
Abstract
We evaluate the evidence of cryptic speciation in Larimus breviceps, a species widely distributed in the western South Atlantic, from the Greater Antilles to Santa Catarina in Brazil. Mitochondrial (COI, Cyt b, and Control Region) and nuclear (IGF1 and Tmo-4C4) sequences were obtained from populations in the western South Atlantic. The analysis revealed two genetically distinct, sympatric lineages with no gene flow, with L. breviceps lineage II (LII) being closer to Larimus pacificus than to the L. breviceps lineage I (LI). The most recent common ancestor (TMRCA) for the L. breviceps LI and L. pacificus/L. breviceps LII clade dates from 12.3 Ma, whereas TMRCA for the L. pacificus and L. breviceps LII dates from 3.4 Ma, indicating that speciation processes may be related to the rise of the Isthmus of Panama. Despite these profound genetic differences, morphometric analyses found only subtle differences between lineages, with specimens of the LI being slightly larger than those of the LII, suggesting the existence of cryptic species. Within each lineage there is a pattern of panmixia within the study area. Therefore, we suggest that it is necessary a taxonomic revision of Larimus from the western Atlantic to validate the species status of such lineages.
Similar content being viewed by others


Introduction
The shorthead drum, Larimus breviceps Cuvier, 1830 is a member of the family Sciaenidae, widely distributed in the western Atlantic from the Antilles and Costa Rica to southern Brazil1,2. This species inhabits coastal waters and estuaries, at depths of up to 60 m on muddy or sandy bottoms3. Larimus breviceps is a small-sized Sciaenidae with total length reaching 32.5 cm4, classified as a marine migrant species, where adults live in the coastal area and uses estuaries for feeding and spawning5,6.
The shorthead drum is a fishery resource caught by artisanal fisheries, mainly as bycatch in Brazilian shrimp fisheries, and a recent study indicated slight overexploitation and overfishing in Northeast Brazil7. However, despite its ecological and economic importance, understanding of the genetic aspects of the species is limited, except for its inclusion in a DNA barcoding study8, and in a phylogeny of the western South Atlantic Sciaenidae9. Both studies have detected two groups with a high degree of genetic divergence, 7,6% for COI in Ribeiro et al.8, and 2.8% for 16S rDNA, 9.2% for COI, and 1% for Tmo-4C4 in Santos et al.9, consistent with species-level differentiation. However, the literature only records L. breviceps in the western South Atlantic1,2, and the biological significance of this genetic divergence remains to be explored. Moreover, no studies have assessed whether these genetic differences suggest the existence of morphological variation between groups of Larimus. In the family Sciaenidae, morphometric studies have detected subtle morphological differences in cryptic species of Macrodon, which inhabits the same area of Larimus10. Thus, geometric morphometrics analysis has become a widespread approach for comparing cryptic species and defining stock units11, particularly for taxa with some degree of threat12 or those of significant economic or social importance10. Therefore, is necessary a systematic study to evaluate the presence of cryptic species of Larimus in the western South Atlantic.
Phylogeographic studies of taxa from the western South Atlantic suggest that climatic fluctuations or physical barriers have driven allopatric speciation10,13,14,15,16. Also, ecological speciation has been suggested to explain divergence with gene flow in marine and estuarine species in the area14,17,18,19.
The western South Atlantic has been influenced by distinct patterns of marine circulation20,21,22, which have created thermal gradients that may have promoted genetic differentiation in the populations of species that inhabit this area. Another relevant factor that may influence population structure in widely distributed species dependent on specific environments, is the habitat discontinuities produced by the variation in the geomorphological conditions of the continental shelf. Areas with predominantly muddy bottoms are found primarily in the vicinity of major rivers, where sediments accumulate from the fluvial discharge, mainly on the northern and southern coasts of Brazil, separated by areas of predominantly rocky bottoms with coral reefs on the northeastern coast of Brazil21. Glaciations have also shaped the genetic structure of population of marine species, by changing ocean circulation patterns, substantially reducing the sea surface temperature and sea level (which decreased by up to 140 m) and causing profound changes in the availability of habitat for marine and estuarine species23,24.
Given its wide distribution, L. breviceps inhabits highly diverse areas with marked differences in circulation patterns, salinity, and sea surface temperatures14,21,22. Additionally, distinct geomorphological conditions of the continental shelf, along with the mouths of large rivers (Amazon, São Francisco and Doce rivers), which discharge freshwater and sediments in some areas, further contribute to these differences14,25. Furthermore, its preference for muddy bottoms associated with the discontinuities of this feature in the western South Atlantic may promote divergent selection, causing population differentiation.
Therefore, considering that the species inhabits areas with marked environmental differences, the present study aimed to evaluate the hypothesis of L. breviceps speciation in the western South Atlantic, sampling populations from Pará on the northern coast of Brazil to Santa Catarina at the southern extreme of the species range on the Brazilian coast, using phylogenetic and population genetic analyses based on a multilocus approach with the mitochondrial (Cytochrome c Oxidase subunit I - COI, the Control Region - CR, and Cytochrome b – Cyt b) and nuclear (Tmo-4C4 and Insulin-like Growth Factor 1 – IGF1) markers. A quantitative morphometric analysis of body shape was also included to assess whether the potential genetic differentiation is reflected in morphological differences between groups.
Results
Phylogenetic analysis, TMRCA estimation, and sequence divergence
For the construction of the Bayesian gene trees (BI) and species tree for the time of the most recent common ancestor (TMRCA) estimates we used an aligned dataset of 3062 bps, including indels, from 79 L. breviceps individuals, composed of fragments of CR (824 bps), COI (624 bps), Cyt b (786 bps), IGF1 (363 bps), and Tmo-4C4 (465 bps), with a total of 495 variable sites. The sequences generated in this study were deposited in GenBank under accession numbers PQ094945 - PQ095298 (for CR), PQ060262-PQ060358 (for COI), PQ095299 - PQ095395 (for Cyt b), PQ095396 - PQ095490 (for Tmo-4C4), and PQ095491 - PQ095568 (for IGF1).
All the trees were concordant and revealed two monophyletic lineages, which were designated as lineages I and II, with high posterior probabilities (Fig. 1; Supplementary Figs. 1–5). These groups coexist within the study area, except in Pará, where all specimens were assigned to lineage I, and in Paraíba, where all individuals were assigned to lineage II. As the analyses produced trees with the same arrangement, only the species tree is provided (Fig. 1). It is noteworthy that, in all analyses, except those based on nuclear markers, lineage II is closer to L. pacificus, from the eastern Pacific, than to lineage I (Fig. 1; Supplementary Figs. 1–5).

