
Abstract
Poly (ADP-ribose) polymerase (PARP) inhibitors have improved the prognosis of homologous recombination deficient (HRD) ovarian cancer (OC), while effective therapeutic strategies for HR-proficient (HRP) OC still need to be established. This study investigates senescence-mediated inflammation as a novel mechanism of action for PARP inhibitors in HRP cancers. Transcriptome analyses were performed in olaparib-treated HeLa cells as a HRP model. Interferon regulatory factor-Lucia luciferase (IRF-Luc) reporter activity was assessed. The effects of PARP inhibitors on senescence-like phenotypes were assessed in seven HRP cancer cell lines, based on morphological changes, senescence-associated β-galactosidase (SA-β-GAL) activity, cellular granularity, and senescence-associated secretory phenotype (SASP)-related gene expression. Peripheral blood mononuclear cell (PBMC) migration assays were also performed with the conditioned medium in treatment with the PARP inhibitor. Transcriptome analyses revealed numbers of inflammatory cytokine- and chemokine-related pathways were significantly upregulated in olaparib-treated HeLa cells, which were confirmed by IRF-Luc reporter assays. The PARP inhibitors induced senescent phenotypes in HRP cancer cell lines: flattened and enlarged morphology, increased SA-β-GAL activity, elevated cellular granularity, and upregulated expressions of SASP-related genes (e.g., IL1B, IL6, and CXCL10). Furthermore, in vitro migration assays revealed that PARP inhibitor-treated HRP cancer cells attracted PBMCs more abundantly, suggesting the potential for recruiting immune cells to HRP cancer cells through senescence-mediated immunological activation. Our findings suggest that PARP inhibitors recruit immune cells to HRP cancer cells, potentially activating immune responses in the tumor microenvironment, providing new insights into the clinical benefits of PARP inhibitors in immunotherapy for patients with HRP OC.
Similar content being viewed by others


Introduction
Ovarian cancer (OC) is a fatal gynecological malignancy worldwide1. BRCA1 and BRCA2 are representative genes for homologous recombination (HR)-repair (HRR)2, and their deleterious mutations are closely associated with hereditary and non-hereditary OC. Approximately 14–18% of patients with OC have germline mutations in BRCA1 or BRCA2, with an estimated OC risk of 40–60% for BRCA1 and 11–27% for BRCA23. These germline BRCA1/2 mutations also increase the risk of other cancers, including breast, prostate, and pancreatic cancers known as hereditary breast and ovarian cancer (HBOC) syndrome4. Somatic mutations in BRCA1/2, on the contrary, are found in approximately 6% of OCs5. Beyond BRCA1/2, mutations in other HRR-related genes, such as RAD51 and PALB2, are also detected in approximately 8% of OCs6. Overall, approximately 50% of advanced OCs are defined as HR-deficiency7.
Owing to the high prevalence of HR deficiency in OCs, the advent of poly (ADP-ribose) polymerase (PARP) inhibitors has revolutionized the therapeutic management of OC. PARP inhibitors target PARP enzymes, primarily PARP1, which is essential for single-strand break (SSB) repair8. PARP1 binds to SSBs and activates and catalyzes poly (ADP-ribose) (PAR) synthesis to recruit DNA repair proteins8. PARP inhibitors disrupt this process, leading to the accumulation of SSBs and subsequent double-strand breaks (DSBs) through replication fork collapse8. Certain PARP inhibitors also exert the effects of “PARP trapping” on SSBs to generate DSBs more intensively9. DSBs are frequently repaired through HRR machineries; thus, HR-deficient (HRD) cancer cells exhibit higher sensitivity to PARP inhibitors, in a process termed “synthetic lethality”8. Supporting their molecular mechanisms of action, several clinical trials have demonstrated significant improvements in the prognosis of patients with primary advanced and recurrent HRD OC treated with PARP inhibitors7,10,11. The introduction of PARP inhibitors has contributed to increasing the 5-year survival rate for OC from 36% in 1975 to 51% in 20191.
