
Abstract
Electron-nuclear coupling plays a crucial role in strong laser induced molecular dissociation dynamics. The interplay between electronic and nuclear degrees of freedom determines the pathways and outcomes of molecular fragmentation. However, a full quantum mechanical treatment of electron-nuclear dynamics is computationally intensive. In this work, we have developed a Strong Laser Induced non-adiabatic Multi-Ionic-Multi-Electric States (SLIMIMES) approach, which contains the electron-laser and electron-nuclear couplings. We validate our model using a showcase example: water dissociation under strong infrared (IR) laser pulses. Our investigation reveals the predominant role of a non-vertical dissociation pathway in the photo-ionization dissociation (PID) process of \(\mathrm {D_{2}O^{2+}}\). This pathway originates from neutral \(\mathrm {D_{2}O}\), which undergoes vertical multi-photon-single-ionization, reaching the intermediate dissociation states of \(\mathrm {D_{I} + OD_{II}^{+} (2^{3}\Sigma )}\) within \(\mathrm {D_{2}O^{+}}\). Subsequently, \(\mathrm {OD_{II}^{+} (2^{3}\Sigma )}\) dissociates into \(\mathrm {O^{+} + D_{II}}\), with both \(\mathrm {D_{I}}\) and \(\mathrm {D_{II}}\) fragments potentially ionizing an electron during interaction with the IR laser. This sequential PID pathway significantly contributes to the dissociation yields of water dication. Our calculations are consistent with recent experimental data, which focus on measuring the branching ratio of water dication dissociation. We aim for our model to provide a deeper understanding and a fresh perspective on the coupling between electron and nuclear dynamics induced by a strong IR laser field.
Similar content being viewed by others

Introduction
The measurement and investigation of photon-induced electron-nuclei coupling dynamics, occurring on the femtosecond (fs) time scale, have long been a target of various time-resolved spectroscopies1,2,3. This coupling effect plays a crucial role in many strong laser-induced physics and chemistry processes, such as Photon-Induced Ionization Dissociation (PID)4,5,6, especially in PID measurement schemes that use femtosecond (fs) strong infrared (IR) laser pulses7,8,9,10,11,12,13,14,15,16,17.
Numerical simulations of molecular dynamics under strong laser fields are essential for understanding ultrafast processes where intense fields drive complex electron-nuclear interactions. In these simulations, approaches like time-dependent density functional theory (TDDFT) coupled with molecular dynamics (MD) or Ehrenfest dynamics are often used to capture the intricate coupling between electronic and nuclear motion. The laser field excites electrons to high energy levels, inducing rapid electronic oscillations that interact with nuclei, affecting bond lengths, angles, and overall molecular geometry. This electron-nuclear coupling can lead to phenomena like bond breaking, ionization, and charge migration on extremely short timescales (femtoseconds to attoseconds). By incorporating both electron dynamics and nuclear motion, simulations can provide a detailed view of energy redistribution and molecular transformations, making them valuable tools for studying strong-field effects in molecules. So far, X-ray time-resolved spectroscopy-based PID measurement techniques such as pump-probe strategies4, transient absorption5, and time-resolved X-ray scattering6 have been employed since these techniques can achieve high temporal resolution signals with probe laser durations as short as a few hundred attoseconds6. On the other hand, in addition to X-ray time-resolved spectroscopy techniques, the use of femtosecond (fs) strong infrared (IR) laser pulses has emerged as a complementary method for imaging structural dynamics since IR pulses are easily generated and do not have limitations on beam time7,8,9,10,11,12. However, fully understanding the detailed insights of PID dynamics induced by IR lasers in the molecular frame remains a challenge, as this process involves both electronic and nuclear motion. Solving the full electron and nuclei time-dependent Schrödinger equation (TDSE) is numerically unachievable so far13,14,15,16,17. This challenge is especially prominent for hydrogen bond dissociation, which can occur in as little as 8 to 10 fs16,17. Within such a small time scale, the laser pulse does not end after dissociation, and the electrons of the fragments still have the possibility to further ionize to higher ionic states. Tunneling ionization and laser coupling between different electron states should not be ignored in this PID process. Experimental and theoretical investigations into this process have already been conducted with small diatomic molecules such as \(\mathrm {H_{2}}\) and \(\textrm{HeH}\)18,19.