Species tree based on Bayesian inference of the nuclear (IGF1 and Tmo-4C4) and mitochondrial data (COI, Cyt b, and CR). The numbers below the nodes indicate the posterior probability, while those above the nodes show the TMRCA and credibility intervals. Node bars depict 95% highest posterior density. The species Sciaenops ocellatus and Nebris microps were used as outgroups.
The TMRCA shows that the separation between Larimus breviceps LI from the clade L. pacificus and L. breviceps LII dates from 12.3 (HPD: 11.1–14.5) Ma, in the middle Miocene, whereas L. breviceps LII split from the L. pacificus at 3.4 (HPD: 1.3–5.4) Ma, during the Pliocene (Fig. 1).
The divergence within the Larimus lineages was low for all markers, ranging from 0.03 to 0.3%, except for IGF1, which showed 1.6% of divergence within lineage I and 2.9% within lineage II (Table 1). The mean divergence between Larimus lineages I and II ranged from 0.5% (for Tmo-4C4) to 10.8% (for CR) (Table 1).
Population, phylogeographic and demographic analyses
For these analyses we used fragments of CR (822 bps), COI (624 bps), Cyt b (786 bps), IGF1 (387 bps), and Tmo-4C4 (465 bps). Only four Tmo-4C4 haplotypes were identified, however, these were sufficient to discriminate between Larimus lineages. Therefore, this marker was included solely in part of the analyses. As the phylogenetic results indicated the presence of two distinct lineages of Larimus, found in most of the sample areas, the analyses were performed to each lineage, independently. Although variation in sample size among different locations may influence the results of genetic analyses, our findings showed no significant differences between populations within lineages I and II. Therefore, populations within each lineage were pooled for population, demographic, and phylogeographic analyses. The number of individuals, haplotypes, and genetic diversity for each marker and lineages are summarized in Table 1.
The levels of haplotype diversity in lineage I varied from 0.581 (for COI) to 0.892 (for IGF1) (Table 1). For the lineage II, the haplotype diversity was lower, and varied from 0.2452 (Cyt b and COI) to 0.661 (IGF1) (Table 1). Conversely, the nucleotide diversity was low in both groups and for all markers, ranging from 0.0003 (for Cyt b in lineage II) to 0.029 (for IGF1 in lineage II) (Table 1). Intriguingly, in general, the genetic diversity was higher in lineage I for all markers.
For all markers, the haplotype networks indicate two groups that corresponds to lineages I and II, previously reported by phylogenetic trees, separated by a minimum of two (Tmo-4C4) and a maximum of 89 (CR) mutations (Fig. 2B, C, D, E, and F). Within each group (except for Tmo-4C4), the network is star-shaped, with a few central and many single peripheral haplotypes. Additionally, within lineages I and II, the most common haplotypes were shared by populations separated by wide geographical distances, indicating the absence of geographically structured groups (Fig. 2B, C, D, E, and F).

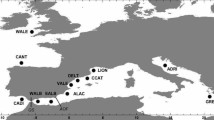
Sampling locations and haplotype genealogy of Larimus breviceps. (A) Sampling locations and the number of individuals analyzed. The haplotype network is based on the maximum likelihood trees for the (B) CR, (C) Cyt b, (D) COI, (E) Tmo-4C4, and (F) IGF1 sequences. Each circle represents a haplotype, and the circle’s size is proportional to its frequency. The dots represent inferred, unsampled or extinct haplotypes. The map shows the geographic distribution of the haplotypes present in lineages I and II.
The Structure analysis confirmed the pattern observed in the remaining analyses, suggesting that k = 2 is the most likely (Fig. 3A). A mixed pattern was found within each lineage, however, indicating a lack of geographic sub-structuring (Fig. 3B and C).

Assignment of individuals to the two genetic lineages using the Structure algorithm, considering K = 2 for (A) Lineages I and II, as defined by the analysis, (B) the populations of lineage I, and (C) the populations of lineage II.
The isolation with migration (IM) model indicated an absence of gene flow between lineages I and II (Fig. 4A). When applied to the analysis of gene flow among the populations within each lineage, the IM model pointed to a bidirectional flow between all pairs of populations in lineage I, except for Bahia-Espírito Santo, where it was unidirectional (Fig. 4B). In lineage II (Fig. 4C), gene flow was unidirectional between pairs of populations (Ceará-Paraíba; Bahia-Paraíba; Bahia-São Paulo).

The posterior probability distributions and population migration rate (2Nm) estimates derived from the Isolation-with-Migration model (IMa2) for pairwise comparisons between (A) Lineages I and II, (B) populations of lineage I, and (C) populations of lineage II. PA = Pará; CE = Ceará; PB = Paraíba; BA = Bahia; ES = Espírito Santo; SP = São Paulo; SC = Santa Catarina; LI = lineage I and LII = lineage II.
In lineage I, Tajima’s D and Fu’s Fs were negative (Table 1), although they were significant only in the case of the CR (both tests), Cyt b and IGF1 (Fs only). In lineage II, all markers (except IGF1) presented significantly negative values for both parameters (Table 1).
For both lineages, analysis of Extended Skyline Bayesian Plot revealed a pattern of population expansion estimated to have occurred around 30 thousand years ago (Fig. 5).

Graphs of Extended Bayesian Skyline Plot based on the five loci analyzed in samples from Larimus breviceps lineage I (A) and Larimus breviceps lineage 2 (B).
Morphometric analysis
The multidimensional scaling analysis (MDS) of the 20 morphometric variables revealed a partial separation of the samples from lineages I and II (Fig. 6). The ANOVA found significant differences between groups in all the dependent variables, with individuals from lineage I having proportionately larger body sizes than those from lineage II (Supplementary Table S1). The PCA confirmed that the individuals of lineage I had head and body dimensions greater than those of lineage II, with head length (measure 1–17) and standard length (measure 1–11) contributing most to the variability in the data (Fig. 7; Supplementary Table S2).

Ordination analysis by the MDS method of the morphological measurements of the genetically defined groups of Larimus breviceps (LI and LII).

Dispersal of the scores of the first and second principal components of the (A) head and (B) body size dimensions data for each genetically-defined lineage of Larimus breviceps.
Discussion
Genetic and morphological differentiation of Larimus lineages
This is the first study that used molecular and morphological analyses to evaluate the speciation hypothesis in L. breviceps, the only valid species of the genus described from the western South Atlantic which is widely distributed from Greater Antilles and Costa Rica to Santa Catarina in Brazilian coast. Although we did not sample the species throughout its entire distribution range, all, phylogenetic, sequence divergence and population genetic analyses indicated the existence of two genetically distinct, sympatric lineages of L. breviceps. The subdivision of populations and speciation in marine ecosystems have been recorded and have been attributed to historical, physical or ecological barriers10,13,14,17,18,26.
