi.org/10.1603/0022-2585(2008)45[337:ihorit]2.0.co;2 (2008).
Artemov, G. N. et al. New cytogenetic photomap and molecular diagnostics for the cryptic species of the malaria mosquitoes Anopheles messeae and Anopheles daciae from Eurasia. Insects 12(9), 835. https://doi.org/10.3390/insects12090835 (2021).
Bertola, M., Mazzucato, M., Pombi, M. & Montarsi, F. Updated occurrence and bionomics of potential malaria vectors in Europe: a systematic review (2000–2021). Parasit. Vectors 15(1), 88. https://doi.org/10.1186/s13071-022-05204-y (2022).
Schmid-Egger, C. et al. DNA barcodes identify 99 per cent of apoid wasp species (Hymenoptera: Ampulicidae, Crabronidae, Sphecidae) from the Western Palearctic. Mol. Ecol. Resour. 19, 476–484 (2019).
Magoga, G., Fontaneto, D. & Montagna, M. Factors affecting the efficiency of molecular species delimitation in a species-rich insect family. Mol. Ecol. Resour. https://doi.org/10.1111/1755-0998.13352 (2021).
Šašić, L. et al. Molecular and morphological inference of three cryptic species within the Merodon aureus species group (Diptera: Syrphidae). PLoS One 11(8), e0160001. https://doi.org/10.1371/journal.pone.0160001 (2016).
Montagna, M. et al. Exploring species-level taxonomy in the Cryptocephalus flavipes species complex (Coleoptera: Chrysomelidae). Zool. J. Linn. Soc. https://doi.org/10.1111/zoj.12445 (2016).
Kajtoch, L., Montagna, M. & Wanat, M. Species delimitation within the Bothryorrhynchapion weevils: Multiple evidence from genetics, morphology and ecological associations. Mol. Phylogenet. Evol. 120, 354–363 (2018).
Dumas, P. et al. Phylogenetic molecular species delimitations unravel potential new species in the pest genus Spodoptera Guenée, 1852 (Lepidoptera, Noctuidae). PLoS One 10(4), e0122407. https://doi.org/10.1371/journal.pone.0122407 (2015).
Wang, Y. et al. Formal nomenclature and description of cryptic species of the Encyrtus sasakii complex (Hymenoptera: Encyrtidae). Sci. Rep. 6, 34372 (2016).
Berrilli, E., Biondi, M., D’Alessandro, P. & Salvi, D. Cryptic, sibling or neither of the two? Integrative species delimitation of Psylliodes flea beetles with overlapping ranges. Zool. Scr. 00, 1–14 (2023).
Li, C., Wilkerson, R. C. Intragenomic rDNA ITS2 variation in the neotropical Anopheles (Nyssorhynchus) albitarsis complex (Diptera: Culicidae). J. Hered. 98(1), 51–59. https://doi.org/10.1093/jhered/esl037 (2007).
Paredes-Esquivel, C., Donnelly, M. J., Harbach, R. E. & Townson, H. A molecular phylogeny of mosquitoes in the Anopheles barbirostris Subgroup reveals cryptic species: implications for identification of disease vectors. Mol. Phylogenet. Evol. 50(1), 141–151. https://doi.org/10.1016/j.ympev.2008.10.011 (2009).
Moreno, M. et al. Phylogeography of the neotropical Anopheles triannulatus complex (Diptera: Culicidae) supports deep structure and complex patterns. Parasit. Vectors 6, 47. https://doi.org/10.1186/1756-3305-6-47 (2013).
Bourke, B. P., Wilkerson, R. C. & Linton, Y. M. Molecular species delimitation reveals high diversity in the mosquito Anopheles tessellatus Theobald, 1901 (Diptera, Culicidae) across its range. Acta Trop. 215, 105799. https://doi.org/10.1016/j.actatropica.2020.105799 (2020).
Beebe, N. W. DNA barcoding mosquitoes: advice for potential prospectors. Parasitology 145(5), 622–633. https://doi.org/10.1017/S0031182018000343 (2018).
Hebert, P. D., Cywinska, A., Ball, S. L., deWaard, J. R. Biological identifications through DNA barcodes. Proc. Biol. Sci. 270(1512), 313–321. https://doi.org/10.1098/rspb.2002.2218 (2003).
Ambrose, L. et al. Population structure, mitochondrial polyphyly and the repeated loss of human biting ability in anopheline mosquitoes from the southwest Pacific. Mol. Ecol. 21(17), 4327–4343. https://doi.org/10.1111/j.1365-294X.2012.05690.x (2012).