In this study, our aim is to advance our understanding by investigating a test case involving \(\mathrm {D_{2}O}\) as the target molecule, revealing the significant role played by ionization and laser coupling in the PID processes of complex molecular systems. Water molecules, along with their cations and dications, are ubiquitous in various natural environments exposed to ionizing radiation, energetic electrons, and other ions, including recent exposure to intense infrared (IR) lasers20,21,22,23. Advancements in vector momentum-resolved coincidence techniques such as velocity map imaging (VMI)1 and cold target recoil ion momentum spectroscopy (COLTRIMS)6 enable the meas urement of kinetic energy release (KER) and branching ratios (BR) of ionic fragments. For instance, Zhao et al.7 observed four dissociation channels of \(\mathrm {D_{2}O^{2+}}\) in their experiment, while Chen et al. only detected three of these channels24. The laser parameters and corresponding BRs are provided in Tables 1 and 2. Despite the presence of the bond rearrangement channel (CIV), it is evident that the dominant dissociation channels are the two-body breakup (CI) in both experiments. However, the BRs of the three-body breakup channels (CII, CIII) exhibit a strong dependence on laser parameters.
In this work, we introduce our SLIMIMES model to elucidate the dependence of BRs on laser parameters observed in these experiments, highlighting the importance of laser coupling between the electron states from different charged molecular states in a molecular dynamics process. Atomic units (a.u.) are used throughout this work, unless mentioned otherwise.
Theory and results
DFT and TDDFT
We adopt time-dependent density functional theory (TDDFT) based Octopus code package25,26,27 to perform most the calculations. The ground states at each molecular geometrical configuration \(\left\{ \textbf{R} \right\}\) can be found by Kohn-Sham equations (KSE):
where \({{V}_{ext}}\left( \textbf{R},\textbf{r} \right)\), \({{V}_{H}}\left( \textbf{R},\textbf{r} \right)\) and \({{V}_{XC}}\left( n;\textbf{R},\textbf{r} \right)\) are the external potential acting on the interacting system, the Hartree potential and the exchange-correlation potential, respectively (details about these potentials can be found in Refs.25,26,27).
\({{\varphi }_{j\sigma }}\) and \({{\varepsilon }_{j\sigma }}\) are the occupied Kohn-Sham (KS) orbitals and corresponding eigenvalues with the spin coordinate is embodied in \(\sigma\) respectively, then the molecular total wavefunction in Octopus code can be expressed as \(\Phi _{j} =\left| \begin{matrix} {{\left| {{\varphi }_{\uparrow 1}} \right| }^{2}} & {{\left| {{\varphi }_{\uparrow 2}} \right| }^{2}} & {{\left| {{\varphi }_{\uparrow 3}} \right| }^{2}} & ...... \\ {{\left| {{\varphi }_{\downarrow 1}} \right| }^{2}} & {{\left| {{\varphi }_{\downarrow 2}} \right| }^{2}} & {{\left| {{\varphi }_{\downarrow 3}} \right| }^{2}} & ...... \\ \end{matrix} \right|\), where the square brackets \(\left| \right|\) DO NOT denote a determinant but indicate the occupation number \({{\left| {{\varphi }_{\sigma j}} \right| }^{2}}\) for different orbitals (detail about this expression method can be found in Octopus user guide27).
The excited states at each molecular geometrical configuration are optimized with Casida module27 from the calculated ground states25,26,27.
The electron-nuclei coupling dynamics under the interaction of strong laser fields to be solved by time dependent KSE:
where \({{V}_{ls}}\left( \textbf{r},t \right) =f\left( t \right) \cos \left( \omega t \right) \hat{\varepsilon }\cdot \textbf{r}\) with the temporal profile f(t), the carrier frequency \(\omega\), and the polarization direction \({\hat{\varepsilon }}\), respectively. In this work, f(t) is defined as \(f\left( t \right) ={{e}_{0}}\exp \left( -{{\left( t-{{t}_{0}} \right) }^{2}}/\left( 2{{\tau }^{2}} \right) \right)\), here \(e_0\) is the electric field amplitude, \(\tau\) is the duration and \(t_0=0\) in our simulation. \({{M}_{\alpha }}\), \({{\textbf{R}}_{\alpha }}\) and \({{\textbf{F}}_{\alpha }}\) is the mass, coordinates, and force27 on nucleus \(\alpha\). If the nuclei are fixed, then \(\frac{{{d}^{2}}{{\textbf{R}}_{\alpha }}}{d{{t}^{2}}}=0\).
