
Abstract
The CDKL5 deficiency disorder (CDD) is a severe neurodevelopmental disorder characterized by early-onset epilepsy, intellectual disability, motor and visual dysfunctions. The causative gene is CDKL5, which codes for a kinase required for brain development. There is no cure for CDD patients; treatments are symptomatic and focus mainly on seizure control. Several pathogenic variants are loss-of-function, but recent studies suggest that the CDD phenotype is sensitive to the CDKL5 gene dosage. Therefore, mRNA-targeted correction strategies that respect the physiological regulation of CDKL5 could be a valid alternative to augmentative gene therapy. Nonsense mutations cause ~ 11% of CDD cases, and these patients might benefit from readthrough therapies. We proved that drug-mediated readthrough efficiently suppresses premature CDKL5 nonsense codons, but the recoded kinase remained highly hypomorphic, curtailing the translational value of this pharmacological approach. In this study we explored if the recently developed Anticodon-edited tRNAs (ACE-tRNAs) offer an alternative readthrough strategy for CDD. Transfecting cells expressing different CDKL5 nonsense variants, we demonstrated that ACE-tRNAs efficiently restore full-length kinase synthesis. The recoded CDKL5 is correctly localized and catalytically active, thereby bringing tRNA-based therapy back into the spotlight for future investigations to assess the efficacy of this approach in correcting the pathological phenotype of CDD.
Similar content being viewed by others


Introduction
Mutations in the X-linked CDKL5gene cause a large variety of neuropsychiatric disorders among which CDKL5 deficiency disorder (CDD) (OMIM 300203) is the most common1. CDD is a severe early infantile epileptic encephalopathy, that, with an incidence of 1 in 40,000–60,000 live births, represents one of the most frequent sources of epilepsy in early childhood; further, it is four times more frequent in females than in males2. More than 90% of the patients suffer from early onset of epilepsy, generally occurring within the first three months of life. Over time CDD patients develop many other clinical symptoms including global psychomotor delay and stereotypical behaviors, profound hypotonia, breathing defects, gastrointestinal problems, and cortical visual impairment2,3.
Consistent with the mainly neurological features associated with CDKL5-related diseases, the human CDKL5gene encodes for a serine-threonine kinase whose molecular functions are involved in proper brain development1,4. CDKL5is a complex gene that through alternative splicing generates 5 different transcriptional isoforms5. Among these, the isoform CDKL5_1, encoding for a protein of 960 amino acids (CDKL5107), is the most abundant in brain5. All CDKL5 protein isoforms share an N-terminal catalytic domain, containing the essential ATP-binding region and serine/threonine kinase active site, and the Thr-Xaa-Tyr (TEY) motif, whose phosphorylation correlates with enzymatic activity6. In contrast, the length and exon composition of the C-terminal domain, which modulates the catalytic activity of the kinase, protein-protein interactions, and subcellular distribution, can subtly change depending on the CDKL5 isoform4,7.
Numerous pathogenic variants have been reported so far, including missense, nonsense, and frameshift alterations. Among them, nonsense mutations account for roughly 11% of the pathogenic cases2,4. Missense mutations mainly localize in the catalytic domain underscoring the importance of CDKL5 enzymatic activity for brain health, while nonsense and frameshift variations are scattered along the whole gene4. Moreover, most of the pathogenic variants lead to the lack or reduction of functional CDKL5, thereby representing loss-of-function mutations. However, some clinical reports suggested that CDKL5duplication can affect neurodevelopment8,9. In possible agreement, we recently suggested that a missense substitution, located in the most distal portion of the CDKL5 C-terminus and leading to a hypermorphic kinase, might be the cause of a mild phenotype identified in a female patient10. Overall, this early genetic evidence led us to consider the maintenance of a tight control of CDKL5 kinase activity in brain as important.
Being aware that the development of efficacious therapies for CDD requires an in-depth comprehension of CDKL5 activity and regulation, as well as of the molecular mechanisms underlying its dysfunctions, several research groups, including our laboratory, have provided important insights. In particular, the specificity of CDKL5 kinase activity was identified revealing the amino acid consensus sequence preferentially recognized by CDKL5, together with a few targets, including the microtubule associated proteins EB2 and MAP1S11,12.
Concerning CDKL5 regulation, its expression is developmentally modulated and increases during neuronal maturation and synaptogenesis7. Further, intracellular localization is tightly tuned; indeed, while CDKL5 appears to be present both in the nucleus and the cytoplasm, the cytoplasmic fraction largely increases throughout development7. An additional level of complexity is given by CDKL5 post-transcriptional control. Indeed, we previously demonstrated that neuronal depolarization induces a rapid increase of CDKL5 levels, mostly mediated by localized protein synthesis13. Whereas this induction is prolonged in young neurons, the activation of a proteasome degradation dependent mechanism leads to a short-lasting increase of the kinase in mature neurons.
