
Abstract
Acute myeloid leukemia (AML) represents a hematological malignancy that arises from the abnormal proliferation of progenitor cells or myeloid hematopoietic stem. The current standard treatments for AML include chemotherapy and hematopoietic stem cell transplantation. However, chemotherapy suffers from high toxicity and a shortage of hematopoietic stem cell donors, which significantly shortens patient survival. A new type of cell death, disulfidptosis, has shown potential in medicine. However, its specific biological mechanism of action in AML is currently unclear. This research developed a prognostic model of disulfidptosis-related long non-coding RNAs (DRLs) based on 132 AML patients with GDC TCGA Acute myeloid leukemia (LAML). In this model, eight DRLs: AL049835.1, EXOC3-AS1, AC009237.14, LINC00944, AP002761.4, LINC00926, AC010247.2, and AC099811.5 were included. Patients with high-risk AML evaluated based on the model had shorter survival, significant infiltration of monocytes and M2 macrophages, and elevated transcriptional levels of immune checkpoint genes. In addition, AML was classified into three subtypes according to the model, and patients in different subtypes showed different overall survival (OS) and drug sensitivity. Overall, we formulated a pioneering prognostic model utilizing DRLs, achieving precise AML outcome predictions. The correlations between the DRL prognostic models and the AML immune microenvironment, drug sensitivity, and tumor subtype were explored. In addition, further studies on the molecular mechanisms of key biomarkers, such as LINC00944 and LINC00926, will greatly contribute to our understanding of AML pathogenesis and drug resistance mechanisms in the future.
Similar content being viewed by others
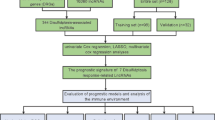


Introduction
Acute myeloid leukemia (AML) occurs at approximately 2.25/100,000, accounting for 80–90% of acute leukemias in adults1. Clinical treatment for AML mainly consists of chemotherapy, hematopoietic stem cell transplantation, and immunotherapy2,3. However, due to the emergence of problems such as chemotherapy intolerance, therapeutic resistance, and the limited number of donors for hematopoietic stem cell transplantation, the five-year survival rate of AML patients is only ~ 29%4. Therefore, exploring new molecular targets for treating AML is essential to improving patient prognosis. In addition, due to the heterogeneity and complexity of AML patients, there is an urgent need to select targeted drugs tailored to their characteristics and enhance their sensitivity in patients.
In recent years, novel therapies that utilize specific cell death mechanisms against cancer cells, such as induced ferroptosis and cuproptosis, have gradually demonstrated their great therapeutic potential5,6. Disulfidptosis is an emerging mode of cell death activated by disulfide stress7. It is characterized by the accumulation of intracellular disulfide, which disrupts the cytoskeletal proteins and F-actin structure, leading to cellular collapse7. In addition, long non-coding RNA (lncRNA) is a single-stranded non-coding RNA that exceeds 200 nucleotides in length and can interfere with a diverse range of cellular functions8. Although these lncRNA molecules do not directly encode proteins, they play critical regulatory roles in cancer initiation, progression, metastasis, and drug resistance9. Several lncRNAs are aberrantly expressed in AML, where they affect cancer cell proliferation, migration, and invasion10. Therefore, many studies have been conducted to construct prognostic models based on the value of lncRNAs in the prognosis of AML and to explore their potential as diagnostic targets11,12. As a new mechanism of cell death, disulfidptosis, exploring the effects of relevant lncRNA alterations in AML has become essential and valuable. Research in this area will help us understand the pathogenesis of AML better and provide essential clues for developing new therapeutic strategies.
In our research, we employed a bioinformatics approach to obtain the expression profiles of disulfidptosis-related lncRNAs (DRLs) based on the GDC TCGA Acute myeloid leukemia (LAML) and GTEX cohorts from the Xena database, combined with the literature search for the collection of disulfidptosis-related genes (DRGs) (Supplementary Tables S1 and S2), and machine learning and other methods were used to screen out DRLs with significant value for AML prognosis and construct a prognostic model. The aim of this research endeavor was to explore the effect of disulfidptosis on the onset and progression of AML and its relationship with patients’ drug sensitivity and tumor microenvironment infiltration. Ultimately, it will provide more prosperous and more accurate predictive tools for the prognostic judgment of AML patients and provide new ideas and support for the progression of AML treatment.