The full information of strong laser induced state transitions, charge migrations, dissociation, and ionizations are all included.
SLIMIMES model
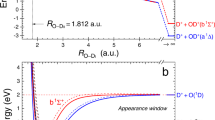
The foundational concept of the SLIMIMES model is illustrated in Fig. 1. In a strong field, the double ionization of \(D_{2}O\) can be achieved through two pathways: Pathway I (PI): Neutral \({{D}_{2}}O\) is initially excited by the laser-electron interaction to a non-dissociative state of \({{D}_{2}}O^{+}\) (such as the ground state of \({{D}_{2}}O^{+}\), denoted as \(1^{2}B{1}\)). Subsequently, \({{D}_{2}}O^{+}\) undergoes further vertical excitation to certain \({{D}_{2}}O^{2+}\) states while maintaining the molecular geometry within the Franck-Condon principle. Finally, dissociation of \({{D}_{2}}O^{2+}\) occurs, as free water dications have never been observed as non-dissociative molecules in vacuum. Pathway II (PII): \({{D}_{2}}O\) is directly vertically excited to an intermediate dissociation state of \({{D}_{2}}O^{+}\). The fragments separate rapidly within the duration of the laser pulse, with each fragment having the potential to be ionized during the photon-ionization-dissociation (PID) process16,17.

Scheme for dissociation pathway I (a) and II (b).
The main Approximations of the SLIMIMES model can be summarized as follows:
-
(1)
Ionization and excitation occur before and at the largest peak of the electric field, \(t<0\). Before the largest laser peak, the nuclei are considered fixed;
-
(2)
Ionization to triple-ionized and higher charged ionic states is disregarded;
-
(3)
The dissociation yield Y of a certain dissociation channel is approximated as the direct sum of all the electron state populations \(C_i\) that contribute to this specific channel: \(Y=\sum \limits _{i}{{{C}_{i}}}\), that is, the quantum interferences between these states are ignored;
In the SLIMIMES model, three main calculation steps are involved: Firstly, we need to get the populations of each electron states of \(D_{2}O\), \({{D}_{2}}O^{+}\) and \({{D}_{2}}O^{2+}\) at \(t=0\) when the electric field reaches its largest peak and this is the key step in SLIMIMES model, just keep in mind that we assume that the molecule is triggered by the laser pulse from the ground state of a neutral water molecule, with the molecular geometry unchanged before \(t=0\), according to our Approximation 1. Usually, as \(D_{2}O\), \({{D}_{2}}O^{+}\) and \({{D}_{2}}O^{2+}\) share different numbers of electrons, directly projecting the specific orbital \({{\varphi }_{j\sigma }}\left( \textbf{R},\textbf{r},t \right)\) on the total wavefunction \(\Psi \left( t \right)\) is numerically difficult to achieve, such calculations can only be roughly estimated by MO-ADK formula28. This is a major challenge in studying the impact of electron dynamics during strong laser-induced dissociation-ionization coupling processes.
In SLIMIMES model, we resolve this difficulty by expanding the total time dependent wavefunction in terms of all electron states of \(D_{2}O\), \({{D}_{2}}O^{+}\) and \({{D}_{2}}O^{2+}\): \(\Psi \left( t \right) =\sum \limits _{j}{{{a}_{j}}\left( t \right) }{{\Phi }_{j}}\left( \textbf{r} \right)\), where \(a_{j}\) and \({{\Phi }_{j}}\) represent the time-dependent amplitude and time-independent wavefunction of state j (j state can be ground or excited electron state of neutral, cation, or diaction molecule). In Octopus code, we have
where \({{\nu }_{\sigma i}}\left( t \right) ={{\left| \left\langle {{\varphi }_{\sigma i}}\left( r,t \right) \right| \left. {{\varphi }_{\sigma i}}\left( r \right) \right\rangle \right| }^{2}}\) is the overlap between the time dependent KS orbital and itself initial orbital, the valve of \({{\nu }_{i\sigma }}\left( t \right)\) corresponding to how many \({{\varphi }_{\sigma i}}\left( r,t \right)\) still stays at its initial position. All KS orbitals overlap \({{\nu }_{\sigma i}}=1\) at the beginning.