To date, no specific therapy is available for CDD patients, and the drugs currently in use aim at improving only main symptoms of CDD, with a particular attention for controlling seizures14. The still limited knowledge of the role that CDKL5 plays in the brain, together with its complex local and developmental regulation, certainly limit the development of therapies. Further, the possible dosage-sensitivity of the CDKL5gene, the necessity to identify regulatory elements governing proper transcriptional and post-transcriptional regulation of CDKL5 levels and cellular distributions, together with the large size of the coding portion of the gene, challenge the development of safe and efficacious approaches of conventional augmentative gene therapy. Recently, we proposed that RNA-based therapeutic approaches that regard the importance of physiological and fine-regulation of CDKL5 levels, might represent convenient alternative approaches15,16. We studied the efficacy of drug-mediated readthrough therapy on CDKL5premature termination codons (PTCs). Although CDKL5 PTCs were efficiently readthrough by aminoglycoside drugs, the restored full-length CDKL5 protein did not recover its catalytic activity, thereby curtailing the potential of this pharmacological approach. Further, less toxic non-aminoglycoside drugs, including PTC124 (Ataluren), were unable to induce readthrough activity on CDKL5 PTCs15. Since we demonstrated that defective catalytic activity of readthrough CDKL5 was not caused by the insertion of missense mutations, we proposed that protracted ribosomal pausing at PTCs might negatively affect CDKL5 folding, thus compromising its functionality.
Suppressor (Sup-)tRNAs or anticodon-engineered tRNAs (ACE-tRNAs) offer an attractive alternative strategy for readthrough therapy17,18. ACE-tRNAs are obtained from natural tRNAs and are characterized by anticodons that have been edited at their anticodon to recognize one of the three stop codons (UGA, UAA, or UAG). Importantly, the tRNA nucleotide substitutions maintain the critical contacts with the endogenous aminoacyl-tRNA synthetases, allowing them to be acylated with their cognate amino acid18. ACE-tRNAs participate in translational elongation at the targeted PTCs, restoring the production of a wild-type, full-length protein with endogenous regulation. Importantly, ACE-tRNA technologies has demonstrated potent PTC suppression in mammalian cells and mice, producing PTC repair in multiple genes (reviewed in refs19,20,21,22). Furthermore, viral transduction of constructs expressing ACE-tRNAs safely and efficiently rescued clinical symptoms of a mouse model of Mucopolysaccharidosis type I, a lysosomal storage disorder23. In accordance with the observed safety, the treatment had minor impact on global readthrough at normal stop codons and did not perturb endogenous tRNA homeostasis. This scientific evidence led us to investigate the potential of ACE-tRNAs to efficiently suppress CDKL5 PTCs and restore kinase subcellular distribution and activity.
Results
Anticodon Engineered tRNAs efficiently suppress premature UGA termination codons
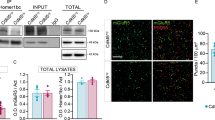
To verify whether the recently developed ACE-tRNAs18 might overcome the limitation of readthrough-inducing drugs, we selected as paradigmatic models three pathogenic human variants resulting from conversion of an arginine (R) codon (CGA) to the UGA PTC. Importantly, among the 18 sense codons that can mutate into termination codons, the selected one has the highest frequency in the human populations, corresponding to 23.7%20. Two of the selected variants (R59X and R134X) are located in the N-terminal catalytic domain, while the R550X PTC is localized in the C-terminus. Notably, all constructs express the most abundant human CDKL5 isoform present in the brain (hCDKL5_1; 107 kDa)5 fused with GFP at the N-terminus. The capacity of ACE-tRNA plasmid to restore the production of full-length CDKL5 was evaluated by transfecting WT GFP-CDKL5 or its PTC variants alone or in combination with plasmids expressing 1X or 4X ACE-tRNAArgUGA transgenes into HEK293T cells. For clarity in ACE-tRNA nomenclature, the superscript indicates the amino acid inserted co-translationally and the subscript indicates the stop codon suppressed. GFP-CDKL5 synthesis was revealed using an α-GFP antibody. As shown in Figs. 1a and 4a, both ACE-tRNA vectors were able to efficiently suppress CDKL5 PTCs. Readthrough efficacy was assessed by calculating the ratio of rescued CDKL5 and the total exogenously expressed kinase, obtained by adding the expression of the full-length protein with that of the corresponding prematurely truncated derivative. Notably, the R134X mutant, which expresses very low levels of the truncated isoform, exhibited a high readthrough efficacy, corresponding to 94% ± 6.2% when co-transfected with 1X ACE-tRNAArgUGA that was comparable with that one showed by the R59X derivative (93% ± 8,51%). In the same experimental conditions, the R550X PTC variant showed the lowest readthrough efficacy, corresponding to 68% ± 18.36%. Transfection with the 4X ACE-tRNAArgUGA vector increased the amount of recovered full-length CDKL5 up to 82% ± 3,86%.