However, the development of therapeutic strategies for HR-proficient (HRP) OC remains challenging. Various combination treatments with PARP inhibitors are being evaluated for HRP cancers, including DNA damage response (DDR) inhibitors, anti-angiogenics, immune checkpoint inhibitors, chemotherapy, and radiotherapy12. Notably, despite the limited efficacy of PARP inhibitors in HRP cancers in vitro13, several clinical trials have revealed some benefits for patients with HRP OC, although the benefits have been inferior to those in HRD cases7,10. These findings indicate potential secondary antitumor mechanisms of PARP inhibitors beyond synthetic lethality.
In this study, we aimed to identify the role of PARP inhibitors in HRP OC patients. We found that PARP inhibitors induce senescence-like phenotypes in HRP cancer cells, upregulating a series of inflammatory cytokine and chemokine genes, called the senescence-associated secretory phenotype (SASP). Furthermore, in vitro migration assays revealed that PARP inhibitor-treated HRP cancer cells attracted peripheral blood mononuclear cells (PBMCs) more abundantly. Our findings suggest that PARP inhibitors recruit immune cells to HRP cancer cells, potentially activating immune responses in the tumor microenvironment.
Materials and methods
Compounds
Olaparib (AZD2281) and niraparib (MK-4827) were purchased from Selleck Chemicals (Houston, TX, USA).
Cell lines
HeLa and A549 cells were purchased from RIKEN BRC, and A549-reporter cells from InvivoGen (CA, USA). The STING knockout cells were generated in the previous study14. OVISE, TYK-nu, and MCAS cells from JCRB Cell Bank, SKOV-3 cells from ATCC, and PBMCs from COSMO BIO CO., LTD. (Tokyo, Japan). Cells were cultured in DMEM or RPMI-1640 with 10% fetal bovine serum (FBS) and 1% penicillin under standard conditions (37℃, 5% CO2). HRP cancer cells were defined as those without alterations in key HRR-associated genes15.
Immunofluorescence
Immunofluorescence was performed as previously described14. Anti-γH2A.X (Ser139) (20E3) (#9718S; Cell Signaling Technology, Danvers, MA, USA), anti-Lamin B1 (#66095–1-Ig; Proteintech, CH, USA) and anti-cGAS (D1D3G) (#15102; Cell Signaling Technology) were used at 10 μg/mL. Images and cell counts were obtained using a BZ-X810 Analyzer (KEYENCE Corp., Osaka, Japan). Cells with five or more γH2A.X foci were considered positive.
CellTiter-Glo luminescent cell viability assay
Cell viability was assessed as previously described14 using the CellTiter-Glo™ 2.0 luminescent assay (Promega Corp., Madison, WI, USA). Luminescence was measured using SpectraMax Paradigm (Molecular Devices, LLC, San Jose, CA, USA).
RNA preparation and quantitative reverse transcription-PCR (qRT-PCR)
RNA preparation and qRT-PCR were performed as previously reported14. Gene expression was normalized to GAPDH. TaqMan probe product IDs were as follows: human GAPDH (Hs99999905_m1), BRCA2 (Hs00609073_m1), IL1B (Hs01555410_m1), IL6 (Hs00174131_m1), CXCL10 (Hs00171042_m1), IFNB1 (Hs00277188_s1), and TNFSF15 (Hs00270802_s1).
Immunoblotting
Immunoblotting followed previously reported protocols14. Primary antibodies (1:1000 dilution) included anti-BRCA2 (D9S6V) (#10741; Cell Signaling Technology), anti-Rad51 (#ab63801; Abcam, Cambridge, UK), and anti-GAPDH (14C10; #2118; Cell Signaling Technology). Proteins were visualized using ECL Select Detection Reagent (Cytiva, Marlborough, MA, USA), and the luminescent images were captured with ImageQuant LAS 4010 (Cytiva, Marlborough, MA, USA). We processed images of blots according to the digital image and integrity policies of Scientific Reports.
siRNA knockdown experiment
siRNA knockdown was performed as previously described16. Human BRCA2 siRNA-SMARTpool (#M-003462–01–0005; Horizon) and Negative Control siRNA (#1022076; Qiagen, Hilden, Germany) were used. The knockdown efficiency was evaluated using qRT-PCR and immunoblotting.