In this step, we start the time prorogation TDDFT calculation when the laser begin interacting with water, and ends at \(t=0\). Then we have the linear equation of \(a_{j}\) at \(t=0\) (or at any time \(t<0\)):
By solving this equation, we obtain the time-dependent coefficient distribution \(a_{j}\) of each possible state to be involved, such as diaction states and the intermediate dissociation states of \({{D}_{2}}{{O}^{+}}\). \(a_j\) gives the information of the j state population at \(t=0\). The electron ionization, excitation as well as any other electron dynamics occurs between different ionic charged electron states are all included in \(a_j\).
In the second step of the SLIMIMES model, our focus shifts to the PII channel, in which the fragments of the \(D_2O^{+}\) and \(D_2O^{2+}\) dissociation states undergoes a possible further ionization. The main purpose of the second step is to find out this further ionization possibilities.
In this step, we perform the TDDFT calculations with the initial electron states setting to be a specific \(D_2O^{+}\) or \(D_2O^{2+}\) dissociation state with nuclei are moving, each calculation with different initial state are performed independently, all the time propagations start at \(t=0\) and terminated a little further than the laser ends time. The ionization possibility of a fragment X can be got by the formula: \(P_{X}=1-{{\rho }{X}}\left( t \right)\), where \({{\rho }{X}}\left( t \right)\) represents the electron charge number of fragment X, calculated utilizing the method proposed in Refs.29,30.
Then the dissociation yield \(Y_j\) of \((XY)^{+}=X^{+}+Y\) channel from the dissociation state j after two SLIMIMES steps is: \({{Y}_{j}}={{a}_{j}}\left[ \left( 1-{{P}_{{{X}^{+}}}} \right) \left( 1-{{P}_{Y}} \right) \right]\), here \(a_j\) is the population of \((XY)^{+}\) at \(t=0\), \({{P}_{{{X}^{+}}}}\) is the ionization possibility of fragment \(X^{+}\) and \({{P}_{Y}}\) is the ionization possibility of fragment Y. In this step, we ignore the ionization to triple-ionized and higher charged ionic states.
The final SLIMIMES step is to collect all possible \(Y_j\) to calculate the total dissociation yield Y of a certain dissociation channel: \(Y=\sum \limits _{j}{{{Y}_{j}}}\) with ignoring the interferences between different dissociation states.
A note is addressed here, although we present water as the show case in Fig. 1 and next Section, SLIMIMES model can easily be expanded to investigate more complex molecular systems, with a higher but still acceptable computational cost (Fig. 2).

(a) All PECs of neutral (one ground state), cation (first 10 states) and diaction (6 states) of D\(_{2}O\) that are involved in SLIMIMES simulation. (b) Four PECs of D\(_{2}\)O\(^{+}\) intermediate dissociation states (13), (15), (16) and (18). (c) PECs of the fragment OD\(^{+}\) from intermediate dissociation state: green curve \({{2}^{3}}\Sigma\) is the fragment from (13) and finally dissociating to \(O{{D}^{+}}\rightarrow D+{{O}^{+}}\). Blue curve \(a^{1}\Delta\) is the fragment from (15), (16) and (18). Meanwhile, the cyan curve \({{X}^{3}}\Sigma\) and red curve are the ground state of \(OD^{+}\) and \(OD^{2+}\), respectively.
Figure 2a presents a series of selectively calculated Potential Energy Curve (PEC) of water from the Octopus code with scanning only one OD bond, the shown states are anticipated to play crucial roles in the PID process (carefully examined for convergence). Detailed properties of these potential curves are provided in Table 3. Each curve is labeled as (qn), where q signifies the charge of the molecule, and n denotes the order of excited states, with \(n=0\) indicating the ground state. The equilibrium distance for OD is 1.8, and the vertical excitation energies from the ground state are indicated. Each orbital \({{\varphi }_{j\sigma }}\) is expressed as \(\varphi \left( {{x}_{i}},{{y}_{i}},{{z}_{i}} \right)\) in terms of grid points in the 3D space. The range for each coordinate is \({{z}_{i}}\in \left[ -200,+200 \right]\), \({{x}_{i}}\in \left[ -200,+200 \right]\) and \({{y}_{i}}\in \left[ -200,+200 \right]\) with \(dx=dy=dz=0.2\). In our simulations, the adiabatic local-density approximation (ALDA) with the parametrization of Perdew and Zunger are used31.