The phylogenetic analyses indicated that L. breviceps is paraphyletic, where lineage II is closer to L. pacificus, which occurs in the eastern Pacific, than to lineage I. It is noteworthy that the ancestors of the lineages diverged in the middle Miocene, approximately 12.3 (HPD: 11.1–14.5) Ma, while the divergence between L. breviceps lineage II and L. pacificus occurred in the Pliocene, 3.4 (HPD: 1.3–5.4) Ma. All population analyses indicate that, although they are sympatric, there are deep and significant differences between lineages I and II, presumably due to the absence of gene flow between them, which corroborates the pattern revealed by the phylogenetic analyses.
Our results are in agreement with the hypothesis of two lineages as suggested by Ribeiro et al.8 and Santos et al.9. Although we did not sample specimens from the northern extreme of the species range, the presence of a lineage closer to the Pacific species (L. pacificus) is similar to the pattern identified by Silva et al.13 in a study that demonstrated speciation in the sciaenid Ophioscion punctatissimus (currently species Stellifer punctatissimus and Stellifer menezesi, see Fricke et al.2). The time of divergence between Larimus lineages (12.3 Ma; HPD: 11.1–14.5 Ma) coincides with the estimated time for the constriction of the Central American Seaway (CAS), which occurred due to the gradual uplift of the Isthmus of Panama, which began around 17 Ma ago and culminated in the closure of the connection between the Atlantic and Pacific around 3 Ma27,28,29. Between 12.9 and 11.8 Ma, the Panama sill rose about 1000 m, due to tectonic disturbances in Northwest South America. This gradual elevation of the Panama sill narrowed the CAS and reduced the flow of water from the Pacific to the Caribbean, which resulted in changes in temperature, salinity, and circulation patterns of marine currents in the Atlantic27,30,31. One of the effects of the reduction of Pacific waters to the Atlantic, in the middle Miocene, was the reversal of the flow of the North Brazil Current (NBC) from southward to northward, its current pattern31, which may have isolated populations of Larimus to the North and South of the area of influence of the NBC in the equatorial Atlantic, promoting divergence between the ancestors of the lineages. However, to make a more comprehensive analysis of the differentiation pattern of these lineages, it is necessary to carry out a sampling throughout the entire range of Larimus in the western Atlantic.
Cladogenesis between L. pacificus and L. breviceps lineage II occurred 3.4 (HPD: 1.3–5.4) Ma, which coincides with the estimated time for the closure of the Pacific-Atlantic connection due to the uplift of the Isthmus of Panama27. Therefore, the isolation of populations in the Atlantic and Pacific, promoted by the closure of the Panama Isthmus, must have prevented gene flow, and allowed them to follow independent evolutionary trajectories, culminating in the process of speciation between L. pacificus and L. breviceps lineage II. Several studies have demonstrated records of geminate species that evolved from isolated populations by the Isthmus of Panama13,26,29,30,32.
The morphometric analysis showed that head and body dimensions were larger in lineage I in comparison to lineage II, which reinforces the hypothesis of two species of Larimus from the western South Atlantic. However, limitations of these analyses must be considered, since multivariate morphometry quantitatively characterizes biological shape by simultaneously analyzing the different levels of variance and covariance between the measurements of an organism, i.e. one of the most difficult problems is to remove the effect of allometric growth in some specimens, especially given that in this study we used individuals collected in different regions, which reflects the richness of the environments and growth performance33,34,35.
Differences in body shape may be associated with ecological adaptations to distinct ecological niches, related to variations in habitat quality and feeding strategies36, although no such differences can be confirmed in the present case. However, variations in behavioral parameters, such as feeding strategies, reproductive patterns, competition, and even the position of the animals in the water column, may contribute to this differentiation. Ecomorphology in fish, as in other organisms, reflects pressures to which species are exposed, whether from relationships with other species or adaptability demands37,38. In Sciaenidae, geometric morphometrics has helped distinguish cryptic species that overlap in most morphological traits10,35, with contrasting patterns associated with distinct habitat use. This reinforces the need for further, detailed studies of the biology and ecology of each group to evaluate the processes that drove morphological differentiation.
Considering the deep genetic divergence and subtle morphological differences, we propose the presence of two cryptic species of Larimus in the western South Atlantic. Thus, a comprehensive revision of the alpha taxonomy of the Larimus species is warranted, including a systematic examination of type specimens or representative material to formally describe the new species within this genus in the western South Atlantic.
Population genetics, phylogeography, and demographic history of Larimus lineages
In general the haplotype diversity was higher in lineage I, while the two lineages showed low nucleotide diversity for all the markers. The higher haplotype diversity in lineage I could be related to sampling, which is greater in this group. However, the two lineages showed low nucleotide diversity for all markers, which may reflect aspects related to the evolutionary history of the groups.
The pattern of high haplotypic and low nucleotide diversities observed in lineage I can be indicative of rapid population expansion and accumulation of mutations after a period of low effective population size caused by bottlenecks39. On the other hand, the low genetic diversity in lineage II may be related to a historical reduction of population size by bottleneck or founder effects39. Both groups are sympatric in regions which went through severe environmental variations due to climatic oscillations during the Pleistocene. During glacial periods, the sea level in the western South Atlantic decreased between 100 and 140 m, sea surface temperature may have reached down to 6oC lower than nowadays temperature and there were changes in the pattern of circulation of marine currents23,24. Therefore, paleoenvironmental changes may have restricted the availability of habitats for coastal and estuarine species, such as Larimus lineages, reducing the effective population size and, consequently their genetic diversity, as proposed for other marine taxa40,41,42. On the other hand, when climatic conditions become favorable, during interglacial periods, there was a demographic growth (discussed below) with a consequent increase in diversity levels. However, considering the different patterns of genetic diversity between both lineages of Larimus, it is likely that lineage II was more affected by the climatic oscillations, although it is necessary a deeper study to test such assumption.