ACE-tRNA therapy restores full-length CDKL5 protein synthesis. (a) Western blot of GFP CTRL, GFP-CDKL5 WT, R59X, R134X and R550X constructs co-transfected in HEK293T cells with or without 1X or 4X ACE-tRNAArgUGA. Fusion proteins were detected using the α-GFP antibody. Black arrowheads indicate full-length GFP-CDKL5. Total protein content was visualized by TGX Stain-Free technology (lower panel). (b,c) Western blot of endogenous VSNL1 (b) and MeCP2 (c) in untransfected HEK293T cells or in HEK293T cells transfected with GFP with or without 1X or 4X ACE-tRNAArgUGA. In panel b, the asterisk indicates a non-specific band recognized by the antibody. Images of original blots can be retrieved in the Supplementary Information file.

ACE-tRNAs stabilize the GFP-CDKL5 R134X transcripts. The graph shows GFP-CDKL5 mRNA levels in transfected HEK293T cells, expressed as a percentage with respect to GFP-CDKL5 WT – transfected cells. Data are shown as mean ± SEM. *p < 0.05, by two-way ANOVA, followed by Sidak’s multiple comparison test.
It has been previously demonstrated that ACE-tRNAs preferentially suppress PTCs, while having limited effects on natural termination codon (NTCs)18,23. Accordingly, Figs. 1a and 4a demonstrate that CDKL5 NTC was correctly recognized, an expected result considering that the NTC corresponds to UAA. As a proxy for NTC readthrough of the translatome, we assessed the impact of different amounts of ACE-tRNAArgUGA on the expression of the two endogenous proteins VSNL1 and MeCP2, whose transcripts harbor an UGA as NTC, that could be a potential off-target of ACE-tRNAArgUGA (Fig. 1b, c). Specifically, readthrough of the VSNL1 or MECP2 NTCs would create C-terminal extensions to the downstream in-frame stop codon, that would increase the molecular weight (MW) by 5 kDa or 3 kDa, respectively. Figure 1b, c and in Supplementary Fig. 1, which shows overexposed images of the same gels, indicate that ACE-tRNAArgUGA expression did not appreciably alter the electrophoretic mobility of the two proteins, thus demonstrating efficient translation termination at the natural stop codon. To strengthen this observation, we compared the amounts of full-length Mecp2 and Vsnl1 present in cells expressing one or four copies of ACE-tRNAArgUGA (normalized to total proteins) with their expression levels in cells not expressing exogenous tRNAs. The average of four independent experiments indicated that in the presence of 1X and 4X ACE-tRNAArgUGA, Mecp2 levels corresponded to 113,81 ± 18,9 and 113,61 ± 9,3 respectively, compared with control cells, and Vsnl1 levels to 97,85 ± 7,9 and 116,44 ± 22,6 providing a first and promising assessment of off-target NTC suppression.
It is well recognized that protein expression also depends on mRNA abundance. Although the highly-efficient readthrough of the analyzed PTCs suggests that the PTC transcript variants do not suffer from instability, we verified their levels in the absence or presence of the plasmid expressing 1X ACE-tRNAArgUGA by quantitative RT-PCR. The results were normalized with respect to GAPDH and transfection levels, obtained by evaluating the amount of GFP DNA contained in each sample through qPCR. Figure 2 shows that levels of the mutated CDKL5 mRNAs are comparable with those of the WT transcripts, with the R134X mRNA showing the lowest levels, a result that appears consistent with the small amount of GFP-CDKL5 134X protein detectable in the absence of ACE-tRNAs (Fig. 1 and ref.15). Furthermore, ACE-tRNA treatment had negligible effects on the abundance of CDKL5transcripts, with the exception of 134X mRNA, which appeared upregulated. Overall these data suggest that, in these experimental conditions, the increase in CDKL5 expression induced by ACE-tRNAs results mainly from efficient PTC suppression and the consequent generation of a full-length protein; however, in the presence of less abundant transcripts, a positive correlation between protein translation rate and mRNA levels can be observed24.

CDKL5 PTC variants recover proper nuclear/cytosolic distribution following delivery of ACE-tRNAArgUGA. (a) Representative immunofluorescence images of GFP-CDKL5 in HEK293T cells transfected with GFP-CDKL5 WT and its truncated derivatives R59X and R550X in the presence or absence of the 1X ACE-tRNAArgUGA expressing vector. (b) Violin plots indicate the median (dashed line) and 25th and 75th percentiles (dotted lines) of the relative distribution in the nucleus and cytoplasm of CDKL5 WT, R59X and R550X variants left untreated (UT) or following treatment with 1X ACE-tRNAArgUGA. Values are expressed as percentages compared to CDKL5 WT. *p < 0.05, ***p < 0.001 and ****p < 0.0001 by Kruskal-Wallis test followed by Dunn’s multiple comparison test. §§p < 0.0001 indicates a difference between R550X treated with 1X ACE-tRNAArgUGA with respect to UT WT. (c) The subcellular distributions of GFP-CDKL5 WT and of its derivative R550X were compared in cells co-transfected or not with the 4X ACE-tRNAArgUGA expressing plasmid. (d) Violin plots were obtained as in a. ****p < 0.0001 and ***p < 0.001 by Kruskal-Wallis test followed by Dunn’s multiple comparison test. §p < 0.05 indicates a difference between R550X treated with 4X ACE-tRNAArgUGA with respect to UT WT.