Bower, J. E., Dowton, M., Cooper, R. D. & Beebe, N. W. Intraspecific concerted evolution of the rDNA ITS1 in Anopheles farauti sensu stricto (Diptera: Culicidae) reveals recent patterns of population structure. J. Mol. Evol. 67(4), 397–411. https://doi.org/10.1007/s00239-008-9161-x (2008).
Ståhls, G. & Savolainen, E. MtDNA COI barcodes reveal cryptic diversity in the Baetis vernus group (Ephemeroptera, Baetidae). Mol. Phylogenet. Evol. 46(1), 82–87 (2008).
Lavinia, P. D. et al. Barcoding the butterflies of southern South America: Species delimitation efficacy, cryptic diversity and geographic patterns of divergence. PLoS One 12(10), e0186845 (2017).
Buckley, T. R., Marske, K. A. & Attanayake, D. Identifying glacial refugia in a geographic parthenogen using palalaeoclimate modelmodelling and phylogeography: the New Zealand stick insect Argosarchus horridus (White). Mol. Ecol. 18, 4650–4663. https://doi.org/10.1111/j.1365-294X.2009.04396.x (2009).
Brunetti, M., Magoga, G., Iannella, M., Biondi, M. & Montagna, M. Phylogeography and species distribution modelmodelling of Cryptocephalus barii (Coleoptera: Chrysomelidae): is this alpine endemic species close to extinction?. ZooKeys 856, 3–25. https://doi.org/10.3897/zookeys.856.32462 (2019).
Mastrantonio, V., Porretta, D., Urbanelli, S., Crasta, G. & Nascetti, G. Dynamics of mtDNA introgression during species range expansion: insights from an experimental longitudinal study. Sci. Rep. 6, 30355 (2016).
Small, S. T. et al. Radiation with reticulation marks the origin of a major malaria vector. Proc. Natl. Acad. Sci. USA 117(50), 31583–31590. https://doi.org/10.1073/pnas.2018142117 (2020).
Ayala, D., Akone-Ella, O., Rahola, N., Kengne, P., Ngangue, M. F., Mezeme, F., Makanga, B. K., Nigg, M., Costantini, C., Simard, F., Prugnolle, F., Roche, B., Duron, O. & Paupy, C. Natural Wolbachia infections are common in the major malaria vectors in Central Africa. Evol. Appl. 12(8), 1583–1594. https://doi.org/10.1111/eva.12804 (2019).
Magoga, G., Sahin, D. C., Fontaneto, D. & Montagna, M. Barcoding of Chrysomelidae of Euro-Mediterranean area: efficiency and problematic species. Sci. Rep. 8(1), 13398. https://doi.org/10.1038/s41598-018-31545-9 (2018).
Funk, D. J. & Omland, K. Species-level paraphyly and polyphyly: frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 34(1), 397–423 (2003).
Nicolescu, G., Linton, Y. M., Vladimirescu, A., Howard, T. M. & Harbach, R. E. Mosquitoes of the Anopheles maculipennis group (Diptera: Culicidae) in Romania, with the discovery and formal recognition of a new species based on molecular and morphological evidence. Bull. Entomol. Res. 94, 525–535 (2004).
Kavran, M. et al. Distribution of Anopheles daciae and other Anopheles maculipennis complex species in Serbia. Parasitol. Res. 117(10), 3277–3287. https://doi.org/10.1007/s00436-018-6028-y (2018).
Lilja, T., Eklöf, D., Jaenson, T. G. T., Lindström, A. & Terenius, O. Single nucleotide polymorphism analysis of the ITS2 region of two sympatric malaria mosquito species in Sweden: Anopheles daciae and Anopheles messeae. Med. Vet. Entomol. 34(3), 364–368. https://doi.org/10.1111/mve.12436 (2020).
Novikov, Iu. M. & Shevchenko, A. I. Inversion polymorphism and the divergence of two cryptic forms of Anopheles messeae (Diptera, Culicidae) at the level of genomic DNA repeats. Russ. J. Genet. 37(7), 754–763 (2001). Translated from Genetika 2001 Jul;37(7):915–25.
Naumenko, A. N. et al. Chromosome and genome divergence between the cryptic Eurasian malaria vector-species Anopheles messeae and Anopheles daciae. Genes (Basel) 11(2), 165. https://doi.org/10.3390/genes11020165 (2020).
Brusentsov, I. I. et al. Patterns of genetic differentiation imply distinct phylogeographic history of the mosquito species Anopheles messeae and Anopheles daciae in Eurasia. Mol. Ecol. 32(20), 5609–5625. https://doi.org/10.1111/mec.17127 (2023).