The population analysis of each Larimus lineage indicates that there is no geographic sub-structuring. The star-shaped haplotype networks of lineages I and II suggest the lack of differentiation within groups, and the Structure analysis did not detect any significant genetic sub-structuring within each group. Furthermore, the migration pattern indicated by IMa, characterized by high levels of gene flow between pairs of populations within each group, suggests that neither circulation patterns, sea surface temperatures nor habitat discontinuities influence the observed genetic structure of the groups. This evidence indicates that each group represents a large panmictic population in the western South Atlantic. High genetic connectivity in marine species generally is attributed to the dispersal capability of adults, eggs and larvae which can be strengthened by marine currents43,44,45. Larimus breviceps is classified as a migrant marine fish46, although there is no information regarding the distance that this species may reach out during dispersion. So, it is likely that there might be a displacement of individuals to adjacent areas which can favor the homogeneization of populations for each lineage. Yet, a deeper understanding of the genetic, biological and ecological aspects are critical for the comprehension of patterns of structuration and the connectivity of groups.
With regard to the demographic history of the two groups, the negative and significant neutrality tests, for most markers, indicate a potential pattern of population expansion in both lineages (Table 1). When the D and Fs values are significantly different from zero, they indicate a deviation from neutrality, and tend to be negative when there is an excess of recent mutations, suggesting deviations caused by population expansion, or other evolutionary processes, such as hitchhiking, background selection, or recombination47. Both, demographic fluctuations and selection have similar signatures in populations, however, contraction or expansion of populations can leave signatures throughout the genome, while selection generally affects genomic regions of functional importance48. Therefore, the analysis of multiple loci allows for distinguishing between stochastic demographic processes and selection49.
The EBSP also suggests population expansion in both lineages of Larimus estimated to have occurred about 30 thousand years ago during the Pleistocene, coinciding with an interglacial period in the southwest Atlantic23,24. During interglacial episodes, there was an increase in marine temperature as well as the sea level increased over the continental shelf, which may have allowed the establishment of new habitats that favored the expansion of Larimus lineages in the western South Atlantic. Demographic expansion in other teleosts occurring in the western South Atlantic has been registered and generally is associated with Pleistocene climate changes10,40,42,43,44,45.
The sum of the evidence from the present study indicates a process of allopatric speciation, resulting in the formation of two monophyletic lineages, which can be considered distinct species, based on the phylogenetic species concept50. These two species subsequently expanded their ranges and now coexist along the western South Atlantic. Despite the clear differentiation found at the molecular level, the two species present subtle morphometric differences and might be thus considered to be cryptic species. As the two groups may occupy similar niches, their morphological similarities are likely due to stabilizing selection, which may reinforce the retention of morphological characteristics that enhance fitness in their respective habitats, irrespective of genetic differentiation, a common pattern observed in cryptic Sciaenidae species, such as Macrodon and Stellifer in the western South Atlantic. However, this hypothesis requires further systematic testing. These findings also reinforce the need for a more comprehensive sample of populations and genetic analyses from the area of occurrence of Larimus in the western Atlantic, as well as behavioural studies, ecological niche modelling, and studies of the biology of the two taxa, to define the geographic limits of the two species and assess potential hidden diversity in Larimus in the northern extreme of the species range. Finally, our results indicate that Larimus lineages should be managed as distinct species, implementing appropiate management strategies to guarantee their genetic integrity, particularly in areas like the Brazilian coast where they are impacted by overexploitation.
Materials and methods
Sampling
A total of 353 specimens were sampled, by artisanal fishing, along the western South Atlantic in the Brazilian states of Pará (N = 35), Ceará (N = 36), Paraíba (N = 53), Bahia (N = 60), Espírito Santo (N = 73), São Paulo (N = 43), and Santa Catarina (N = 53) (Fig. 2). The tissue of Larimus pacificus (N = 1) was donated by the Fish Collection of the Zoology Museum of the University of Costa Rica (UCR). The specimens were identified morphologically by consulting the specialized literature3, and samples of muscle tissue were preserved in absolute ethanol and/or frozen until processing in the laboratory.
In Brazil, the samples were purchased from artisanal fishermen, and permission to undertake collection, handling, transportation, and DNA extraction was obtained by Dr. Simoni Santos from the Brazilian Environment Ministry (Permit number 18401-3). The sample from Costa Rica was collected under permissions of the Sistema Nacional de Áreas de Conservación (Permit number R-SINAC-SE-DT-PI-003-2021) and the Comisión Nacional para la Gestión de la Biodiversidad (Permit number R056-2015-OT-CONAGEBIO), accessed through Resolución No. 377 of the Vicerectoría de Investigación of the UCR.
DNA extraction, amplification, and sequencing of genomic regions
The total DNA was extracted using a DNeasy kit (QIAGEN) following the manufacturer’s protocol. To check the quality of the material obtained, the DNA was electrophoresed in agarose gel (1%) stained with Gel Red (Biotium Inc., CA), with the runs being viewed under ultraviolet light.
The fragments of the Control Region (CR), Cytochrome C Oxidase subunit I (COI), Cytochrome b (Cyt b), Tmo-4C4, and intron 2 of the Insulin-like Growth Factor 1 (IGF1) were amplified by Polymerase Chain Reaction (PCR) using the specific primers and amplification protocols described in Table 2. The PCR reactions were run in a total volume of 25 µL containing 4 µL dNTPs (1.25 mM), 2.5 µL buffer (10X), 2 µL MgCl2 (25 mM), 0.25 µL of each primer (200 ng/µl), 1–1.5 µL genomic DNA (100 ng/µL), 1 U of Taq DNA Polymerase (5 U/µL), and purified water. The amplified products were purified with the ExoSAP-IT enzyme (Amersham Pharmacia Biotech, Inc., UK) according to the manufacturer’s instructions and sequenced using the Sanger method with a BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems), following the manufacturer’s instructions. Electrophoresis was conducted in an ABI 3500 XL (Applied Biosystems).
Phylogenetic analysis, divergence time estimates, and sequence divergence
The phylogenetic analyses were conducted using a dataset comprising all nuclear and mitochondrial markers derived from 79 individuals: 50 from Larimus lineage I and 29 from Larimus lineage II. Specimens were selected based on haplotypes observed in the Control Region, for which sequences of all markers were available, except IGF1, which was not sequenced in 12 specimens (02 from lineage I and 10 from lineage II). The sequences were aligned in CLUSTAL W with default parameters59 and edited in BioEdit 7.2.560. Degenerated bases were used in the heterozygous sites of nuclear datasets. The following sciaenids species were used as outgroups, as they showed high homology score with Larimus lineages in BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and that are common to most sequence data used in the phylogenetic analyses, Nebris microps Cuvier, 1830 (COI - NC029875, Cyt b - KP722653, Tmo-4C4 - JX904046, CR - NC034350) a taxon closely related to Larimus61, and Sciaenops ocellatus Linnaeus, 1766 (COI - MF004315, Cyt b - KP722687, CR - NC016867, Tmo-4C4 - KC130636, and IGF1 - GU799603).