ACE-tRNAs overcome the limitations observed for CDKL5 PTCs with the drug-mediated readthrough therapy
Since CDKL5 functions appear to mainly occur in the cytoplasm25, we initially validated functional rescue arising from CDKL5 readthrough by comparing the subcellular localization of truncated derivatives with their corresponding readthrough products. Experiments were performed on the R59X and R550X mutants and their distribution between the nucleus and cytoplasm was assessed using the α-GFP antibody. As shown in Fig. 3a and b, both truncated derivatives displayed a significantly increased nuclear localization with respect to full-length CDKL5, particularly for the R550X variant. Interestingly, while the expression of 1X ACE-tRNAArgUGA was sufficient to rescue the abnormal sub-localization of the R59X variant, this was not the case for the R550X mutant (Fig. 3a, b). In fact, the significant defect in the subcellular distribution was maintained, despite the evident amelioration. This result, together with the previous indication that the 4X ACE-tRNAArgUGA plasmid results in more efficient PTC readthrough (Fig. 1), led us to assess whether the presence of 4X ACE-tRNAArgUGA was more efficacious in reducing the GFP nuclear signal of the R550X derivative (Fig. 3c, d). Obtained results demonstrated that although slightly ameliorated the defective subcellular distribution was not fully recovered (Fig. 1). Importantly, in all conditions, ACE-tRNAs did not affect the subcellular distribution of WT GFP-CDKL5.

Phosphorylation of the exogenous GFP-CDKL5 TEY motif and endogenous EB2 are restored following ACE-tRNAArgUGAdelivery. (a, c) Representative western blots for CDKL5 pTyr171 (a) and EB2 pSer222 (c) in HEK293T co-transfected with GFP, WT GFP-CDKL5 or -R59X, -R134X, -R550X constructs with or without 1X or 4X ACE-tRNAArgUGA. CDKL5 phosphorylation at Tyr171 was revealed together with full-length CDKL5 (through an anti-GFP antibody). White arrowheads show the premature truncated GFP-CDKL5 R550X derivative that was not subjected to readthrough. Original images of blots in a are shown in the Supplementary Information file. (b,d) Histograms depict the percentages of full-length GFP-CDKL5 phosphorylation at Tyr171(b) and of EB2 phosphorylation at Ser222 (d) with respect to untreated (UT) GFP-CDKL5 WT. For analysis of phosphorylated EB2, for each sample data were normalized to the total levels of corresponding protein. Data are expressed as mean ± SEM. *p < 0.05 by Kruskal-Wallis test followed by Dunn’s multiple comparison test.
To assess the enzymatic activity of the rescued CDKL5 proteins, phosphorylation of the TEY motif was examined, as it represents a good marker of the kinase activity6. CDKL5 PTC variants were transfected into HEK293T cells with or without 1X ACE-tRNAArgUGA, and CDKL5 phosphorylation of Tyr171 was assessed by western blot (Fig. 4a, b). Obtained data, normalized to the amount of full-length GFP-CDKL5, demonstrated that ACE-tRNAs were highly effective in restoring CDKL5 catalytic activity.
The recovery of the enzymatic activity of CDKL5 proteins subjected to readthrough was confirmed by analyzing the phosphorylation state of two endogenous bona fidetargets: the microtubule associated proteins EB2 and MAP1S11,12 (Fig. 4c, d and Suppl. Figure 2). Of note, while EB2 phosphorylation (pEB2) was normalized to total EB2 (Fig. 4c, d), MAP1S phosphorylation was normalized to total protein extracts (visualized through the TGX stain free technology; lower panel in Sup. Figure 2) after having verified that comparable levels of full-length CDKL5 were present after readthrough. As expected, in untreated R59X and R134X samples, the lack of exogenous kinase activity resulted in a significant reduction in EB2 phosphorylation compared to cells transfected with the WT kinase; these levels were comparable to those observed in cells transfected with GFP. Importantly, with delivery of ACE-tRNAArgUGA, the observed defects in EB2 phosphorylation were no longer present, and the protein post-translational modification was significantly increased with 4X ACE-tRNA in R59X-transfected cells. Similar results were obtained analyzing MAP1S phosphorylation (Suppl. Figure 2) thereby confirming functional restoration of the CDKL5 protein.