Notably, the PEC from (13) to (19) cluster near 30 eV for \({{D}_{2}}O^{+}\), and from (20) to (24) also exhibit proximity. These curves are pivotal for the double ionization of \({{D}_{2}}O\). In \({{D}_{2}}O^{+}\), the first three levels (10), (11), and (12) represent pure levels with one electron removed from \({{D}_{2}}O\). For the higher levels from (13) to (19), configuration mixing is significant, as shown in Table 3. For the curves (20) to (25), only removal of two electrons from the HOMO, HOMO-1, and HOMO-2 are involved. Table 3 also depicts the vertical excitation energy of each curve at the equilibrium distance of \(D_{2}O\), the symmetry in \(C_{2v}\), and the dissociation limit. The five potential curves in \({{D}_{2}}O^{2+}\) do not support long-lived bound states; the molecule would directly dissociate via two-body breakup into \({{D}^{+}}/O{{D}^{+}}\), or by three-body breakup into \({{D}^{+}}/{{D}^{+}}/O\). Although (25) can dissociate into \({{D}^{+}}/D/{{O}^{+}}\), its vertical excitation energy is much higher, making this channel open only at higher photon energy.
Based on Table 3, it becomes evident that direct removal of two electrons from the \({{D}_{2}}O\) or \({{H}_{2}}O\) molecules from the outer shells will result in two-body breakup into \(D^{+}/OD^{+}\) and three-body breakup into \({{D}^{+}}/{{D}^{+}}/O\). This is consistent with experimental results from Reedy et al.21. using a synchrotron light source at 57 eV. Furthermore, we discern that (13), (15), (16), and (18) are the crucial intermediate dissociation states. (13) would lead to three-body breakup: \({{D}_{2}}{{O}^{+}}\rightarrow D+D+{{O}^{+}}\), while (15), (16), and (18) would result in two-body breakup: \({{D}_{2}}{{O}^{+}}\rightarrow D+O{{D}^{+}}\), which are shown in Fig. 3b and c.
We can summarize the dissociation yield CI, CII and CIII of \(D_{2}O^{2+}\) from above analysis as:
where \(Y_{CI}\), \(Y_{CII}\) and \(Y_{CIII}\) corresponding to the dissociation yield of \({{D}^{+}}/O{{D}^{+}}\), \({{D}^{+}}/D/{{O}^{+}}\) and \({{D}^{+}}/{{D}^{+}}/O\), respectively. \(P_{Dqn}\) and \(P_{Oqn}\) are the probability of ionizing the neutral D or neutral O from the state [qn] from second step of SLIMIMES. \(C_{qn}\) is the population of state [qn] which comes from calculation of first step of SLIMIMES. It’s clear to identify that the \(C_{2n}\) terms are from PI pathway while \(C_{1n}\) terms are from the non vertical pathway PII.
In the TDDFT calculations, \(D_{2}O\) is planar on the xz-plane. The time-step is chosen to be 0.08, or about 2 attoseconds. Such small step size is need since typical electronic time scale is about 10-20 attoseconds. An absorbing potential is applied when each coordinate is -5.0 away from the boundary surface. The water geometry in first step of SLIMIMES is set to be the \(C_{2v}\) symmetry with both \(OD=1.8\) and \(\angle DOD\) is 104\(^{\circ }\).
To account for the effect of molecular nuclear vibrations on the final results, we used the Wigner phase space distribution to sample the initial conditions of the classical trajectories:
\(W\left( Q,P \right) =\frac{1}{{{\left( \pi \hbar \right) }^{3N-6}}}\underset{j=1}{\overset{3N-6}{\mathop {\Pi }}}\,\exp \left[ -\frac{{{\omega }_{j}}}{\hbar }Q_{j}^{2}-\frac{P_{j}^{2}}{\hbar \omega } \right]\)
Where \(Q_j\), \(P_j\), \(\omega _j\) are the normal-mode coordinates, momenta and frequency defined in terms of the eigenvectors and eigenvalues of the mass weighted Hessian. Detail of the Wigner phase space distribution can be found in Ref.20. The final results at specific polar and azimuthal angles \(Y\left( \theta ,\varphi \right)\) were then statistically averaged \(Y\left( \theta ,\varphi \right) =\frac{{{Y}_{k}}{{W}_{k}}}{\sum \limits _{k}{{{W}_{k}}\left( Q,P \right) }}\). Here \(Y_k\) and \(W_k\) corresponding the branching ration calculated from classical trajectory k which has an initial condition \(W_k\).