To infer the evolutionary relationships of L. breviceps lineages, we perform the Bayesian analysis (BI) for each marker in Mr. Bayes 3.2.762. Optimal nucleotide substitution models were selected for each partition in PartitionFinder v2.1.163 based on the Bayesian Information Criterion (BIC). The HKY + Gamma model was selected for the markers Cyt b and CR, the HKY + I model for COI, the F81 model for IGF1, and K81UF model for Tmo-4C4. The BI analyses were conducted with four simultaneous Markovian Monte Carlo chains (MCMC), running for 1,000,000 generations with parameters defined by each model as starting values, and sampled every 1,000 generations, with a 25% burn-in applied to ensure robust convergence of the phylogenetic trees. Convergence diagnostics were assessed through effective sample size (ESS) values in Tracer 1.7.164 and by checking the Potential Scale Reduction Factor (PSRF) criterion provided by Mr. Bayes. Upon completion, consensus trees were generated with posterior probability values providing node support.
We performed a Bayesian analysis, using StarBEAST365 to estimate the divergence time of Larimus lineages in BEAST v. 2.7.766. The analysis was performed using the HKY + I + G for partitions, using the relaxed clock (log-normal) method, assuming independent rates on different branches67. The prior trees were modeled according to the Yule process of speciation, while all other prior information was based on the default values available in BEAST 2.7.7. The time calibration was estimated using sciaenid fossils, Larimus henrici and Larimus steurbauti from the Early Miocene Cantaure Formation (i.e., 23–16 Ma)68, used to constrain the TMRCA of the clade Larimus. Two independent runs were conducted with a chain length of 100 million being sampled every 100 iterations through the speciation process based on the Yule model and 10% burn-in. The results were checked using Tracer 1.7.164 and then transferred to TreeAnnotator 1.869 for the inclusion of calibration times in the branches.
All the trees were visualized in the FigTree 1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/) and edited in the Inkscape 1.1 (https://inkscape.org/pt-br/).
We estimated the mean of uncorrected p genetic divergence between the Larimus lineages I and II from each dataset on MEGA 1170.
Population, phylogeographic and demographic history analyses
These analyses were based on 353 individuals for the CR, 96 for COI and Cyt b, 94 for Tmo-4C4, and 45 for IGF1. For the nuclear markers, the gametic phase for the heterozygous sequences was defined in the PHASE algorithm71, using the default parameters, and the haplotypes with probabilities of less than 0.8 were not included in the analyses. The sequences were edited and aligned in BioEdit, and the haplotypes were defined using the DNAsp5 program72.
The haplotype and nucleotide diversity for each marker (except Tmo-4C4), was obtained using ARLEQUIN 3.573.
The genealogy and distribution of haplotypes among populations were evaluated by generating the haplotype network for each marker in the software Haploviewer74, based on unrooted Maximum Likelihood (ML) trees.
The alignments of the different markers were processed, and the informative sites were identified in the Fasta2Structure software75, which converts this information into the input format for the Structure software. The Structure 2.3.476 was used to infer the number of genetic demes in the samples, based on an admixture model with uncorrelated allele frequencies and without prior information on sample population membership, a burn-in period of 500,000, and a run length of 106 iterations. One to four clusters or demes were tested, and for each value of k, 10 independent simulations were run. The most likely k was defined by comparing the log-likelihood and Evanno’s ∆K using the softwate Structure Harvester77.
Migration patterns, using asymmetric rates, between groups and among populations within the same group were assessed including all the genetic markers using the isolation with migration approach implemented in the IMa 2.0 program78. In the run, 10 MCMCMC were used with linear heating mode. Multiple preliminary runs were conducted for prior adequacy and the mixing of chains. Different runs using different seeds were conducted to confirm the results, all of which used the same conditions, 50,000,000 steps and 10% burn-in. Both convergence and ESS values (> 200) were checked to determine the quality of the runs.
Tests of selective neutrality, D79 and Fs47, were run in ARLEQUIN, to check for deviations from neutrality in all the markers (except Tmo-4C4), and to provide inferences on their demographic history.
The demographic history of the L. breviceps populations was evaluated using five markers with Extended Bayesian Skyline Plot method (EBSP) method80, implemented in BEAST66. We used the GTR + I + G model for the partitions as suggested by the PartitionFinder 2.1 software. A relaxed clock with an uncorrelated lognormal distribution was assumed, and the markers’ ucld.stdev values were found to be close to zero, indicating that the hypothesis of a uniform replacement rate was not rejected. Consequently, a strict clock with an evolutionary substitution rate of 6.2% per million years was used, considering a minimum rate of 3.6% and a maximum rate of 8.8% for the control region81,82. After the analysis was completed, runs with ESS values greater than 200 were verified using TRACER 1.7.164, and the graph was generated using a Python script provided by Heled83.
Morphometrics analysis
All measurements were taken from the left side of the body, following84, who define 20 truss length measurements for the definition of body shape and the proportions in relation to structures such as head and abdomen (including the caudal peduncle) (Fig. 8). To reduce the error due to the positioning of the specimens on the image recording screen, the specimens were carefully measured with a caliper and then the measurement was confirmed using images. A total of 160 individuals used in the molecular analyses, representing all populations (except Espírito Santo), were measured, irrespective of sex, with the values being recorded in spreadsheets for subsequent statistical treatment.

Adapted from Parsons et al. 84.
The 20 truss lengths measured on each specimen for the morphometric analysis. (A) body measurements; (B) head measurements. 1–2: length of the snout to the origin of the dorsal fin, 1–3: length of snout to the origin of the pelvic fin, 1–11: standard length, 2–3: body depth, 2–9: dorsal base length, 4–5: length of pectoral fin, 3–7: length from the origin of the pelvic fin to the posterior margin of the anal fin, 6–9: length from the origin of the anal fin to the posterior of the dorsal fin, 2–7: length from the anterior margin of the dorsal fin to the posterior margin of the anal fin, 9 − 7: depth of the anterior caudal peduncle, 8–10: depth of the posterior caudal peduncle, 9–10: length of the dorsal caudal peduncle, 7–8: length of the ventral caudal peduncle, 9 − 8: distance from the dorsal anterior to the ventral posterior caudal peduncle, 7–10: distance between the ventral anterior and dorsal posterior caudal peduncle, 1–12: snout length, 12–13: eye diameter, 1–17: head length 14–15: cheek depth, 15–16: lower jaw length.