Discussion
Mutations in the X-linked CDKL5gene are associated with a wide spectrum of neuropsychiatric disorders that remain untreatable. The still limited knowledge of the main molecular functions of the CDKL5 kinase and the consequences of its deficiency delay the development of therapies1,4. Furthermore, the complex structure of the encoding gene, its tight post-transcriptional and spatial regulation and the evidence suggesting the necessity to preserve this fine regulation, might limit or postpone the development of classical augmentative gene therapy approaches5,7,9,10,13,26,27. For this reason, in recent years we have been interested in testing whether correction approaches acting at the post-transcriptional level, and thereby preserving the physiological regulation of CDKL5, could be effective, thus offering the opportunity for personalized therapies for CDD. In particular, we focused on splicing and readthrough technologies15,16. Although splicing technologies revealed interesting results, we realized that since CDKL5 patients are rare, and recurrent mutations are very few, these personalized treatments may be too expensive for clinical translation. On the contrary, if efficient, readthrough therapies would offer the possibility of using few molecules for all patients carrying nonsense variants in CDKL5(almost 11%), thus reducing the costs of treatment. Furthermore, the same drug could be used for multiple diseases, increasing its practicality as a therapy. Accordingly, many studies have investigated the value of small readthrough molecules for the treatment of different disorders, and some of these reached the clinic28,29,30. These concepts provided the rationale for our previous study, testing the in vitro efficacy of aminoglycoside and non-aminoglycoside drugs on CDKL5PTCs15. Although aminoglycosides manifested strong readthrough activity on all tested nonsense mutations, CDKL5 proteins products resulting from aminoglycoside-dependent readthrough revealed strongly impaired kinase activity. Notably, the observed functional defect was independent of the involved protein domain and not strictly related to the insertion of a missense mutation in place of the PTC. These findings, together with the unsuccessful clinical trial performed with Ataluren on CDD patients31, discouraged a preclinical study in a mouse model of CDD. In our previous publication, we suggested that a protracted pausing of ribosomes at CDKL5 PTCs during drug-induced readthrough might have negatively affected CDKL5 folding and activity32. This evidence encouraged us to investigate whether the induction of CDKL5 readthrough with exogenously expressed ACE-tRNAs leading to the generation of WT protein could overcome the limitations encountered.
We find that ACE-tRNAs induce efficient suppression of CDKL5 PTCs restoring its fundamental catalytic activity and recovering, or significantly ameliorating, its abnormal subcellular distribution. Furthermore, an initial quantitative evaluation of off-target NTC suppression, performed on two endogenous proteins with a natural UGA stop codon, did not reveal readthrough of native stop codons. However, as proteins extended to the carboxy-terminus can be degraded by cellular surveillance mechanisms, the safety of this approach will be further investigated in the future by performing Ribo-seq analyses that interrogate global readthrough at the mRNA levels23,33.
Interestingly, the most effective readthrough results were found with the two PTCs located towards the 5’ end of the transcript, affecting the N-terminal catalytic domain. Although at the molecular level, this result might find a logical explanation in the capacity of sustained protein synthesis to stabilize the transcript, thus considerably increasing the levels of full-length recoded CDKL5 compared to those of the truncated protein, quantitative real time data do not seem to support this hypothesis. An intriguing alternative might be that the translation termination activity promoted by polyadenylate binding proteins disfavors readthrough at PTCs closer to the natural stop codon34. Although this result might suggest that some CDD patients might gain more from a readthrough therapy based on ACE-tRNAs, conclusions in this regard will be drawn only after repeating the study in an endogenous genomic context. Indeed, it is well known that readthrough therapy is sensitive to levels of the target mRNA and, conversely, half-life of PTC-bearing transcripts could be affected by parameters or mechanisms that might be not present or might be altered in a cDNA context, such as non-sense mediated mRNA decay, splicing and the length of 3’UTR35.
In conclusion, we believe that our results have revived the possibility of treating CDD patients with a readthrough therapy based on ACE-tRNAs. To further prove its efficacy in the near future, we will assess efficacy of ACE-tRNAs to restore the synthesis of a full-length and catalytically active CDKL5 kinase in a native human and mouse genomic context, in which the chromatin environment, native transcriptional and post-transcriptional regulation and processing are preserved. Further, by administering ACE-tRNAs in a Cdkl5 mouse model featuring a nonsense mutation, we will establish the safety of this approach and its efficacy in achieving sufficient readthrough to significantly improve behavioral defects that typically affect mouse models of CDD.
If successful, these results will be useful for many other neurodevelopmental disorders in which conventional gene therapy is restricted by the necessity to deliver gene sequences that may exceed viral packaging limits or cause transgene-related toxicities36.
Methods
Plasmids
Plasmids expressing both the WT CDKL5 and R59X, R134X and R550X variants, fused to a GFP at the N-terminus, were generated by site-directed mutagenesis, as described in ref. 15. A plasmid expressing only GFP was used as control. Plasmids expressing 1 (1X) or 4 (4X) copies of arginine UGA suppressing ACE-tRNA transgenes (ACE-tRNAArgUGA) were generated as described previously18. For this study, the parent arginine tRNA sequence engineered to suppress UGA stop codons is tRNA-Arg-TCT-3-1 (tRNA-Arg-TCT→UCA-3-1).