Vaulin, O. V. & Novikov, Y. M. Polymorphism and interspecific variability of cytochrome oxidase subunit I (COI) gene nucleotide sequence in sibling species of A and B Anopheles messeae and An. Beklemishevi (Diptera: Culicidae). Russ. J. Genet. Appl. Res. 2, 421–429 (2012).
Bellin, N. et al. Unsupervised machine learning and geometric morphometrics as tools for the identification of inter and intraspecific variations in the Anopheles Maculipennis complex. Acta Trop. 233, 106585. https://doi.org/10.1016/j.actatropica.2022.106585 (2022).
Christin, S., Hervet, E. & Lecomte, N. Applications for deep learning in ecology. Methods Ecol. Evol. 10(10), 1632–1644. https://doi.org/10.1111/2041-210X.13256 (2019).
Dong, X., Yan, N. & Wei, Y. Insect sound recognition based on convolutional neural network. In 3rd IEEE International Conference on Image, Vision and Computing, ICIVC, 855–859. https://doi.org/10.1109/ICIVC.2018.8492871 (2018).
Kim, D., DeBriere, T. J., Cherukumalli, S., White, G. S. & Burkett-Cadena, N. D. Infrared light sensors permit rapid recording of wingbeat frequency and bioacoustic species identification of mosquitoes. Sci. Rep. 11(1), 1–9. https://doi.org/10.1038/s41598-021-89644-z (2021).
Porretta, D. & Canestrelli, D. The ecological importance of hybridization. Trends Ecol. Evol. 38(11), 1097–1108. https://doi.org/10.1016/j.tree.2023.07.003 (2023).
Calzolari, M. et al. Enhanced West Nile Virus Circulation in the Emilia-Romagna and Lombardy Regions (Northern Italy) in 2018 Detected by Entomological Surveillance. Front. Vet. Sci. 5(7), 243. https://doi.org/10.3389/fvets.2020.00243 (2020).
Calzolari, M. et al. Mosquitoes of the Maculipennis complex in Northern Italy. Sci. Rep. 11(1), 6421. https://doi.org/10.1038/s41598-021-85442-9 (2021).
Jalali, S., Ojha, R. & Venkatesan, T. DNA barcoding for identification of agriculturally important insects. In New Horizons in Insect Science: Towards Sustainable Pest Management (ed Chakravarthy, A.) (Springer, 2015).
Marinucci, M., Romi, R., Mancini, P., Di Luca, M. & Severini, C. Phylogenetic relationships of seven palearctic members of the maculipennis complex inferred from ITS2 sequence analysis. Insect Mol. Biol. 8(4), 469–480 (1999).
Ratnasingham, S., Hebert, P. D. N. The Barcode of Life Data System (http://www.barcodinglife.org). Mol. Ecol. Notes 7(3), 355–364. https://doi.org/10.1111/j.1471-8286.2007.01678.x (2007).
Benson, D. A., Cavanaugh, M., Clark, K., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J. & Sayers, E. W. GenBank. Nucleic Acids Res. 41(Database issue), D36–D42. https://doi.org/10.1093/nar/gks1195 (2013).
Di Luca, M., Boccolini, D., Marinucci, M. & Romi, R. Intrapopulation polymorphism in Anopheles messeae (An. maculipennis complex) inferred by molecular analysis. J. Med. Entomol. 41(4), 582–586. https://doi.org/10.1603/0022-2585-41.4.582 (2004).
Linton, Y. et al. The Anopheles (Anopheles) maculipennis complex (Diptera: Culicidae) in Greece. J. Nat. Hist. 41, 2683–2699. https://doi.org/10.1080/00222930701403255 (2007).
Ahrens, D. et al. Rarity and incomplete sampling in DNA-based species delimitation. Syst. Biol. 65(3), 478–494. https://doi.org/10.1093/sysbio/syw002 (2016).
Katoh, K. & Toh, H. Improved accuracy of multiple ncRNA alignment by incorporating structural information into a MAFFT-based framework. BMC Bioinform. 9, 212 (2008).
Leigh, J. W. & Bryant, D. PopART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 6(9), 1110–1116. https://doi.org/10.1111/2041-210X.12410 (2015).
Ryder, O. A. Species conservation and systematics: The dilemma of the subspecies. Trends Ecol. Evol. 1, 9–10 (1986).
Puillandre, N., Lambert, A., Brouillet, S. & Achaz, G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol. Ecol. 21(8), 1864–1877. https://doi.org/10.1111/j.1365-294X.2011.05239.x (2012).
Fujisawa, T. & Barraclough, T. G. Delimiting species using single-locus data and the Generalized Mixed Yule Coalescent approach: a revised method and evaluation on simulated datasets. Syst. Biol. 62(5), 707–724. https://doi.org/10.1093/sysbio/syt033 (2013).