Methods
Data acquisition
Transcriptomic and clinical information for the LAML cohort (132 AML samples) and the GTEX cohort (337 non-tumor samples) were retrieved from the Xena database (http://xena.ucsc.edu/). Somatic mutation data for AML patients were sourced from the downloads available on the TCGA database, accessible at http://portal.gdc.cancer.gov/. This study focused on differential genes at the mRNA level between AML and non-tumor samples. Based on the existing research literature, 24 DRGs were selected (Supplementary Table S1)13. By correlation analysis, 432 DRLs were extracted (Supplementary Table S2). These DRLs were selected based on their strong correlation with DRGs, and the correlation threshold was set at ≥ 0.5.
Construction and validation of the DRLs prognosis model
The cohort of 132 AML samples was evenly split, with half (n = 66) allocated to a training subset and the remaining half (n = 66) constituting a validation subset. Using Univariate COX regression analysis, 52 DRLs significantly correlated with AML outcome were meticulously screened out in the training subset. Then, LASSO-Cox regression analysis evaluated the practical worth of DRLs in predicting the outcome of AML patients and effectively prevented overfitting. According to the training subset, multivariate Cox regression analysis was employed to construct an AML prognosis prediction model that integrated specific coefficients of DRLs to improve the accuracy of prognosis prediction. The AML prognosis prediction model incorporated individualized coefficients for the following DRLs: AL049835.1 (coefficient: − 0.95937069086329), EXOC3-AS1 (coefficient: 2.11922674500076), AC009237.14 (coefficient: 1.0479274303187), LINC00944 (coefficient: 2.8220862848859), AP002761.4 (coefficient: − 0.685608529068001), LINC00926 (coefficient: − 0.660371802480452), AC010247.2 (coefficient: 0.593311215883076), AC099811.5 (coefficient: − 1.78360735203649). In accordance with the risk score, patients were classified into high- and low-risk cohorts, and Kaplan–Meier survival analysis was employed to contrast overall survival (OS) between cohorts. The performance of the model was evaluated through ROC curves, and the area under the curve (AUC) was quantified for predicting 1-year, 2-year, and 3-year survival outcomes.
Nomogram construction
Using the “rms” package in R, visual nomograms of clinical variables and risk scores were created14. Based on the total score, these nomograms accurately predicted the probability of survival at one, two, and three years. The rigorous evaluation of the model’s survival prediction performance, employing both calibration and ROC curve analyses, emphasizes the meticulousness of our study approach.
Comprehensive assessment of genomic variations, functional pathways, and drug responsiveness
Somatic mutations in AML patients were analyzed utilizing the “maptools” package in R. To visually compare the mutational profiles of high-risk and low-risk patient groups, waterfall plots were utilized15. Enriched features within each group were identified through GSEA. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses elucidated functional annotations and pathways enriched by differentially expressed DRLs between the two cohorts. The oncoPredict algorithm estimated IC50 values for commonly prescribed drugs in different AML patient populations to calculate drug sensitivities and assess potential treatment outcomes.
Classification of tumors by prognostic model DRL expression
The robust classification of AML samples into 3 subtypes according to DRL expression levels was achieved with the “ConsensusClusterPlus” package in R16. Data analysis was conducted using R.ggplot 2, and OS differences among subtypes were examined using Kaplan–Meier analysis17. The calculation of immune cells and stromal cells in AML samples was performed using R. estimate and R. CIBERSORT, and the enrichment of immune cells in AML was assessed using predefined gene sets and ssGSEA18.
Statistical analysis
This study used mean values with corresponding standard deviations for data presentation. Additionally, bioinformatics analyses were performed exclusively using R software, specifically version 4.3.3. Statistical significance was determined when P < 0.05, indicated by asterisks as follows: * for p < 0.05, ** for p < 0.01, and *** for p < 0.001.
Results
Discovering of DRLs linked to AML outcome
The existing literature has reported 24 DRGs (Table S1)13. To explore the correlation between lncRNA and mRNA expression in AML samples, we used the “limma” package in R, with p < 0.001 and correlation coefficient > 0.5 as thresholds19. This analysis revealed 432 DRLs. Differential analysis revealed 266 differently expressed DRLs in AML (Fig. 1A), with the top 185 genes showing upregulation (Fig. 1B) being the most significant expression differences in AML samples. Furthermore, 52 DRLs that were substantially correlated with the prognosis of AML patients were found by univariate Cox regression analysis of the training set (p < 0.05). Of these, 23 DRLs were found to be poor prognostic variables (hazard ratio > 1) (Fig. 1C). Figure 1D displays the expression levels of these DRLs in both normal and AML samples.

Screening for prognostically relevant DRLs. (A) Volcano plot showing the distribution of differentially expressed DRLs. (B) Heatmap visualizing differentially expressed DRL expression levels in each sample. (C) Forest plot showing the hazard ratio of prognostically relevant DRLs. (D) Heatmap showing each sample’s expression levels of prognostically relevant DRLs. (E, F) Lasso regression illustrates the overfitting for varying numbers of genes, as well as the degree of overfitting at different counts.