Cell cultures and transfection
HEK293T were cultured in DMEM (Dulbecco’s modified Eagle’s medium, #D5671, Merk), containing 10% FBS (#F0804, Life Technologies), 1% L-glutamine (#G7513, Merk) and 1% penicillin/streptomycin (#P0781, Merk) and maintained at 37 °C with 5% CO2 in 100 mm dishes. When confluent, cells were detached and seeded in 12 well plates for western blot analyses or on coverslips in 24 well plates for immunofluorescence experiments. At 70–80% of confluence, HEK293T were transfected with 1.2 µg of DNA (1:1 ratio of GFP-CDKL5 derivatives and 1X or 4X ACE-tRNAArgUGA; in the absence of ACE-tRNAs, WT GFP-CDKL5 was co-transfected with an unrelated vector) using the calcium phosphate method. After 24 h, transfection efficiency was evaluated under a fluorescent microscope and cells exhibiting comparable levels of transfection were employed for subsequent experiments.
Western blot
Cells were lysed using sample buffer (200 µl/well; #1610747, Bio-Rad) containing 2% β-mercaptoethanol (#M6250, Merk) and sonicated for 10 s at 30% amplitude. Samples were then heated at 95 °C for 5 min or at 70 °C for 10 min to preserve phosphorylation. After separation on a 4–15% or 4–20% TGX™ precast gel (Bio-Rad), proteins were transferred on a nitrocellulose membrane using a semidry transfer apparatus (Trans-blot SD; Bio-Rad).
Membranes were incubated 1 h in blocking solution (5% BSA in Tris-buffered saline containing 0.1% Tween-20 - TBST) or 5 min in EveryBlot blocking buffer (#12010020, Bio-Rad) and subsequently incubated with primary antibodies overnight at 4 °C. The following primary antibodies were used at 1:1000 dilution: mouse monoclonal anti-GFP (#11814460001, Roche Diagnostics), sheep polyclonal anti-CDKL5 phospho-Tyr171 (MRC PPU Reagents and Services, University of Dundee, UK), sheep anti-MAP1S p-Ser900 (MRC PPU Reagents and Services, University of Dundee, UK), rabbit polyclonal anti-EB2 phospho-Ser222 (#00117741, Covalab) and rabbit polyclonal anti-EB2 (#orb234827, Biorbyt).
The HRP-conjugated secondary antibody (1:5000 in 5% BSA in TBS-T or EveryBlot buffer) was then added for 1 h. Immunocomplexes were visualized using the ECL substrate kits Westar sun (Cyanagen), Antares (Cyanagen) or Clarity™ Western ECL Substrate (Bio-Rad) and the ChemiDoc™ system (Bio-Rad) or the Uvitec system (Cleaver Scientific). Quantification of bands was performed using the Image Lab 5.2.1 Software (Bio-Rad) or the Uvitec software (Cleaver Scientific). When appropriate, blots were cut prior to hybridization with the antibody. Data are expressed as a percentage of controls, arbitrarily set at 100%, and corresponding to untreated WT GFP-CDKL5. The readthrough efficiency of GFP-CDKL5 was measured by dividing the intensity of the full-length GFP-CDKL5 protein signal by the sum of the intensities of detected GFP-CDKL5 bands. TEY phosphorylation was measured by dividing the intensity of Tyr171 phosphorylation of readthrough GFP-CDKL5 derivatives with that of full-length WT GFP-CDKL5. EB2 phosphorylation was quantified by dividing the signal intensity of pEB2 with that of total EB2. MAP1S phosphorylation was measured by dividing the signal intensity of pMAP1S with that of TGX.
Quantitative RT-PCR
To investigate the impact of ACE-tRNAArgUGA on the RNA stability of exogenously expressed CDKL5 mRNAs, total RNA was extracted from 4/5 of the aqueous phase of the PureZOL (#7326890, Bio-Rad) volume added to transfected cells. RNA quantification was performed, after DNA removal by DNase I digestion (#D5307, Merk), using a NanoDrop spectrophotometer and its integrity was assessed by agarose gel electrophoresis. 500 ng of RNA was reverse-transcribed using the iScript cDNA Synthesis Kit (#1708891, Bio-Rad) according to the manufacturer’s instructions. The resulting cDNA served as a template for qRT-PCR using the SYBR Green Master Mix (#4472908, Applied Biosystem) and the following primers: human CDKL5 forward 5ʹ-TGGTTGGCATGTGTCCTCTG-3ʹ, reverse 5ʹ-ATTTGGCCCACTTTTGGCAC-3ʹ; human GAPDH forward 5ʹ-CCACATCGCTCAGACACCAT-3ʹ, reverse 5ʹ-CCAGGCGCCCAATACG-3ʹ. From the same samples, DNA was isolated starting from 1/5 of the aqueous phase and adding glycogen (50 µg/ml), 0.3 M sodium acetate (pH = 5) and 100% ethanol to favor DNA precipitation. DNA pellet was obtained by centrifuging samples at 18,000 g for 30 minutes and washing it with 70% ethanol. The resulting DNA was resuspended in ddH2O and used for qPCR with the following GFP primers: forward 5’-ATATCATGGCCGACAAGCA-3’, reverse 5’-GTTCTGCTGGTAGTGGTCG-3’. qPCRs were run in a QuantStudio 5 Real-Time PCR System (Thermo Fisher Scientific). The melting curves showed a single product peak, indicating good product specificity. Fold change in gene expression was calculated by the 2-ΔΔCt method, using GAPDH as an internal standard; obtained values were corrected for the transfection efficiency estimated from the real time conducted on the GFP gene.