Dual reporter assay
A549-reporter cells (5.0 × 103/well) were seeded in 96-well plates and treated with DMSO or olaparib (0.01–10 μM) for 120 h. Luciferase activity was measured with QUANTI-Luc (InvivoGen, #rep-qlc) and normalized to cell viability assessed by crystal violet staining as previously reported17. Luminescence was measured using a SpectraMax Paradigm (Molecular Devices).
Transcriptome analysis
HeLa cells (1.0 × 105/dish) were seeded on 10 cm dishes and treated with 10 μM DMSO or olaparib for 72 h. Total RNA was extracted using the RNeasy kit (Qiagen), and libraries prepared by Rhelixa were sequenced on the Illumina platform. Data curation was performed as follows: quality control (QC) checks by FastQC (version 0.11.9), trimming by trimmomatic (version 0.39), ribosomal RNA removal by bowtie (version 2.5.2), read mapping to a reference genome using STAR (version 2.7.6), extraction of unique mapped reads by Samtools (version 1.19), and read counting using featureCounts (version 2.0.3). A comparative analysis using baySeq was performed in the TCC library (version 1.40.0) using R (version 4.3.3). Enrichment analysis was performed using gene set enrichment analysis (GSEA) tools.
Senescence-associated β-galactosidase (SA-β-GAL) activity assay
Cells (1.0 × 104/well) were seeded in 6-well plates and treated with 10 μM DMSO or PARP inhibitors (olaparib or niraparib) for 72 h. SA-β-GAL staining was performed at pH 6.0 using a Senescence β-Galactosidase Staining Kit (Cell Signaling Technology). Images were captured using a BZ-X810 Analyzer (KEYENCE Corp.), and SA-β-GAL-positive cells were manually counted.
Fluorescence-activated cell sorting (FACS) analysis
Cells were fixed with 70% ethanol, washed with phosphate-buffered saline (PBS) containing 4% FBS, and stained with 20 μg/mL propidium iodide (Thermo Fisher Scientific, Waltham, MA, USA) and 100 μg/mL RNase A (Nippon Gene, Tokyo, Japan). Ten thousand cells were analyzed using a FACSCanto™ II flow cytometer (Becton–Dickinson, Franklin Lakes, NJ, USA). FlowJo software (v.10.7.1) was used for data analysis.
PBMC migration assay
PBMC migration was assessed as previously described14. Conditioned medium from cells treated with 10 μM DMSO or olaparib for 72 h was used. After 4 h incubation, migratory PBMCs fixed to the membrane were stained with crystal violet. Images were captured using a BZ-9000 Analyzer (Keyence Corp.).
Statistical analysis
Statistical analyses were performed using Excel (Microsoft Office) and GraphPad Prism (version 10.0.3). A two-sided Student’s t test and two-way ANOVA with Sidak’s multiple comparisons test were applied (*P < 0.05, **P < 0.01, ****P < 0.0001; n.s., not significant).
Results
Effect of PARP inhibitors on cell viability in HRP cancer cells is limited
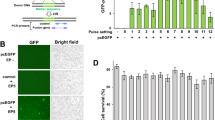
We determined the biological effects of PARP inhibitors on HRP OC cells in vitro. We first investigated the DNA-damaging activity of olaparib in HeLa cells, selected for their suitability in various in vitro assays14,18. γH2 A.X foci, an index of DNA damage, was measured. Based on human pharmacokinetic data from clinical trials19, the treatment of olaparib was set at 10 μM or lower. Immunofluorescence revealed a substantial increase in γH2 A.X foci-positive cells treated with olaparib for 72 h compared with DMSO-treated controls (Fig. 1A, Supplementary Fig. 1, and Supplementary Table 1). These results indicate that olaparib caused DNA damage, even in HRP cancer cells. Next, the antiproliferative activities of PARP inhibitors were determined in four HRP OC cell lines (OVISE, SKOV3, TYK-nu, and MCAS). Using CellTiter-Glo, cell viability was assessed 48 h post-treatment. Minimal growth inhibition was observed in PARP inhibitor-treated cells at concentrations up to 1 μM, and none of them reached a survival rate below 50%, even at 10 μM (Fig. 1B–C). To confirm the contribution of HR proficiency to these outcomes, we knocked down BRCA2 in HeLa and SKOV3 cells using siRNA (siBRCA2). qRT-PCR revealed that BRCA2 siRNA achieved 84% knockdown (Fig. 1D), and western blotting confirmed effective inhibition of BRCA2 protein expression in BRCA2-depleted HeLa cells (Fig. 1E and Supplementary Fig. 2). CellTiter-Glo assays in BRCA2-depleted HeLa and SKOV3 cells (HRD cells) treated with PARP inhibitors for 72 h revealed significant growth inhibition at ≥ 0.3 μM. Survival rates in BRCA2-depleted HeLa cells dropped below 50% at ≥ 3 μM for olaparib and ≥ 1 μM for niraparib, while in BRCA2-depleted SKOV3 cells, a similar reduction was observed at ≥ 3 μM for both olaparib and niraparib (Fig. 1F–G). These results suggest that BRCA2 depletion markedly increases PARP inhibitor sensitivity in HRP cancer cells. Collectively, these results indicated that while PARP inhibitors could induce DNA damage, their antiproliferative activities in HRP cancer cells were limited, suggesting a divergence of the biological effects of PARP inhibitors between DNA damage and antiproliferation in HRP cancer cells.