Developing of a prognostic model based on DRLs in AML
Through LASSO regression analysis, the most promising DRLs were selected from a pool of 52 prognostic-related DRLs to prevent overfitting (Fig. 1E and F). Subsequently, using multivariate Cox regression analysis, eight DRLs were determined to be strongly linked with outcome in AML patients. A risk scoring model for AML patients was established based on these DRLs.
Following is the calculation of the risk score: risk score = (− 0.95937069086329) × AL049835.1 expression + 2.11922674500076 × EXOC3-AS1 expression + 1.0479274303187 × AC009237.14 expression + 2.8220862848859 × LINC00944 expression + (− 0.685608529068001) × AP002761.4 expression + (− 0.660371802480452) × LINC00926 expression + 0.593311215883076 × AC010247.2 expression + (− 1.78360735203649) × AC099811.5 expression. In these DRLs, there were notable distinctions between AML samples and normal samples (Fig. 2A). Among them, EXOC3-AS1, AC009237.14, LINC00944, and AC010247.2 were significantly associated with a poor prognosis (Fig. 2B).

Expression of DRLs prognostic model genes in AML and their impact on prognosis. (A) Box plot using LAML cohort data shows the expression of eight DRLs in the model. (B) Survival analysis showing a correlation between lncRNA expression levels and OS in the LAML cohort.
The patients in the training group were categorized into high- or low-risk groups following the median risk score value in order to assess the predictive accuracy of the risk model. Figure 3A and B present the distribution and survival status of AML patients in the training group, and Fig. 3C exhibits the expression profiles of DRLs used in model development, comparing patients in the high-risk and low-risk groups. High-risk scores were correlated with increased death rates, according to Kaplan–Meier survival analysis (Fig. 3D). The DRLs model’s ROC curve showed good efficacy, with AUC values for 1, 3, and 5 years, respectively, of 0.874, 0.897, and 0.982. Additionally, a dual validation of the model was conducted through the analysis of distribution plots, heatmaps, survival curves, and ROC curves in both the training and testing groups (Fig. 3E–O).

Construction and Validation of the DRLs Prognostic Model. (A) Distribution of risk scores and outcomes in the training set. (B) Distribution of risk scores, survival time, and survival status in the training set. (C) Expression distribution of eight DRL models in patients of the training set. (D) Survival curves of patients with different risk scores in the training set. (E) ROC curves and area under the curve (AUC) of the model in the training set. The characteristics of the test set (F–J) and all patients (K–O) were analyzed similarly.
Relationship between clinical factors and DRLs prognostic models and development and validation of nomograms
By univariate and multivariate Cox regression analyses, the risk score (p < 0.001, hazard ratio = 1.062, 95%CI = 1.036–1.089) and age (p < 0.001, hazard ratio = 1.030, 95%CI = 1.013–1.046) independently predicted OS in AML patients (Fig. 4A and Fig. 4B). Furthermore, the DRLs model predicted OS in AML patients better than other clinical features (AUC = 0.790) (Fig. 4C). To better quantify the survival outcomes of AML patients, column-line plots were constructed using the risk score and three clinical factors (gender, AML-FAB, and age) (Fig. 4D). The calibration curve and C-index of 0.706 (95% CI 0.645–0.766) indicate that the model has a near-moderate predictive accuracy for 1-year, 3-year, and 5-year survival outcomes in AML patients (Fig. 4E). In addition, a gender and age subgroup analysis was carried out. The risk score demonstrated excellent predictive power for prognosis in AML patients across three subgroups: those aged < = 65, males, and females. However, in the age group > 65, there was a trend suggesting an association between high-risk scores and poor prognosis, which did not reach statistical significance (p = 0.336) (Fig. 4F).

Association of prognostic models with clinical factors. (A) Univariate Cox regression analysis demonstrating the effect of risk scores and other clinical factors on AML prognosis. (B) Multivariate Cox regression analysis assessing whether risk scores and other clinical factors can serve as independent prognostic factors for AML. (C) Multivariate ROC curves assessing the predictive accuracy of risk scores and other clinical factors for AML prognosis. (D) Nomogram diagrams assessing the specificity of the AML prognostic scoring criteria. (E) C-index curve assessing the validity of the nomogram. (F) Survival curves based on risk scores for different clinical subgroups.