In the experimental setup, water molecules are randomly oriented, necessitating the averaging of the polar and azimuthal angles \(\theta\) and \(\varphi\) of the dissociation yield in our simulation, denoted as \({{{\bar{Y}}}_{i}}=\int _{-\pi }^{\pi }{\int _{0}^{2\pi }{{{Y}_{i}}\left( \theta ,\varphi \right) \sin \theta d\theta d\varphi }}\). Each angle is sampled 100 times, totaling 10,000 samples in all.
We intend to reproduce the experimental BR in Tables 1 and 2 and our simulation results are also presented in Tables 1 and 2. We observe good agreement between our model and the experimental data from both studies, except for the absence of the CIV channel in Zhao et al’s work7. An analytical explanation is provided for deeper understanding the influence of laser conditions on the experimental BR. According to the Eq. (5), \(\frac{Y2}{Y3}=\frac{{{C}_{25}}\left[ 1-{{P}_{D25}} \right] +{{C}_{13}}\cdot 2{{P}_{D13}}\cdot \left[ 1-{{P}_{D13}} \right] }{{{C}_{24}}\cdot \left[ 1-{{P}_{O24}} \right] +{{C}_{23}}\cdot \left[ 1-{{P}_{O23}} \right] }\approx \frac{{{C}_{13}}}{{{C}_{24}}}{{P}_{D13}}\), where \({{P}_{D13}}\) represents the ionization probability from the (13) state. If we neglect the contribution from the (25) state due to its low population and assume \(P_{O23}=P_{O24}=P_{D13}\), then \(\frac{Y2}{Y3}\) can be approximated as \(\frac{{{C}_{13}}}{{{C}_{24}}}{{P}_{D13}}\). As laser conditions change from the setup described in Ref.7 to that in Ref.24, \(P_{D13}\) decreases rapidly, resulting in a sharp decline in \(\frac{Y2}{Y3}\).
Similarly, considering the ratio \(\frac{Y2}{Y1}\), we have\(\frac{Y2}{Y1}=\frac{{{C}_{26}}\left[ 1-{{P}_{D26}} \right] +2{{C}_{13}}\cdot {{P}_{D13}}\cdot \left[ 1-{{P}_{D13}} \right] }{{{C}_{20}}+{{C}_{21}}+{{C}_{22}}+{{C}_{15}}{{P}_{D15}}+{{C}_{16}}{{P}_{D16}}+{{C}_{18}}{{P}_{D18}}}\approx \frac{2\delta \cdot {{P}_{D}}\cdot \left[ 1-{{P}_{D}} \right] }{3+3\delta \cdot {{P}_{D}}}\). Here, \(\delta =\frac{{{C}_{13}}}{{{C}_{20}}}\), and we ignore the contribution from the (25) state as before. In this equation, \({{C}_{13}}\cdot {{P}_{D}}\) corresponds to the probability of diaction ionization from the (13) state, comparable to \(C_{20}\). Hence, the term \(\delta \cdot {{P}_{D}}=\frac{{{C}_{13}}\cdot {{P}_{D}}}{{{C}_{20}}}\) can be treated as a constant. Consequently, we approximate \(\frac{Y2}{Y1}\) as \(\frac{2\delta \cdot {{P}_{D}}\cdot \left[ 1-{{P}_{D}} \right] }{3+3\delta \cdot {{P}_{D}}}\approx \frac{2C}{3+3C}\times \left[ 1-{{P}_{D}} \right]\). With increasing peak intensity and duration, \(\frac{Y2}{Y1}\) grows larger. This analytical model aligns well with the experimental data.