The effect of PARP inhibitors on cell viability in HRP cancer cells is limited. (A) The effect of olaparib on γH2A.X foci formation in HeLa cells. Hela cells were treated with olaparib at 10 μM for 72 h. Red and blue signals indicate γH2A.X and DAPI (DNA), respectively. Scale bars indicate 50 μ m. (B) (C) The effect of PARP inhibitors on cell viability in HRP OC cells. The HRP OC cell lines were treated with olaparib (B) and niraparib (C) at the indicated concentrations for 72 h. (D) (E) Knockdown efficiencies of siBRCA2 in HeLa cells. Hela cells were treated with siBRCA2 and siNS for 24 h. The knockdown efficiencies were evaluated with qRT-PCR (D) and immunoblotting (E). Cropped immunoblot images are shown. Original blots are presented in Supplementary Fig. 2. (F) (G) Effect of BRCA2 knockdown on sensitivities to the PARP inhibitors in HeLa and SKOV3 cells. HeLa (left) and SKOV3 (right) cells were treated with siNS (black) and siBRCA2 (red), followed by treatment with olaparib (F) and niraparib (G) at the indicated concentrations for 72 h. Data in B–D are shown as means ± SD (n = 3–8).
Inflammatory cytokine-related pathways are transcriptionally upregulated in olaparib-treated HeLa cells
To explore the mechanisms underlying PARP inhibitors’ effects in HRP cancer cells, transcriptome analyses were performed in comparison between olaparib- and DMSO-treated HeLa cells. Cells were treated with 10 μM olaparib or DMSO for 72 h, where moderate anti-proliferation with considerable DNA damage was observed, and were subjected to RNA sequencing (RNA-seq) (Fig. 2A). In total, 17,118 and 16,786 transcripts were detected in olaparib- and DMSO-treated cells, respectively. Analysis of differentially expressed genes (DEGs) revealed 866 genes, including 503 upregulated and 363 downregulated genes in olaparib-treated cells (Fig. 2B–C and Supplementary Table 2–3). Volcano plots indicated the upregulation of inflammatory cytokine-related genes (Fig. 2C). Enrichment analyses using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database20,21 identified inflammation-related pathways as prominently upregulated in olaparib-treated cells, with five of the top 10 pathways involving inflammation-related genes (Fig. 2D). Dot plot analyses also confirmed the upregulation of inflammatory hallmarks, such as TNFA signaling via NFκB, inflammatory response, IL2-STAT5 signaling and interferon gamma response, in olaparib-treated cells (Fig. 2E). Furthermore, the GSEA determined significant enrichment of inflammatory cytokine-related KEGG pathways, including cytokine–cytokine receptor interaction, JAK-STAT signaling, and Rig I like receptor signaling pathways, as well as inflammatory hallmarks such as TNFA signaling via NFκB, interferon alfa response, and interferon gamma response in olaparib-treated cells (Fig. 2F).