Biological function and pathway analysis of the high- or low-risk patients
Differential analysis was performed to explore the molecular mechanisms underlying the poor prognosis of high-risk patients, which revealed the presence of 271 up-regulated and 139 down-regulated genes in high-risk AML patients (Fig. 5A). In addition, Fig. 5B shows the expression of the top 49 differential genes (log2FC ≥ 1 or ≤ − 1). These findings reveal the molecular features and potential mechanisms of action that contribute to the high-risk phenotype of AML, providing us with a deeper understanding of AML. KEGG enrichment analysis revealed significant enrichment of pathways associated with disulfidptosis, including the B-cell receptor signaling pathway shown and the hematopoietic cell lineage (Fig. 5C)20. GO enrichment analysis illustrated the role of differentially expressed genes in positive regulation of cytokine production, positive regulation of leukocyte cell–cell adhesion, and inhibitory MHC class I receptor activity, among other essential physiological processes and molecular functions (Fig. 5D and E). In addition, GSEA enrichment analysis revealed significant enrichment of pathways, including CELL_ADHESION_MOLECULES_CAMS, in high-risk cohort (Fig. 5F). In contrast, low-risk cohort showed significant enrichment of the BUTANOATE_METABOLISM and PROTEIN_EXPORT pathways (Fig. 5G). After extensive functional analysis, it is clear that DRLs may play an essential role in regulating disulfide death through known pathways or physiological mechanisms.

Functional enrichment analysis based on risk score. (A) Distribution of differentially expressed genes in high-risk and low-risk groups. (B) Distribution of differentially expressed genes in high-risk and low-risk populations. (C) KEGG enrichment analysis reveals enriched KEGG pathways for differentially expressed genes. (D) The bar plot displays enriched GO pathways for differentially expressed genes. (E) The circos plot visualizes the upregulated or downregulated status of differentially expressed genes in GO pathways. (F) GSEA analysis shows highly expressed pathways in the high-risk group. (G) GSEA analysis showing low-expressed pathways in the low-risk group. (H) Waterfall plots showing mutated genes and their frequencies in the high-risk group. (I) Waterfall plots showing mutated genes and their frequencies in the low-risk group. (J) Survival curves for the high- and low-TMB groups. (K) Combined survival analysis of TMB and risk score curves.
Genomic mutation analysis in AML patients
The somatic mutation data were compared between the high-risk and low-risk cohorts. RUNX1 and NPM1 exhibited the highest mutation frequency among genes in high-risk AML patients. The mutation rate of RUNX1 was significantly elevated in the high-risk cohort, reaching 11%, in contrast to just 3% in the low-risk cohort, suggesting that RUNX1 may contribute to the worse outcome of high-risk patients (Fig. 5H and I). In addition, Fig. 5J shows that AML patients with low TMB have a tendency for poorer prognosis compared to those with high TMB (p = 0.092). To further explore this relationship, we combined TMB levels with risk scores to conduct an in-depth analysis of the survival status of AML patients in four different groups. The research results show that, regardless of high or low TMB, patients in the high-risk cohort exhibit shorter OS compared to those in the low-risk group (Fig. 5K). AML patients exhibiting both high TMB and low-risk scores were observed to have a significantly extended OS. This comprehensive analysis strongly suggests that, compared to TMB levels, risk scores have a closer relationship with the prognosis of AML patients and are more effective in predicting their prognosis.
Exploration of immune infiltration, immune checkpoints and immunotherapy in high-and low-risk AML patient cohorts
We study the relationship between immune infiltration and the DRLs model score in AML. After analyzing immune cell infiltration with different algorithms, it was found that the expression levels of macrophage M2 and monocyte cells were higher in the high-risk group (Fig. 6A). This finding suggests a possible correlation between monocyte infiltration, macrophage M2, and high-risk scores. Assessment of immune cell infiltration in survival outcomes revealed that macrophage M1, macrophage M2, and monocyte infiltration were correlated with worse clinical outcomes (Fig. 6B). In contrast, mast cell infiltration correlated with a better outcome (Fig. 6B). The immune function scores and immune cell enrichment analysis showed significant differences between the low-risk and high-risk cohorts in 23 immune characteristics (including ten immune cells and 13 immune function scores). Figure 6C shows that the high-risk cohorts tended to have significantly higher levels of these immune characteristics. Further analysis revealed a lower percentage of plasma cells and a higher percentage of CD8 T cells in the high-risk group (Fig. 6D). Elevated expression of immune checkpoint genes (ICGs) was detected within the high-risk cohort, suggestive of immune dysregulation (Fig. 6E). In the sensitivity assessment of immune-targeted and chemotherapeutic agents, the high-risk cohort was resistant to treatment with 5-fluorouracil, temozolomide, dabrafenib, MK-2206, and dactylitis but had an entinostat, AZD5991, and sepantronium bromide therapy with increased sensitivity (Fig. 6F). Conversely, the low-risk cohort exhibited a contrasting trend in terms of drug sensitivity.