To discern the contributions from PI and PII under different laser conditions, we present the SLIMIMES simulation for other four distinct laser conditions, as outlined in Table 4. We can find that CIII solely arises from the PI pathway, while CII predominantly originates from the PII pathway, owing to the significantly higher vertical energy of the (25) state compared to others, rendering its contribution negligible. In scenarios of low intensity (e.g., \(4\times {{10}^{14}}, \text {W}/\text {cm}^2\)), the contributions from PI and PII to CI are comparable. This parity stems from the fact that at low intensities, the ionization of the D atom remains low, resulting in a relatively small PII component. However, as intensity and duration increase, \(P_{D}\) escalates, causing the PII pathway to predominate in CI. These simulations underscore the crucial role played by PII in the dissociation process, necessitating its inclusion for comprehensive modeling. This analysis illuminates the intricate interplay between different pathways under varying laser conditions, highlighting the necessity of considering both PI and PII to accurately capture the dynamics of the dissociation process.
To further extend the applicability of our model, we also calculated the KER of the dissociation dynamics of doubly ionized CH\(_3\)OH. The initial state was set as the ground state of the CH\(_3\)OH dication, and 1000 classical trajectories were sampled based on the Wigner distribution for the initial conditions. We collected the kinetic energy of the dissociation fragments at intervals of 1 eV. The histogram represents our model, while the dotted line represents the results from32. As shown, our results agree very well with those reported in the literature.

Comparison of KER from our model and from Ref.32.
Conclusion
In summary, we have introduced a coupled electron-nuclear dynamics model to elucidate the laser-dependence of dissociation Branching Ratios. Our analysis delineates two distinct dissociation pathways: a vertical transition pathway and a non-vertical transition pathway. Significantly, we underscore the pivotal role played by the non-vertical pathway in the PID process. The robustness of our approach is underscored by its alignment with experimental findings and numerical simulations. While our methodology has been exemplified in the context of the sequential fragmentation of heavy water following ionization by a strong IR laser, its applicability extends beyond this specific scenario. We anticipate that our methodology can be effectively applied to a diverse array of polyatomic molecules. By enhancing our capabilities for exploring molecular reaction dynamics, particularly those involving intermediate molecular dissociation states leading to sequential fragmentation, our approach opens new avenues for advancing our understanding of complex chemical processes. The identification of non-vertical transitions in molecular dissociation reactions underscores the significance of ionization and laser coupling in the interaction between strong lasers and molecular systems. This observation adds further depth to our comprehension of molecular dynamics under intense laser fields.
Data availability
The data of finding this work is available from the corresponding author upon reasonable request.
References
Yin, Z. et al. Femtosecond proton transfer in urea solutions probed by X-ray spectroscopy. Nature 619, 749. https://doi.org/10.1038/s41586-023-06182-6 (2023).
Severt, T. et al. Step-by-step state-selective tracking of fragmentation dynamics of water dications by momentum imaging. Nat. Commun. 13, 5146 (2022).
Ridente, E. et al. Femtosecond symmetry breaking and coherent relaxation of methane cations via x-ray spectroscopy. Science 380, 713 (2023).
Marangos, J. P. The measurement of ultrafast electronic and structural dynamics with X-rays. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 377, 20170481 (2019).
Geneaux, R., Marroux, H. J. B., Guggenmos, A., Neumark, D. M. & Leone, S. R. Transient absorption spectroscopy using high harmonic generation: A review of ultrafast X-ray dynamics in molecules and solids. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 377, 20170463 (2019).
Jahnke, T. et al. Inner-shell-ionization-induced femtosecond structural dynamics of water molecules imaged at an X-ray free-electron laser. Phys. Rev. X 11, 041044 (2021).
Zhao, S. et al. Strong-field-induced bond rearrangement in triatomic molecules. Phys. Rev. A 99, 053412 (2019).
Crane, S. W. et al. Nonadiabatic coupling effects in the 800 nm strong-field ionization-induced Coulomb explosion of methyl iodide revealed by multimass velocity map imaging and ab initio simulation studies. J. Phys. Chem. A 125, 9594 (2021).
Cheng, C. et al. Multiparticle cumulant mapping for coulomb explosion imaging. Phys. Rev. Lett. 130, 093001 (2023).
Allum, F. et al. Multi-particle three-dimensional covariance imaging: “Coincidence’’ insights into the many-body fragmentation of strong-field ionized D2O. J. Phys. Chem. Lett. 12, 8302 (2021).