Transcriptome analyses in PARP inhibitor-treated HeLa cells. (A) Experimental scheme for transcriptome analyses. (B) Heatmap of clustered DEGs in DMSO- and olaparib-treated HeLa cells. Red and blue indicate upregulated and downregulated gene sets, respectively. (C) Volcano plot of DEGs in olaparib-treated HeLa cells. Red and blue indicate significantly upregulated and downregulated gene sets in olaparib-treated cells compared with DMSO-treated cells. The representative genes in cytokine–cytokine receptor interaction pathways are highlighted. (D) Top ten enriched gene sets and their normalized enrichment scores (NES) in olaparib-treated HeLa cells. Yellow indicates inflammatory cytokine-related pathways. (E) Dot plots of the NES of the enriched hallmark pathways in olaparib-treated HeLa cells. The dot color and size indicate adjusted p-values and counts, respectively. Inflammatory-associated hallmarks are highlighted. (F) GSEAs of representative inflammation-related terms. GSEAs were performed in comparison between olaparib and DMSO treatments. (G) Dose-dependent effect of olaparib on cell viability (black line) and IRF reporter activity (red bars). A549-reporter cells were treated with olaparib at the indicated concentrations for 120 h. Data are shown as means ± SD (n = 6).
To confirm the upregulation of inflammatory response genes, reporter assays were performed in A549-based interferon regulatory factor (IRF)-Lucia luciferase (Luc) reporter cells (A549 reporter cells). Treatment with olaparib for 120 h showed significant dose-dependent increases in IRF-Luc activities, 2- and 3-fold with olaparib treatment at 1 μM and 10 μM, respectively (Fig. 2G, red bars), whereas the antiproliferative activities were limited, with survival rates above 50% (Fig. 2G, black line). Collectively, our findings demonstrate that PARP inhibitors upregulate inflammation-related pathways in HRP cancer cells despite limited antiproliferative activities.
PARP inhibitors induce cellular senescence in HRP cancer cells
Considering the close correlation between DNA damage-induced inflammatory activation and a senescence-like phenotype, termed SASP22, we next determined whether olaparib-treated HRP cancer cells exhibited senescence-like phenotypes based on morphological alteration, SA-β-GAL activity, and cellular granularity in seven cell lines: four OC cell lines (OVISE, SKOV3, TYK-nu, and MCAS) and three other cancer cell lines (HeLa, A549, and A549-reporter). The olaparib-treated cells flattened and enlarged 24 h after treatment, and their cytomegalic phenotype progressed until 72 h, with enlarged nuclei or grape-like multinuclear clusters, which are hallmarks of DNA damage-induced senescence-like cells23 (Fig. 3A and Supplementary Fig. 3). To further assess the senescence-like phenotypes, we determined SA-β-GAL activity and cytoplasmic granules, which are common senescence markers23,24. The number of SA-β-GAL-positive cells considerably increased in all seven cell lines treated with olaparib compared to controls (Fig. 3B and Supplementary Table 4). Similar to olaparib treatment, niraparib also increased SA-β-GAL activity in HeLa, OVISE, and SKOV3 cells, along with morphological alterations of senescence-like phenotype (Fig. 3C–D and Supplementary Table 4). Next, we identified cytoplasmic granules in olaparib-treated cells using FACS. Cellular granularity is evaluated using side-scatter (SSC) values, reflecting intracellular complexity, compared to forward-scatter (FSC) values24. In HeLa cells, treatment with 10 μM olaparib for 72 h increased both SSC and FSC (Fig. 3E, Q2: 6.57% in olaparib and 2.92% in DMSO), with SSC increase being more substantially increased (Fig. 3E, Q1: 8.07% in olaparib and 1.03% in DMSO), indicating an enlarged morphology with intracellular granular accumulation. Similar increases in SSC were also observed in the niraparib-treated HeLa cells (Fig. 3F). Furthermore, elevated SSC values were observed in olaparib-treated A549, A549-reporter and MCAS cells, as well as in the niraparib-treated SKOV3 cells (Fig. 3G–H). These results suggest that PARP inhibitor-induced DNA damage causes cellular senescence or senescence-like phenotypes, with elevated inflammatory gene expression in HRP cancer cells, despite limited antiproliferative effects.