Immune status and drug sensitivity in different risk-scoring groups. (A) Quantitative analysis of immune cell infiltration in AML using different algorithms such as XCELL, TIMER, QUANTISEQ, MCPCOUNTER, EPIC, CIBERSORTABS, and CIBERSORT. (B) Survival curves based on different immune cell infiltration scores. (C) Immune function status in different risk score groups. (D) Immune cell infiltration patterns in different risk score groups. (E) Expression of immune checkpoints in different risk score groups. (F) Drug sensitivity profile in different risk scoring groups.
Profiling of AML molecular subtypes utilizing DRLs
This study’s goal was to use the DRLs model to classify AML samples. Among the tested values, κ = 3 shows a flatter CDF distribution and is close to the maximum (Fig. 7A–C). Therefore, we chose κ = 3 to classify AML samples into three subtypes: cluster 1 (C1), cluster 2 (C2), and cluster 3 (C3) (Fig. 7D). Figure 7E showed that C1 and C2 were predominantly patients with high-risk scores, whereas C3 were predominantly patients with low-risk scores. The survival analysis indicated that C1 and C2 had a poorer prognosis compared to C3 (P = 0.012) (Fig. 7F). The accuracy of the DRLs model in classifying AML was validated through the application of PCA and tSNE analyses, as evidenced by the results presented in Fig. 7G and H. In addition, Fig. 7I and J clearly distinguish between the high- and low-risk cohorts. This suggests the DRLs model can be used for AML classification and risk stratification.

Classification of AML subtypes based on the DRLs model. (A) Sample distribution for different numbers of classifications. (B) CDF curves for different numbers of classifications. (C) Consensus CDF for different numbers of classifications. (D) Consensus matrix of the three classifications. (E) Sankey diagram illustrating the correspondence between AML subtypes and high/low-risk score groups. (F) Survival curves of the different AML subtypes. (G) PCA analysis showing the distribution of samples in different AML subtypes. (H) t-SNE analysis showing the distribution of samples with different AML subtypes. (I) PCA analysis showing the distribution of samples in different risk score groups. (J) t-SNE analysis showing the distribution of samples in different risk score groups.
Immunotherapy analysis in patients with DRL-based molecular subtypes
Immune checkpoints were analyzed in this study to determine suitable targets for personalized immune therapies in AML patients (Fig. 8A). In addition, the sensitivity of patients with three AML subtypes to different drugs was evaluated (Fig. 8B–D). C1 patients showed better sensitivity to cytarabine, dasatinib, nilotinib, ibrutinib, and vorinostat; C2 patients exhibited increased sensitivity to 5-fluorouracil; and C3 patients showed better sensitivity to cytarabine and dasatinib (Fig. 8E). Together, molecular subtyping based on DRLs provides suggestions for a deeper understanding of the immune microenvironment of AML, enlightening the field of leukemia and immunotherapy. This model shows excellent potential for the realization of precision immunotherapy, providing a new level of insight into AML treatment strategies.

Immune status and drug sensitivity of different AML subtypes. (A) Quantitative analysis of immune cell infiltration in AML using different algorithms such as XCELL, TIMER, QUANTISEQ, MCPCOUNTER, EPIC, CIBERSORTABS, and CIBERSORT. (B) ESTIMATE score of different AML subtypes. (C) StromalScore of different AML subtypes. (D) ImmuneScore of different AML subtypes. (E) Drug sensitivity profiles of different AML subtypes.
Discussion
Targeted therapies and immunotherapies have played an essential role in treating AML patients. However, the 5-year survival rate of AML patients still needs to improve due to the heterogeneity and high relapse rate21. The 5-year overall survival rate of adult AML patients is only 26%, and the 5-year overall survival rate of patients older than 60 years is less than 10%22. Disulfidptosis is a new mode of cell death that has recently been discovered23. Regulation of cancer cell death, such as disulfidptosis and ferroptosis, has significant therapeutic potential and application as a new cancer therapy24. Although the mechanism of induction of disulfidptosis has been studied in various cancers, its specific connection to the drug sensitivity and immune microenvironment in AML requires further investigation. In addition, lncRNAs, as multifunctional molecules, play an essential role in tumorigenesis and development25. Therefore, we identified DRLs based on DRGs and developed an assay containing eight DRLs (AL049835.1, EXOC3-AS1, AC009237.14, LINC00944, AP002761.4, LINC00926, AC010247.2, and AC099811.5) of AML prognostic models.