The measurements were analyzed individually with a one-way ANOVA (α = 5%), considering the genetically defined L. breviceps lineages I and II as categorical variables, including validation of the residuals using the normality (Shapiro Wilks) and heteroscedasticity (Levene) tests. A quadratic matrix was constructed considering the Bray Curtis distance squared between numerical variables, where multidimensional scaling (MDS) and principal components analysis (PCA) have been applied. The MDS method spatially distributes measures considering a ranking of distance, which confirms the similarity between the categories tested85,86. The stress value was used as a representative measure of the groups, where values < 0.20 were considered acceptable87. Likewise, PCA is a factorial model in which the factors are based on variance, which is explained according to vectors arranged in two or more axes. For this analysis, the first two axes were used, and their percentage of explanation of the variance was shown in tables. Before starting the calculation, the data were normalized (Z = (x - µ) / σ) to reduce stress. All the statistical analyses in this study were carried out using the PAST software88.
Data availability
The data that support the findings of this study will be openly available in GenBank at https://www.ncbi.nlm.nih.gov/genbank/, under the accession numbers PQ094945 - PQ095298 (for CR), PQ060262-PQ060358 (for COI), PQ095299 - PQ095395 (for Cyt b), PQ095396 - PQ095490 (for TMO-4C4), and PQ095491 - PQ095568 (for IGF1).
References
Parenti, P. An annotated checklist of fishes of the family Sciaenidae. J. Anim. Divers. 2, 1–92 (2020).
Fricke, R., Eschmeyer, W. N. & van der Laan, R. Eschmeyer’s catalog of fishes: genera, species, references (2024). http://researcharchive.calacademy.org/research/ichthyology/catalog/fishcatmain.asp
Cervigón, F. Los Peces Marinos de Venezuela. V II 2rd edn (Fundación Científica Los Roques, Caracas, 1993).
Aparecido, K. C., Negro, T. D., Tutui, S. L. S., Souza, M. R. & Tomás, A. R. G. Larimus breviceps Cuvier 1830 at the inner continental shelf of São Paulo and Paraná states in Growth in Fisheries Resources from the Southwestern Atlantic (eds. Vaz-Dos-Santos, A. M. & Rossi-Wongtschowski, C. L. D. B.) 162–164 (Instituto Oceanográfico – Universidade de São Paulo, 2019).
Santos, L. V. et al. Reproductive biology of the shorthead drum Larimus breviceps (Acanthuriformes: Sciaenidae) in northeastern Brazil. Reg. Stud. Mar. Sci. 48, 102052. https://doi.org/10.1016/j.rsma.2021.102052 (2021).
Santos, L. V. et al. Trophic ecology and ecomorphology of the shorthead drum Larimus breviceps (Acanthuriformes: Sciaenidae), from the northeastern Brazil. Thalassas 38, 1–11 (2022).
Santos, L. et al. Stock assessment of Larimus breviceps, a bycatch species exploited by artisanal beach seining in Northeast Brazil. Fish. Manag. Ecol. 31, e12647. https://doi.org/10.1111/fme.12647 (2024).
Ribeiro, A. O. et al. DNA barcodes identify marine fishes of São Paulo State, Brazil. Mol. Ecol. Resour. 12, 1012–1020 (2012).
Santos, S., Gomes, M. F., Ferreira, A. R. S., Sampaio, I. & Schneider, H. Molecular phylogeny of the western South Atlantic Sciaenidae based on mitochondrial and nuclear data. Mol. Phylogenet. Evol. 66, 423–428 (2013).
Santos, S., Hrbek, T., Farias, I. P., Schneider, H. & Sampaio, I. Population genetic structuring of the king weakfish, Macrodon ancylodon (Sciaenidae), in Atlantic coastal waters of South America: deep genetic divergence without morphological change. Mol. Ecol. 15, 4361–4373 (2006).
Cadrin, S. X., Maunder, M. N. & Punt, A. E. Spatial structure: theory, estimation and application in stock assessment models. Fish. Res. 229, 105608. https://doi.org/10.1016/j.fishres.2020.105608 (2020).
Aminan, A. W., Kit, L. L. W., Hui, C. H. & Sulaiman, B. Morphometric analysis and genetic relationship of Rasbora spp. in Sarawak, Malaysia. Trop. Life Sci. Res. 31, 33–49 (2020).
Silva, T. F. et al. Phylogeny of the subfamily Stelliferinae suggests speciation in Ophioscion Gill, 1863 (Sciaenidae: Perciformes) in the western South Atlantic. Mol. Phylogenet. Evol. 125, 51–61 (2018).
Martins, N. T., Macagnan, L. B., Cassano, V. & Gurgel, C. F. D. Brazilian marine phylogeography: a literature synthesis and analysis of barriers. Mol. Ecol. 31, 5423–5439 (2022).
Da Silva, T. F. et al. Species delimitation by DNA barcoding reveals undescribed diversity in Stelliferinae (Sciaenidae). PLOS One. 18, e0296335. https://doi.org/10.1371/journal.pone.0296335 (2023).
Sales, J. B. L. et al. The vicariant role of Caribbean formation in driving speciation in American loliginid squids: the case of Doryteuthis pealeii (Lesueur 1821). Mar. Biol. 171, 82. https://doi.org/10.1007/s00227-024-04391-9 (2024).
Beheregaray, L. B. & Levy, J. A. Population genetics of the silverside Odontesthes argentinensis (Teleostei, Atherinopsidae): evidence for speciation in an estuary of southern Brazil. Copeia 2, 441–447 (2000).
Beheregaray, L. B. & Sunnucks, P. Fine-scale genetic structure, estuarine colonization and incipient speciation in the marine silverside fish Odontesthes argentinensis. Mol. Ecol. 10, 2849–2866 (2001).
García, G. et al. Promiscuous speciation with gene flow in silverside fish genus Odontesthes (Atheriniformes, Atherinopsidae) from south western Atlantic ocean basins. PLOS One. 9, e104659. https://doi.org/10.1371/journal.pone.0104659 (2014).
Schmid, C., Schäfer, H., Podestá, G. & Zenk, W. The Vitória eddy and its relation to the Brazil Current. J. Phys. Oceanogr. 25, 2532–2546 (1995).
Castro, B. M. & de Miranda, L. B. Physical oceanography of the western Atlantic continental shelf located between 4º N and 34º S coastal segment (4, W) in The Sea VII (eds Robinson, A. R. & Brink, K. H.) 209–251 (John Willey and Sons Inc., New York, 1998).
Casey, K. S. & Cornillon, P. A comparison of satellite and in situ-based sea surface temperature climatologies. J. Clim. 12, 1848–1863 (1999).