Kapli, P. et al. Multirate Poisson tree processes for single-locus species delimitation under maximum likelihood and Markov chain Monte Carlo. Bioinformatics 33(11), 1630–1638. https://doi.org/10.1093/bioinformatics/btx025 (2017).
Montagna, M. et al. Molecular species delimitation of the Asian chestnut gall wasp biocontrol agent released in Italy. Insect Syst. Evol. 50(3), 327–345. https://doi.org/10.1163/1876312X-00002188 (2019).
Montagna, M., Mereghett, V., Lencioni, V. & Rossaro, B. Integrated taxonomy and DNA barcoding of Alpine midges (Diptera: Chironomidae). PLoS One 11, e0149673 (2016).
Bouckaert, R., Vaughan, T. G., Barido-Sottani, J., Duchêne, S., Fourment, M., Gavryushkina, A., Heled, J., Jones, G., Kühnert, D., De Maio, N., Matschiner, M., Mendes, F. K., Müller, N. F., Ogilvie, H. A., du Plessis, L., Popinga, A., Rambaut, A., Rasmussen, D., Siveroni, I., Suchard, M. A., Wu, C. H., Xie, D., Zhang, C., Stadler, T. & Drummond, A. J. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 15(4), e1006650. https://doi.org/10.1371/journal.pcbi.1006650 (2019).
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T. & Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34(3), 772–773. https://doi.org/10.1093/molbev/msw260 (2017).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol., syy032. https://doi.org/10.1093/sysbio/syy032 (2018).
Paradis, E., Claude, J. & Strimmer, K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 20, 289–290. https://doi.org/10.1093/bioinformatics/btg412
Dujardin, J. P., Kaba, D. & Henry, A. B. The exchangeability of shape. BMC Res. Notes 22(3), 266. https://doi.org/10.1186/1756-0500-3-266 (2010).
Adams, D. C., Collyer, M. L., Kaliontzopoulou, A. & Baken, E. K. Geomorph: Software for geometric morphometric analyses. R package version 4.0.4. https://cran.r-project.org/package=geomorph (2022).
Šlenker, M., Koutecký, P. & Marhold, K. MorphoTools2: an R package for multivariate morphometric analysis. Bioinformatics 38(10), 2954–2955 (2022).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, 2016).
Schlager, S. Morpho and Rvcg—shape analysis in R. In Statistical Shape and Deformation Analysis (eds Zheng, G., Li, S., Szekely, G.) 217–256. ISBN 9780128104934. https://doi.org/10.1093/bioinformatics/btac173 (Academic Press, 2017).
Kuhn, M. Building predictive models in R using the caret Package. J. Stat. Softw. 28(5), 1–26. https://doi.org/10.18637/jss.v028.i05 (2008).
Blomberg, S. P., Garland, T. Jr. & Ives, A. R. Testing for phylogenetic signal in comparative data: behavioral traits are more labile. Evolution 57(4), 717–745. https://doi.org/10.1111/j.0014-3820.2003.tb00285.x (2003).
Wright, E. S. Using DECIPHER v2.0 to analyse big biological sequence data in R. R J. 8(1), 352–359 (2016).
Keck, F., Rimet, F., Bouchez, A. & Franc, A. phylosignal: an R package to measure, test, and explore the phylogenetic signal. Ecol. Evol. 6(9), 2774–2780. https://doi.org/10.1002/ece3.2051 (2016).
Galili, T. dendextend: an R package for visualizing, adjusting, and comparing trees of hierarchical clustering. Bioinformatics https://doi.org/10.1093/bioinformatics/btv428 (2015).
Acknowledgements
This research was supported by the PRC2015017 (E88C16000190001) and by EU funding within the NextGenerationEU-MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases (Project no. PE00000007, INF-ACT). This work has benefited from the equipment and framework of the COMP-R Initiatives, funded by the “Departments of Excellence” program of the Italian Ministry for University and Research (MUR, 2023–2027). This work was also supported by the National Science Foundation (NSF) EPSCoR RII Track-2 FEC (OIA 1736026) to Riccardo Papa.
Author information
Authors and Affiliations
Contributions
M.C. collected the samples. M.C., N.B., M.Dott, M.DiL., V.R., G.M., M.M. analysed the data. N.B. performed morphometric analysis. G.M. and M.M. performed species delimitation analysis. M.C., N.B., G.M., M.M. wrote the manuscript. All authors contributed to the final version of the paper.
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Calzolari, M., Bellin, N., Dottori, M. et al. Integrated taxonomy to advance species delimitation of the Anopheles maculipennis complex. Sci Rep 14, 30914 (2024). https://doi.org/10.1038/s41598-024-81767-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-81767-3