Some DRLs involved in model construction have been reported previously and are closely associated with tumorigenesis and progression: LINC00944 functions as an oncogene in renal cell carcinoma and has prognostic value in breast cancer26,27. In addition, Guo-Jian Shi et al. demonstrated that LINC00926 inhibits breast cancer through the PGK1-mediated Warburg effect to inhibit breast cancer proliferation, migration, and invasion28. However, their specific mechanism of action in AML remains elusive and requires deeper exploration. Research on other DRLs is limited, but in spite of scarce information, their importance is evident.
Utilizing the model’s risk score, AML patients were stratified into distinct subsets of high- and low-risk categories. Bioinformatic analysis corroborated the prognostic significance of the model in AML. According to the Kaplan–Meier survival curve, the model accurately predicted the prognosis of AML, as demonstrated by the shorter OS in the high-risk set (Fig. 3). The effectiveness of the model in predicting AML prognosis was further confirmed by ROC curve analysis (Fig. 3), and the results of the ROC curves demonstrated AUC values of 0.790, 0.802, and 0.920 for 1-year, 3-year, and 5-year risk scores. A study by Fangmin Zhong et al. used DRGs to construct an AML prognostic model. In their study, the AUC values achieved for predicting 1-year, 3-year, and 5-year survival were 0.779, 0.714, and 0.778, which were lower than those of our prognostic model29. The 2022 ELN risk classification can only provide a general categorization based on genetic abnormality (favorable, intermediate and adverse) and cannot accurately quantify the risk level among different patients using numerical data30. It’s possible that the model built using the eight DRLs from our study will forecast AML patients’ prognoses with more accuracy and dependability. Therefore, our model is expected to be a good prognostic tool for AML patients.
The increased expression levels of macrophage M2 and monocyte cells in the high-risk cohort may be linked to the progression of AML (Fig. 6A). Both monocytes and macrophages play essential roles in the immune response and tumor microenvironment31. M2 macrophages have an anti-inflammatory effect that can inhibit the body’s anti-tumor immune response to a certain extent32. Meanwhile, monocytes can inhibit lymphocyte activation under certain conditions, which in turn creates favorable conditions for tumor progression33. Therefore, the prognosis of high-risk patients may be improved by inhibiting M2 macrophage polarization, suppressing monocyte aberrant activation, or modulating the differentiation of monocytes into macrophage types with anti-tumor functions34,35.
Sensitivity assessment of immune-targeted drugs and chemotherapeutic agents demonstrated significant resistance to drugs such as dabrafenib and MK-2206 in high-risk AML patients (Fig. 6F). In comparison, drugs such as AZD5991 and sepantronium bromide may have superior therapeutic efficacy in these patients. This finding has important implications for guiding individualized treatment strategies for high-risk AML patients (Fig. 6F).
Currently, most studies tend to divide AML patients into high-risk and low-risk groups when using prognostic models. However, this crude categorization makes it challenging to fully reveal each AML patient’s clinical heterogeneity and limits the ability to develop targeted drugs for these characteristics. To better address the diversity of AML, we have identified three AML subtypes associated with disulfidptosis that differ in OS and sensitivity to immunotherapy. The C3 subtype has a better outcome and is sensitive to cytarabine and dasatinib. In contrast, the C1 and C2 subtypes had a poor outcome, with C1 displaying better sensitivity to cytarabine, dasatinib, nilotinib, ibrutinib, and vorinostat, and C2 showing better sensitivity to 5-fluorouracil. These new findings highlight the great potential of personalized immunotherapy prediction according to the DRL risk score or AML subtype, which could revolutionize AML treatment in the future.
Conclusion
In conclusion, we have successfully created a prognostic risk model based on DRLs to predict the survival of AML patients. The model provides personalized treatment guidance for patients and significantly improves prognostic prediction. Our thorough exploration of the strong correlation between the model and tumor subtype, immune microenvironment, and AML immunotherapy provides a comprehensive understanding that can further improve the therapeutic efficacy of AML through precise, targeted therapeutic strategies.
Limitation
It is undeniable that the results of our analyses are subject to certain unavoidable limitations. On the one hand, the incidence of disulfidptosis among the 132 AML patients we collected cannot be accurately determined. On the other hand, the primary cells from 132 AML patients were not available, so cellular experiments were not conducted for verification.
Data availability
The datasets analyzed during the current study are available in the Xena database (http://xena.ucsc.edu/) and the TCGA database (http://portal.gdc.cancer.gov/).