Rabassa, J., Coronato, A. M. & Salemme, M. Chronology of the late Cenozoic Patagonian glaciations and their correlation with biostratigraphic units of the Pampean region (Argentina). J. South. Amer. Earth Sci. 20, 81–103 (2005).
Rabassa, J., Coronato, A. & Martínez, O. Late Cenozoic glaciations in Patagonia and Tierra del Fuego: an updated review. Biol. J. Linn. Soc. Lond. 103, 316–335 (2011).
Tosetto, G., Bertrand, E., Neumann-Leitão, S. & Nogueira Júnior, M. The Amazon River plume, a barrier to animal dispersal in the Western Tropical Atlantic. Sci. Rep. 12, 537. https://doi.org/10.1038/s41598-021-04165-z (2022).
Rocha, L. A., Lindeman, K. C., Rocha, C. R. & Lessios, H. A. Historical biogeography and speciation in the reef fish genus Haemulon (Teleostei: Haemulidae). Mol. Phylogenet. Evol. 48, 918–928 (2008b).
Duque-Caro, H. Neogene stratigraphy, paleoceanography and paleobiogeography in northwest South America and the evolution of the Panama Seaway. Palaeogeogr. Palaeoclimatol. Palaeoecol. 77, 203–234 (1990).
Steph, S. et al. Changes in Caribbean surface hydrography during the Pliocene shoaling of the Central American Seaway. Paleoceanography 21, PA4221. https://doi.org/10.1029/2004PA001092 (2006).
O’Dea, A. et al. Formation of the Isthmus of Panama. Sci. Adv. 2, e1600883. https://doi.org/10.1126/sciadv.1600883 (2016).
Lessios, H. A. The great American schism: divergence of marine organisms after the rise of the Central American Isthmus. Annu. Rev. Ecol. Evol. Syst. 39, 63–91 (2008).
Heinrich, S. & Zonneveld, K. A. F. Influence of the amazon river development and constriction of the central american seaway on middle/late miocene oceanic conditions at the Ceara Rise. Palaeogeogr. Palaeoclimatol. Palaeoecol. 386, 599–606 (2013).
Lima, F. D., Strugnell, J. M., Leite, T. S. & Lima, S. M. Q. A biogeographic framework of octopod species diversification: the role of the Isthmus of Panama. PeerJ 8, e8691. https://doi.org/10.7717/peerj.8691 (2020).
Duarte, M. R. et al. Genetic and morphometric evidence that the jacks (Carangidae) fished off the coast of Rio de Janeiro (Brazil) comprise four different species. Biochem. Syst. Ecol. 71, 78–86 (2017).
Monteiro, L. R. & Reis, S. F. Princípios de Morfometria Geométrica 1st edn (Holos, São Paulo, 1991).
Andrade-Santos, J., Rosa, R. S. & Ramos, T. P. A. Spotting mistakes: reappraisal of spotted drum Stellifer punctatissimus (Meek & Hildebrand, 1925) (Teleostei: Sciaenidae) reveals species misidentification trends and suggests latitudinal sexual dimorphism. Zool. 165, 126180. https://doi.org/10.1016/j.zool.2024.126180 (2024).
Currens, K. P., Sharpe, C. S., Hjort, R., Schreck, C. B. & Li, H. W. Effects of different feeding regimes on the morphometrics of chinook salmon (Oncorhynychus tshawytscha) and rainbow trout (O. mykiss). Copeia 3, 689–695 (1989).
Schwab, J. A. et al. Evolutionary ecomorphology for thetwenty-first century: examples from mammalian carnivores. Proc. R. Soc. B 290, 20231400. https://doi.org/10.1098/rspb.2023.1400 (2023).
de França, V. F. C. & Severi, W. Ecomorphological relationships and dissimilarities of Engraulidae juveniles in a Brazilian tropical surf-zone environment. Thalassas 40, 1179–1191 (2024).
Grant, W. S. & Bowen, B. W. Shallow population histories in deep evolutionary lineages of marine fishes: insights from sardines and anchovies and lessons for conservation. J. Hered. 89, 415–426 (1998).
Fernández-Iriarte, P. J., Pía Alonso, M., Sabadin, D. E., Arauz, P. A. & Iudica, C. M. Phylogeography of weakfish Cynoscion guatucupa (Perciformes: Sciaenidae) from the southwestern Atlantic. Sci. Mar. 75, 701–706 (2011).
Ludt, W. B. & Rocha, L. A. Shifting seas: the impacts of Pleistocene sea-level fluctuations on the evolution of tropical marine taxa. J. Biogeogr. 42, 25–38 (2015).
Silva, D. et al. Genetic differentiation in populations of lane snapper (Lutjanus synagris – Lutjanidae) from Western Atlantic as revealed by multilocus analysis. Fish. Res. 198, 138–149 (2018).
Da Silva, R. et al. High levels of genetic connectivity among populations of yellowtail snapper, Ocyurus chrysurus (Lutjanidae-Perciformes), in the western South Atlantic revealed through multilocus analysis. PLOS One. 10, e0122173. https://doi.org/10.1371/journal.pone.0122173 (2015).
Da Silva, R., Sampaio, I., Schneider, H. & Gomes, G. Lack of spatial subdivision for the snapper Lutjanus purpureus (Lutjanidae-Perciformes) from southwest Atlantic based on multi-locus analyses. PLOS One. 11, e0161617. https://doi.org/10.1371/journal.pone.0161617 (2016).
Veneza, I. et al. Genetic connectivity and population expansion inferred from multilocus analysis in Lutjanus alexandrei (Lutjanidae–Perciformes), an endemic snapper from Northeastern Brazilian coast. PeerJ 11, e15973. https://doi.org/10.7717/peerj.15973 (2023).
Passarone, R. et al. Ecological and conservation aspects of bycatch fishes: an evaluation of shrimp fisheries impacts in Northeastern Brazil. Braz. J. Oceanogr. 67, e19291. https://doi.org/10.1590/S1679-87592019029106713 (2019).
Fu, Y. X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147, 915–925 (1997).
Nielsen, R. Molecular signatures of natural selection. Annu. Rev. Genet. 39, 197–218 (2005).
Fahey, A. L., Ricklefs, R. E. & Dewoody, J. A. DNA-based approaches for evaluating historical demography in terrestrial vertebrates. Biol. J. Linn. Soc. 112, 367–386 (2014).
Cracraft, J. Species concepts and speciation analysis in Current Ornithology. Vol. 1, 159–187 (ed. Johnston, R. F.) (Plenum Press, New York, 1983).