Immunofluorescence and image analysis
Cells seeded on coverslips were fixed with 4% paraformaldehyde in PBS for 10 min at room temperature and then washed four times with PBS. Cells were permeabilized in PBS containing 0.2% Triton X-100 at 4 °C and washed three times with PBS containing 0.2% BSA. Following a 15-minute incubation in blocking solution (4% BSA in PBS) at room temperature, immunofluorescence staining was carried out by incubating the primary anti-GFP antibody (1:100 in blocking solution; #11814460001, Roche Diagnostics) overnight at 4 °C. After 3 washes in 0.2% BSA in PBS, cells were incubated with anti-mouse Alexa Fluor 488 secondary antibody (1:500 in blocking solution) for 1 h at room temperature. Nuclei were stained with DAPI (1:1000 in PBS; #62248, Thermo Fisher Scientific) and coverslips were mounted using Fluoromount-G reagent (#00–4958-02, Thermo Fisher Scientific). Images were acquired from at least 3 coverslips for each experimental group. For the analysis of the nuclear/cytosolic distribution, images were acquired using a confocal microscope with a 63X oil immersion objective (Zeiss LSM 800). Pinhole size was set to 1 Airy Unit (AU) and the laser intensity was kept constant, while the gain was adjusted to prevent saturated pixels. Image analysis and cell counting were performed using Fiji software. Fluorescence values were expressed as Integrated Density corrected for the background.
Statistical analysis
The data are presented as means ± standard error (SEM) and were analyzed using GraphPad Prism 8 software. The presence of outliers was assessed by ROUT test (Q = 1%) or Grubb’s test (a = 0.05%). Before statistical analysis, normality distribution was assessed by D’Agostino and Pearson tests, and Shapiro-Wilk test. Statistical significance was analysed by proper ANOVA test, depending on the experimental setting. A p-value of less than 0.05 was considered statistically significant.
Data availability
All data are available upon request to corresponding author.
References
Van Bergen, N. J. et al. CDKL5 deficiency disorder: molecular insights and mechanisms of pathogenicity to fast-track therapeutic development. Biochem. Soc. Trans. 50, 1207–1224 (2022).
Jakimiec, M., Paprocka, J. & Śmigiel, R. CDKL5 Deficiency Disorder-A Complex Epileptic Encephalopathy. Brain Sci. 10, 107 (2020).
Mangatt, M. et al. Prevalence and onset of comorbidities in the CDKL5 disorder differ from Rett syndrome. Orphanet J. Rare Dis. 11, 39 (2016).
Kilstrup-Nielsen, C. et al. What we know and would like to know about CDKL5 and its involvement in epileptic encephalopathy. Neural Plast. 728267 2012 (2012).
Hector, R. D. et al. Characterisation of CDKL5 transcript isoforms in Human and Mouse. PLoS One. 11, e0157758 (2016).
Bertani, I. et al. Functional consequences of mutations in CDKL5, an X-linked gene involved in infantile spasms and mental retardation. J. Biol. Chem. 281, 32048–32056 (2006).
Rusconi, L. et al. CDKL5 expression is modulated during neuronal development and its subcellular distribution is tightly regulated by the C-terminal tail. J. Biol. Chem. 283, 30101–30111 (2008).
Szafranski, P. et al. Neurodevelopmental and neurobehavioral characteristics in males and females with CDKL5 duplications. Eur. J. Hum. Genet. 23, 915–921 (2015).
Sismani, C. et al. 9 mb familial duplication in chromosome band Xp22.2-22.13 associated with mental retardation, hypotonia and developmental delay, scoliosis, cardiovascular problems and mild dysmorphic facial features. Eur. J. Med. Genet. 54, e510–e515 (2011).
Frasca, A. et al. Not just loss-of-function variations: identification of a hypermorphic variant in a patient with a CDKL5 missense substitution. Neurol. Genet. 8, e666 (2022).
Muñoz, I. M. et al. Phosphoproteomic screening identifies physiological substrates of the CDKL5 kinase. EMBO J. 37, e99559 (2018).
Baltussen, L. L. et al. Chemical genetic identification of CDKL5 substrates reveals its role in neuronal microtubule dynamics. EMBO J. 37, e99763 (2018).
La Montanara, P. et al. Synaptic synthesis, dephosphorylation, and degradation: a novel paradigm for an activity-dependent neuronal control of CDKL5. J. Biol. Chem. 290, 4512–4527 (2015).
Olson, H. E. et al. Current neurologic treatment and emerging therapies in CDKL5 deficiency disorder. J. Neurodev Disord. 13, 40 (2021).
Fazzari, M., Frasca, A., Bifari, F. & Landsberger, N. Aminoglycoside drugs induce efficient read-through of CDKL5 nonsense mutations, slightly restoring its kinase activity. RNA Biol. 16, 1414–1423 (2019).