References
Zhong, G. et al. MAGEA1 and hTERT peptide treatment improves the potency of the dendritic cell-cytotoxic T lymphocytes (DC-CTL) immunotherapy in DAC treated acute myeloid leukemia. J. Cancer 13, 1252–1260. https://doi.org/10.7150/jca.66501 (2022).
Shi, H. et al. Quercetin induces apoptosis via downregulation of vascular endothelial growth factor/akt signaling pathway in acute myeloid leukemia cells. Front. Pharmacol. 11, 534171. https://doi.org/10.3389/fphar.2020.534171 (2020).
Zou, Y., Huang, Y. & Ma, X. Phenylhexyl isothiocyanate suppresses cell proliferation and promotes apoptosis via repairing mutant P53 in human myeloid leukemia M2 cells. Oncol. Lett. 18, 3358–3366. https://doi.org/10.3892/ol.2019.10620 (2019).
Valiulienė, G., Vitkevičienė, A., Skliutė, G., Borutinskaitė, V. & Navakauskienė, R. Pharmaceutical drug metformin and MCL1 Inhibitor S63845 exhibit anticancer activity in myeloid leukemia cells via redox remodeling. Molecules https://doi.org/10.3390/molecules26082303 (2021).
Tang, B. et al. Integrative analysis of the molecular mechanisms, immunological features and immunotherapy response of ferroptosis regulators across 33 cancer types. Int. J. Biol. Sci. 18, 180–198. https://doi.org/10.7150/ijbs.64654 (2022).
Yun, Y., Wang, Y., Yang, E. & Jing, X. Cuproptosis-related gene—SLC31A1, FDX1 and ATP7B—polymorphisms are associated with risk of lung cancer. Pharmgenom. Pers. Med. 15, 733–742. https://doi.org/10.2147/pgpm.S372824 (2022).
Zheng, T., Liu, Q., Xing, F., Zeng, C. & Wang, W. Disulfidptosis: A new form of programmed cell death. J. Exp. Clin. Cancer Res. 42, 137. https://doi.org/10.1186/s13046-023-02712-2 (2023).
Yao, N. et al. Long noncoding RNA PVT1 promotes chondrocyte extracellular matrix degradation by acting as a sponge for miR-140 in IL-1β-stimulated chondrocytes. J. Orthop. Surg. Res. 17, 218. https://doi.org/10.1186/s13018-022-03114-4 (2022).
Cheng, L. et al. Melatonin regulates cancer migration and stemness and enhances the anti-tumour effect of cisplatin. J. Cell. Mol. Med. 27, 2215–2227. https://doi.org/10.1111/jcmm.17809 (2023).
Li, J. J., Zhu, J. F., Zhang, F., Zhang, P. P. & Zhang, J. J. Upregulation of lncRNA CCAT2 predicts poor prognosis in patients with acute myeloid leukemia and is correlated with leukemic cell proliferation. Int. J. Clin. Exp. Pathol. 11, 5658–5666 (2018).
Ding, W., Ling, Y., Shi, Y. & Zheng, Z. DesA prognostic risk model of LncRNAs in patients with acute myeloid leukaemia based on TCGA data. Front. Bioeng. Biotechnol. 10, 818905. https://doi.org/10.3389/fbioe.2022.818905 (2022).
Zhao, C. et al. A prognostic autophagy-related long non-coding RNA (ARlncRNA) signature in acute myeloid leukemia (AML). Front. Genet. 12, 681867. https://doi.org/10.3389/fgene.2021.681867 (2021).
Li, D., Li, X., Lv, J. & Li, S. Creation of signatures and identification of molecular subtypes based on disulfidptosis-related genes for glioblastoma patients’ prognosis and immunological activity. Asian J Surg https://doi.org/10.1016/j.asjsur.2024.02.041 (2024).
Li, C. et al. Exploration of epithelial-mesenchymal transition-related lncRNA signature and drug sensitivity in breast cancer. Front Endocrinol (Lausanne) 14, 1154741. https://doi.org/10.3389/fendo.2023.1154741 (2023).
Mayakonda, A., Lin, D. C., Assenov, Y., Plass, C. & Koeffler, H. P. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res 28, 1747–1756. https://doi.org/10.1101/gr.239244.118 (2018).
Wilkerson, M. D. & Hayes, D. N. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573. https://doi.org/10.1093/bioinformatics/btq170 (2010).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis. https://ggplot2.tidyverse.org (Springer-Verlag New York, 2016).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12, 453-457, https://doi.org/10.1038/nmeth.3337 (2015).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucl. Acids Res. 43, e47–e47 (2015).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucl. Acids Res. 51, D587-d592. https://doi.org/10.1093/nar/gkac963 (2023).