PARP inhibitors induce cellular senescence in HRP cancer cells. (A) Phase-contrast images of HRP cancer cells in treatment with olaparib. The indicated HRP cancer cell lines were treated with olaparib at 10 μM for 72 h treatment. Scale bars indicate 50 μm. (B) Representative images of SA-β-GAL staining in olaparib-treated HRP cancer cells. The indicated HRP cancer cell lines were treated with olaparib at 10 μM for 72 h treatment, followed by SA-β-GAL staining. Scale bars indicate 50 μm. (C) Phase-contrast images of HRP cancer cells in treatment with niraparib. The indicated HRP cancer cell lines were treated with olaparib at 10 μM for 72 h treatment. Scale bars indicate 50 μm. (D) Representative images of SA-β-GAL staining in niraparib-treated HRP cancer cells. The indicated HRP cancer cell lines were treated with olaparib at 10 μM for 72 h treatment, followed by SA-β-GAL staining. Scale bars indicate 50 μm. (E) Flow cytometry in Hela cells treated with olaparib. Forward-scatter (FSC) and side-scatter (SSC) histograms are shown. Black and red indicate cell treated with DMSO and olaparib at 10 μM for 72 h, respectively. (F) Flow cytometry in Hela cells treated with niraparib. FSC and SSC histograms are shown. Black and red indicate cell treated with DMSO and niraparib at 10 μM for 72 h, respectively. (G) SSC histograms in A549, A549-reporter, and MCAS cells in treatment with olaparib at 10 μM for 72 h. (H) SSC histograms in SKOV3 cells in treatment with niraparib at 10 μM for 72 h.
cGAS-STING signaling pathway is involved in cellular senescence induced by the PARP inhibitor in HRP cancer cells
Senescent cells upregulate the expression of inflammatory cytokines and chemokines, termed SASP22. Next, we determined the SASP-related gene expression in olaparib-treated HRP cancer cells. IL-1B, a representative SASP factor, was significantly upregulated in a dose-dependent manner in olaparib-treated HeLa cells (by 2.7-fold at 3 μM, 19.3-fold at 10 μM, and 77.1-fold at 30 μM; Fig. 4A) and A549-reporter cells (by 2.3-fold at 1 μM, 17.9-fold at 3 μM, 17.1-fold at 10 μM, and 59.5-fold at 30 μM; Fig. 4B). Other SASP factors, IL-6 and CXCL10, also increased after treatment with olaparib at concentrations ranging from 3 μM to 30 μM in both HeLa and A549-reporter cells (Fig. 4A–B). In three HRP OC cells (OVISE, SKOV3, and MCAS) and A549 cells, IL1B expression also significantly increased in a dose-dependent manner after olaparib treatment (Fig. 4C). In addition, IFNB1 and TNFSF15, which are representative genes in type-I interferon and TNF-α pathways that activate cellular senescence25,26, were also significantly upregulated in olaparib-treated OVISE, SKOV3, and MCAS cell lines (Fig. 4D). Multiple studies have demonstrated the involvement of cGAS-STING pathway in PARP inhibitor-associated inflammation27,28,29. Immunofluorescence revealed that olaparib treatment profoundly generated micronuclei in both A549 and SKOV3 cells, where cGAS protein was intensively localized (Fig. 4E). Furthermore, knockout of STING drastically suppressed the reporter activity of Luc-IRF in olaparib-treated A549-reporter cells (Fig. 4F), indicating that the cGAS-STING pathway plays critical roles in inflammatory responses in the PARP inhibitor-treated cells.