Schubart, C. D. Mitochondrial DNA and decapod phylogenies: the importance of pseudogenes and primer optimization in Decapod Crustacean Phylogenetics (eds. Martin, J. W., Crandall, K. A., Folder, D. F.) 47–65 (CRC Press, 2009).
Schubart, C. D. & Huber, M. G. J. Genetic comparisons of German populations of the stone crayfish, Austropotamobius torrentium (Crustacea: Astacidae). Bull. Fr. Peche Piscic. 380–381, 1019–1028 (2006).
Santa Brígida, E. L. et al. Population analysis of Scomberomorus cavalla (Cuvier, 1829) (Perciformes, Scombridae) from the Northern and Northeastern coast of Brazil. Braz. J. Biol. 67, 919–924 (2007).
Gomes, G. et al. Can Lutjanus purpureus (south red snapper) be legally considered a red snapper (Lutjanus campechanus)? Genet. Mol. Biol. 31, 372–376 (2008).
Santos, S., Schneider, H. & Sampaio, I. Genetic differentiation of Macrodon ancylodon (Sciaenidae, Perciformes) populations in Atlantic coastal waters of South America as revealed by mtDNA analysis. Genet. Mol. Biol. 26, 151–161 (2003).
Smith, M. F. & Patton, J. L. The diversification of South American murid rodents: evidence from mitochondrial DNA sequence data for the Akodontine tribe. Biol. J. Linn. Soc. Lond. 50, 149–177 (1993).
Streelman, J. T. & Karl, S. A. Reconstructing labroid evolution with single-copy nuclear DNA. Proc. R. Soc. B. 264, 1011–1020 (1997).
Rodrigues-Filho, L. F. S. Identificação e filogeografia de tainhas do gênero Mugil e avaliação do estado taxonômico das espécies Mugil liza Valenciennes, 1836 e Mugil platanus Günther, 1880. (Universidade Federal do Pará. Doctoral Thesis, 2011).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 (1994).
Hall, T. A. BioEdit: a user friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids. Symp. Ser. 41, 95–98 (1999).
Lo, P-C. et al. A multi-gene dataset reveals a tropical New World origin and Early Miocene diversification of croakers (Perciformes: Sciaenidae). Mol. Phylogenet. Evol. 88, 132–143 (2015).
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T. & Calcott, B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773 (2017).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Douglas, J., Jiménez-Silva, C. L. & Bouckaert, R. StarBeast3: adaptive parallelized bayesian inference under the multispecies coalescent. Syst. Biol. 71, 901–916 (2022).
Bouckaert, R. et al. BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 15, e1006650. https://doi.org/10.1371/journal.pcbi.1006650 (2019).
Drummond, A. J., Ho, S. Y. W., Phillips, M. J. & Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 4, 699–710. https://doi.org/10.1371/journal.pbio.0040088 (2006).
Nolf, D. & Aguilera, O. Fish otoliths from the Cantaure Formation (Early Miocene of Venezuela). Bull. de L’institut Royal des. Sci. Nat. de Belgique Sci. de la. Terre. 68, 237–262 (1998).
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Stephens, M., Smith, N. J. & Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 68, 978–989 (2001).
Librado, P. & Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinform. 25, 1451–1452 (2009).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Salzburger, W., Ewing, G. B. & Von Haeseler, A. The performance of phylogenetic algorithms in estimating haplotype genealogies with migration. Mol. Ecol. 20, 1952–1963 (2011).
Bessa-Silva, A. Fasta2Structure: a user-friendly tool for converting multiple aligned FASTA files to STRUCTURE format. BMC Bioinform. 25, 73. https://doi.org/10.1186/s12859-024-05697-7 (2024).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Earl, D. A. & Vonholdt, B. M. Structure Harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2012).
Hey, J. Isolation with migration models for more than two populations. Mol. Biol. Evol. 27, 905–920 (2010).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
Heled, J. & Drummond, A. J. Bayesian inference of population size history from multiple loci. BMC Ecol. Evol. 8, 289. https://doi.org/10.1186/1471-2148-8-289 (2008).
Donaldson, K. A. & Wilson, R. R. Jr. Amphi-panamic geminates of snook (Percoidei: Centropomidae) provide a calibration of the divergence rate in the mitochondrial DNA control region of fishes. Mol. Phylogenet. Evol. 13, 208–213 (1999).
Sturmbauer, C., Baric, S., Salzburger, W., Rüber, L. & Verheyen, E. Lake level fluctuations synchronize genetic divergences of cichlid fishes in African lakes. Mol. Biol. Evol. 18, 144–154 (2001).
Heled, J. Extended Bayesian skylineplot tutorial http://beast.bio.ed.ac.uk/Tutorials (2010).
Parsons, K. J., Robinson, B. W. & Hrbek, T. Getting into shape: an empirical comparison of traditional truss-based morphometric methods with a newer geometric method applied to New World cichlids. Environ. Biol. Fishes. 67, 417–431 (2003).
Clarke, K. R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 18, 117–143 (1993).
Legendre, P. & Legendre, L. Numerical Ecology 2nd edn. (Elsevier, Amsterdam, 1998).
Clarke, K. R. & Warwick, R. M. Similarity-based testing for community pattern: the two-way layout with no replication. Mar. Biol. 118, 167–176 (1994).
Hammer, Ø., Harper, D. A. T. & Ryan, P. D. PAST: paleontological statistics software package for education and data analysis. Palaeont. Electr. 4, 1–9 (2001).
Acknowledgements
We would like to thank the Brazilian Federal Agency for Higher Graduate Education (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – CAPES) for the concession of a graduate (masters) scholarship (Finance Code 001) to SA. We thank Alfredo Carvalho Filho, Arturo Angulo, João Bráullio de Luna Sales, Rosa Rodrigues, Yrlene Ferreira, and Wemerson Silva for their help with the collection of samples. We thank to Jeferson Carneiro for his help with some of the data analyzes in this study.
Author information
Authors and Affiliations
Contributions
The study conception and design were performed by SA and SS. Material preparation, data collection and analysis were performed by SA, BB, IS, TFS, MV, ACCS, ARB and SS. The first draft of the manuscript was written by SA and SS, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The approval by the ethics committee was not requested because the fish were purchased from artisanal fishermen and were already dead at the time of collection.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Alencar, S., Bentes, B., Sampaio, I. et al. Deep genetic divergences and few morphological changes support the cryptic speciation in Larimus breviceps (Sciaenidae, Acanthuriformes) from the western South Atlantic. Sci Rep 14, 31541 (2024). https://doi.org/10.1038/s41598-024-83196-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-83196-8