De, S. et al. Following dynamic nuclear wave packets in N2, O2, and CO with few-cycle infrared pulses. Phys. Rev. A 84, 043410 (2011).
Li, X. et al. Coulomb explosion imaging of small polyatomic molecules with ultrashort x-ray pulses. Phys. Rev. Res. 4, 013029 (2022).
Pan, S. et al. Manipulating parallel and perpendicular multiphoton transitions in H2 molecules. Phys. Rev. Lett. 130, 143203 (2023).
Howard, A. et al. Strong-field ionization of water: Nuclear dynamics revealed by varying the pulse duration. Phys. Rev. A 103, 043120 (2021).
Sanderson, J. H. et al. Geometry modifications and alignment of H2O in an intense femtosecond laser pulse. Phys. Rev. A 59, R2567 (1999).
Chen, J. et al. Nonadiabatic molecular dynamics simulation of in a strong laser field. Chin. Phys. B 29, 113202 (2020).
Wolter, B. et al. Ultrafast electron diffraction imaging of bond breaking in di-ionized acetylene. Science 345, 308 (2016).
Qiang, J. et al. Laser-driven charge migration in a molecular electrophore. Phys. Rev. Lett. 132, 103201 (2024).
Ursrey, D., Anis, F. & Esry, B. D. Multiphoton dissociation of HeH+ below the He+(1s)+H(1s) threshold. Phys. Rev. A 85, 023429 (2012).
Streeter, Z. L. et al. Dissociation dynamics of the water dication following one-photon double ionization. I. Theory. Phys. Rev. A 98, 053429 (2018).
Reedy, D. et al. Dissociation dynamics of the water dication following one-photon double ionization. II. Experiment. Phys. Rev. A 98, 053430 (2018).
Inhester, L., Burmeister, C. F., Groenhof, G. & Grubmüller, H. Auger spectrum of a water molecule after single and double core ionization. J. Chem. Phys. 136, 144304 (2012).
Inhester, L., Hanasaki, K., Hao, Y., Son, S.-K. & Santra, R. X-Ray multiphoton ionization dynamics of a water molecule irradiated by an X-ray free-electron laser pulse. Phys. Rev. A 94, 023422 (2016).
C. Chen, titleKansas state university seminar and private discussion (2021).
Andrade, X. et al. Time-dependent density-functional theory in massively parallel computer architectures: the octopus project. J. Phys. Condens. Matter 24, 233202 (2012).
Castro, A. et al. octopus: a tool for the application of time-dependent density functional theory. Phys. Status Solidi B 243, 2465 (2006).
Marques, M. A. L., Castro, A., Bertsha, G. F. & Rubio, A. octopus: a first-principles tool for excited electron-ion dynamics. Comput. Phys. Commun. 151, 60 (2003).
Tong, X. M., Zhao, Z. X. & Lin, C. D. Theory of molecular tunneling ionization. Phys. Rev. A 66, 033402. https://doi.org/10.1103/PhysRevA.66.033402 (2002).
Troullier, N. & Martins, J. L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 43, 1993 (1991).
Hirshfeld, F. L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 44, 129 (1977).
Perdew, J. P. & Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 23, 5048 (1981).
Luzon, I., Livshits, E., Gope, K., Baer, R. & Strasser, D. Making sense of coulomb explosion imaging. J. Phys. Chem. Lett. 10, 1361. https://doi.org/10.1021/acs.jpclett.9b00576 (2019).
Acknowledgements
This project was supported by the National Key Research and Development Program of China (Grant Nos. 2022YFE134200, the Fundamental Research Funds for the Central Universities (Grants No.GK202207012), QCYRCXM-2022-241, the Natural Science Foundation of Jilin Province, China (Grant No. 20220101016JC), National Natural Science Foundations of China (NSFC) under Grants No. 12334011 and No. 61973317,62405054.
Author information
Authors and Affiliations
Contributions
All authors contributed substantially to this work. JW and SNG performed the simulation, AHL, LHH and XZ developed the initial theoretical ideas. LHH and XZ supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no cometing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, J., Gao, S.N., Liu, A. et al. Non vertical ionization-dissociation model for strong IR induced dissociation dynamics of \({{D}_{2}}O^{2+}\). Sci Rep 15, 117 (2025). https://doi.org/10.1038/s41598-024-83209-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-83209-6