cGAS-STING signaling pathway is involved in the PARP inhibitor-induced cellular senescence in HRP cancer cells. (A) (B) Gene expression of of IL1B, IL6, and CXCL10 in the PARP inhibitor-treated cells. HeLa cells (A) and A-549-reproter cells (B) were treated with olaparib at the indicated concentrations for 72 h. qRT-PCR for IL1B, IL6, and CXCL10 was performed. (C) IL1B gene expression in olaparib-treated HRP cells. A549, OVISE, SKOV3, and MCAS cells were treated with olaparib at the indicated concentrations for 72 h. qRT-PCR for IL1B was performed. (D) IFNB1 and TNFSF15 gene expressions in olaparib-treated HRP OC cells. OVISE, SKOV3, and MCAS cells were treated with olaparib at 10 μM for 72 h. qRT-PCR for NFB1 and TNFSF15 was performed. (E) Immunofluorescence of cGAS in olaparib-treated cells. A549 and SKOV3 cells were treated with olaparib at 10 μ M for 72 h. Red, green, and blue signals indicate cGAS, Lamin B1, and DAPI, respectively. White arrows indicate the localization of cGAS in micronuclei. Scale bars indicate 50 μm. (F) IRF-Luc activity in STING-knockout (KO) cells in treatment with olaparib. The STING KO (gray bars) and negative KO (black bars) A549-reporter cells were treated with DMSO and olaparib at 10 μM for 72 h, followed by IRF reporter assay. Data are shown as means ± SD (n = 3).
SASP produced in olaparib-treated HRP cancer cells recruits PBMCs in vitro migration assays
Finally, we determined whether the PARP inhibitor-induced senescence-like cells could attract PBMCs via secretory factors. In vitro migration assays were performed with PBMCs and conditioned medium from olaparib-treated HRP cancer cells, using HeLa and MCAS cells treated with 10 μM DMSO or olaparib for 72 h (Fig. 5A). The chemoattractant activities were assessed by quantifying PBMCs that migrated through the membrane, stained with crystal violet. The number of PBMCs on the membrane profoundly increased in the conditioned medium from olaparib-treated cells compared to that from controls in both HeLa and MCAS cell lines (Fig. 5B), suggesting that olaparib-treated HRP cancer cells could immunologically activate PBMCs through secreted chemoattractants. Collectively, our findings demonstrate that PARP inhibitors induce cellular senescence with less cell death activity in HRP cancer cells, potentially leading to immunological activation to attract blood cells to HRP cancer cells (Fig. 5C).

PBMC migration assays with conditioned media from olaparib treated cells. (A) Experimental scheme for PBMC migration assays with conditioned media from olaparib treated cells. (B) Representative images of migrated PBMCs through the membrane. The membranes used for migration assays were stained with crystal violet. Scale bars indicate 100 μm. (C) Proposed mechanism of inflammatory activation induced by PARP inhibitors in HRP cancer cells.
Discussion
In this study, we demonstrated that PARP inhibitors induce senescence-like phenotypes to produce SASP factors in HRP cancer cells, although their antiproliferative effects are limited. In vitro migration assays confirmed that conditioned medium from PARP inhibitor-treated HRP cells attracted PBMCs more profoundly than conditioned medium from control-treated cells. These findings suggest that PARP inhibitors enhance immune cell recruitment to HRP cancer cells through senescence-mediated immunological activation.
PARP inhibitors have dramatically improved the poor prognosis of OC owing to the high prevalence of HRD in advanced cases, known as “synthetic lethality.” In contrast, treatment strategies for HRP OC have not been fully established yet. In some clinical trials, unexpectedly, PARP inhibitors exhibited marginal efficacy even in patients with HRP OC7,10, hinting at potential antitumor activity that is mechanistically distinct from cytotoxicity by “synthetic lethality.” Recent studies have demonstrated that PARP inhibitors induce cellular senescence through the accumulation of DNA damage30,31. The p53/p21 and p16/RB pathways are involved in senescence-associated antiproliferative effects22. Furthermore, even in p53-mutant OC cell lines, PARP inhibitors induce p53-independent but p21-mediated cellular senescence accompanied by DNA damage response (DDR) activation30, suggesting diverse mechanisms underlying cellular senescence in cancer cells. We found that PARP inhibitors induced senescence-like phenotypes in OC and other HRP cancer cells, which was confirmed using various senescence biomarkers: morphological alterations, SA-β-GAL activity, cellular granularity, and SASP-related gene expression (Fig. 3A–H, Fig. 4A–D). Transcriptome analyses revealed a robust upregulation of gene sets and pathways associated with inflammatory cytokines and chemokines in PARP inhibitor-treated HRP cancer cells, strongly indicating senescence induction in HRP OC and other cancer types.