Yang, Y. et al. Establishment and validation of a carbohydrate metabolism-related gene signature for prognostic model and immune response in acute myeloid leukemia. Front. Immunol. 13, 1038570. https://doi.org/10.3389/fimmu.2022.1038570 (2022).
Yu, H. et al. Venetoclax enhances DNA damage induced by XPO1 inhibitors: A novel mechanism underlying the synergistic antileukaemic effect in acute myeloid leukaemia. J. Cell Mol. Med. 26, 2646–2657. https://doi.org/10.1111/jcmm.17274 (2022).
Hong, S. et al. Disulfidptosis-related lncRNAs signature predicting prognosis and immunotherapy effect in lung adenocarcinoma. Aging 16, 9972–9989. https://doi.org/10.18632/aging.205911 (2024).
Xu, K. et al. Disulfidptosis-related lncRNA signatures assess immune microenvironment and drug sensitivity in hepatocellular carcinoma. Comput. Biol. Med. 169, 107930. https://doi.org/10.1016/j.compbiomed.2024.107930 (2024).
Yang, L. et al. 5mC and H3K9me3 of TRAF3IP2 promoter region accelerates the progression of translocation renal cell carcinoma. Biomark Res. 10, 54. https://doi.org/10.1186/s40364-022-00402-3 (2022).
Chen, C. & Zheng, H. LncRNA LINC00944 promotes tumorigenesis but suppresses Akt phosphorylation in renal cell carcinoma. Front. Mol. Biosci. 8, 697962. https://doi.org/10.3389/fmolb.2021.697962 (2021).
de Santiago, P. R. et al. Immune-related IncRNA LINC00944 responds to variations in ADAR1 levels and it is associated with breast cancer prognosis. Life Sci 268, 118956. https://doi.org/10.1016/j.lfs.2020.118956 (2021).
Shi, G. J., Zhou, Q., Zhu, Q., Wang, L. & Jiang, G. Q. A novel prognostic model associated with the overall survival in patients with breast cancer based on lipid metabolism-related long noncoding RNAs. J. Clin. Lab. Anal. 36, e24384. https://doi.org/10.1002/jcla.24384 (2022).
Zhong, F. et al. Development and validation of a disulfidptosis-related scoring system to predict clinical outcome and immunotherapy response in acute myeloid leukemia by integrated analysis of single-cell and bulk RNA-sequencing. Front. Pharmacol. 14, 1272701. https://doi.org/10.3389/fphar.2023.1272701 (2023).
Döhner, H. et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 140, 1345–1377. https://doi.org/10.1182/blood.2022016867 (2022).
Futami, M. et al. The novel multi-cytokine inhibitor TO-207 specifically inhibits pro-inflammatory cytokine secretion in monocytes without affecting the killing ability of CAR T cells. PLoS One 15, e0231896. https://doi.org/10.1371/journal.pone.0231896 (2020).
Cao, L. et al. M2 macrophage infiltration into tumor islets leads to poor prognosis in non-small-cell lung cancer. Cancer Manag. Res. 11, 6125–6138. https://doi.org/10.2147/cmar.S199832 (2019).
Yamanouchi, K. et al. Integrated evaluation of inflammatory, nutritional, and sarcopenia markers to predict survival in metastatic breast cancer patients. In Vivo 37, 811–817. https://doi.org/10.21873/invivo.13146 (2023).
Li, X., Lei, Y., Wu, M. & Li, N. Regulation of macrophage activation and polarization by HCC-Derived exosomal lncRNA TUC339. Int. J. Mol. Sci. https://doi.org/10.3390/ijms19102958 (2018).
Yuan, X. et al. Prognostic value of systemic inflammation response index in nasopharyngeal carcinoma with negative Epstein-Barr virus DNA. BMC Cancer 22, 858. https://doi.org/10.1186/s12885-022-09942-1 (2022).
Funding
This is supported by the grant of Shandong Provincial Natural Science Foundation of China (ZR2020MH379).
Author information
Authors and Affiliations
Contributions
YYZ: Writing–original draft, Writing–review and editing. H-EC: Writing–review and editing, Writing–original draft . JFW: Writing–review and editing. YKZ: Writing–original draft. ZJL: Data curation, Writing-original draft. YHS: Formal Analysis, Writing–review and editing. YLS: Writing–original draft, Writing–review and editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study does not involve any human participants or the use of human cells.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, Y., Cheng, He., Wang, J. et al. Disulfidptosis-related lncRNA signature to assess the immune microenvironment and drug sensitivity in acute myeloid leukemia. Sci Rep 14, 32015 (2024). https://doi.org/10.1038/s41598-024-83560-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-83560-8