Balestra, D. et al. Splicing mutations impairing CDKL5 expression and activity can be efficiently rescued by U1snRNA-Based therapy. Int. J. Mol. Sci. 20, 4130 (2019).
Temple, G. F., Dozy, A. M., Roy, K. L. & Kan, Y. W. Construction of a functional human suppressor tRNA gene: an approach to gene therapy for beta-thalassaemia. Nature 296, 537–540 (1982).
Lueck, J. D. et al. Engineered transfer RNAs for suppression of premature termination codons. Nat. Commun. 10, 822 (2019).
Porter, J. J. et al. Therapeutic promise of engineered nonsense suppressor tRNAs. Wiley Interdiscip Rev. RNA. 12, e1641 (2021).
Coller, J. & Ignatova, Z. RNA therapeutics for genetic diseases. Nat. Rev. Drug Discov. 23 (2), 108–125 (2024).
Anastassiadis, T. & Köhrer, C. Ushering in the era of tRNA medicines. J. Biol. Chem. 299, 105246 (2023).
Morais, P., Zhang, R. & Yu, Y. T. Therapeutic nonsense suppression modalities: from small molecules to nucleic acid-based approaches. Biomedicines 12, 1284 (2024).
Wang, J. et al. AAV-delivered suppressor tRNA overcomes a nonsense mutation in mice. Nature 604, 343–348 (2022).
Wu, Q. & Bazzini, A. A. Translation and mRNA Stability Control. Annu. Rev. Biochem. 92, 227–245 (2023).
Chen, Q. et al. CDKL5, a protein associated with Rett syndrome, regulates neuronal morphogenesis via Rac1 signaling. J. Neurosci. 30, 12777–12786 (2010).
Sudhakar, V. & Richardson, R. M. Gene Therapy for neurodegenerative diseases. Neurotherapeutics 16, 166–175 (2019).
Zhang, Y. & Wu, Z. Y. Gene therapy for monogenic disorders: challenges, strategies, and perspectives. J. Genet. Genomics. 51, 133–143 (2024).
Spelier, S., van Doorn, E. P. M., van der Ent, C. K. & Beekman, J. M. Koppens, M.A.J. Readthrough compounds for nonsense mutations: bridging the translational gap. Trends Mol. Med. 29, 297–314 (2023).
Li, S. et al. Pharmaceuticals promoting premature termination Codon Readthrough: Progress in Development. Biomolecules 13, 988 (2023).
Lombardi, S., Testa, M. F., Pinotti, M. & Branchini, A. Molecular insights into determinants of translational readthrough and implications for nonsense suppression approaches. Int. J. Mol. Sci. 21, 9449 (2020).
Devinsky, O., King, L., Bluvstein, J. & Friedman, D. Ataluren for drug-resistant epilepsy in nonsense variant-mediated dravet syndrome and CDKL5 deficiency disorder. Ann. Clin. Transl Neurol. 8, 639–644 (2021).
Rodnina, M. V. The ribosome in action: tuning of translational efficiency and protein folding. Protein Sci. 25, 1390–1406 (2016).
Hashimoto, S., Nobuta, R., Izawa, T. & Inada, T. Translation arrest as a protein quality control system for aberrant translation of the 3’-UTR in mammalian cells. FEBS Lett. 593, 777–787 (2019).
Ivanov, A. et al. Polyadenylate-binding protein-interacting proteins PAIP1 and PAIP2 affect translation termination. J. Biol. Chem. 294, 8630–8639 (2019).
Martins-Dias, P. & Romão, L. Nonsense suppression therapies in human genetic diseases. Cell. Mol. Life Sci. 78, 4677–4701 (2021).
Palmieri, M., Pozzer, D. & Landsberger, N. Advanced genetic therapies for the treatment of Rett syndrome: state of the art and future perspectives. Front. Neurosci. 17, 1172805 (2023).
Acknowledgements
We thank Dr. Alessandro Arcari for his technical help. We are grateful to all members of N.L. laboratory for helpful discussions. Eventually we are indebted to the two Italian association of parents, Pro RETT Ricerca and L’Albero di Greta, that always inspire our research. This study was funded partially by NIH grant R01 HL153988 (to J.D.L) and Pro RETT Ricerca and L’Albero di Greta (to N.L.).
Author information
Authors and Affiliations
Contributions
Conceptualization: N.L.; methodology: N.L.; formal analysis: A.F.; investigation: S.P., A.M., G.F., P.A.; resources: J.J.P., J.D.L, A.F. and N.L; writing (original draft): N.L.; writing (Review & Editing): M.P, A.F., A.B., J.J.P. and J.D.L; visualization: S.P., A.M. and A.F.; supervision: N.L.; funding acquisition: J.D.L and N.L.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pezzini, S., Mustaccia, A., Aboa, P. et al. Engineered tRNAs efficiently suppress CDKL5 premature termination codons. Sci Rep 14, 31791 (2024). https://doi.org/10.1038/s41598-024-82766-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-82766-0