The effects of senescence on tumor development, including tumor-promoting or tumor-suppressive effects, have been reported to be controversial. Senescent cells produce secretory proteins termed SASP factors, including inflammatory cytokines, chemokines, growth factors, and MMP22,32. SASP factors facilitate tumor progression by promoting tumor growth, epithelial–mesenchymal transition (EMT), and immunosuppression32. Based on the concepts of “tumor-promotive effects of senescence,” novel therapeutic strategies of senolytics, which eliminates senescent cells, or senomorphics, which blocks SASP factors, are under development32. Conversely, SASP factors also contribute to tumor regression by activating immune surveillance, enhancing the infiltration of natural killer (NK) cells, macrophages, and T cells, and facilitating the clearance of senescent tumor cells32. In our study, PARP inhibitor-treated HeLa cells showed a significant increase in the expression of CXCL10 and other chemokines (Fig. 4A), which promoted T-cell infiltration33. KEGG-based enrichment analysis of DEGs revealed that NK cell-mediated cytotoxicity pathways were significantly enriched in the PARP inhibitor-treated cells (Fig. 2D). In addition to these senescent phenotypes and gene expression profiles, conditioned medium from PARP inhibitor-treated HRP cancer cells profoundly attracted PBMCs in the in vitro migration assays (Fig. 5B). Collectively, PARP inhibitors are expected to boost immunological activation in the TME by recruiting blood cells via the induction of senescence in cancer cells. These inflammatory effects of PARP inhibitors may contribute to the antitumor efficacy of HRP in OC.
In conclusion, we demonstrated that PARP inhibitors induce cellular senescence in HRP and other cancer cells, upregulating inflammatory cytokines and chemokines to attract PBMCs in vitro. These findings suggest the potential for recruiting immune cells to HRP cancer cells through senescence-mediated immunological activation. Further studies will provide new insights into the clinical benefits of PARP inhibitors with immunotherapy for patients with HRP OC.
Data availability
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
References
Siegel, R. L., Giaquinto, A. N. & Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 74, 12–49. https://doi.org/10.3322/caac.21820 (2024).
Yoshida, K. & Miki, Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 95, 866–871. https://doi.org/10.1111/j.1349-7006.2004.tb02195.x (2004).
Lheureux, S., Braunstein, M. & Oza, A. M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA: A Cancer J. Clin. https://doi.org/10.3322/caac.21559 (2019).
Daly, M. B. et al. NCCN Guidelines® Insights: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 2.2024. J. Natl. Compr. Cancer Netw. 21, 1000–1010. https://doi.org/10.6004/jnccn.2023.0051 (2023).
Arcieri, M. et al. How BRCA and homologous recombination deficiency change therapeutic strategies in ovarian cancer: A review of literature. Front. Oncol. 14, 1335196. https://doi.org/10.3389/fonc.2024.1335196 (2024).
Pennington, K. P. et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 20, 764–775. https://doi.org/10.1158/1078-0432.CCR-13-2287 (2014).
González-Martín, A. et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N. Engl. J. Med. 381, 2391–2402. https://doi.org/10.1056/nejmoa1910962 (2019).
Lord, C. J. & Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158. https://doi.org/10.1126/science.aam7344 (2017).
Murai, J. et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 72, 5588–5599. https://doi.org/10.1158/0008-5472.CAN-12-2753 (2012).
Monk, B. J. et al. A randomized, phase III trial to evaluate rucaparib monotherapy as maintenance treatment in patients with newly diagnosed ovarian cancer (ATHENA–MONO/GOG-3020/ENGOT-ov45). J. Clin. Oncol. 40, 3952–3964. https://doi.org/10.1200/jco.22.01003 (2022).
Pujade-Lauraine, E. et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 18, 1274–1284. https://doi.org/10.1016/S1470-2045(17)30469-2 (2017).
Xie, Y. et al. Combined strategies with PARP inhibitors for the treatment of BRCA wide type cancer. Front. Oncol. https://doi.org/10.3389/fonc.2024.1441222 (2024).
Farmer, H. et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. https://doi.org/10.1038/nature03445 (2005).
Morita, T. Y. et al. CDC7 inhibition induces replication stress-mediated aneuploid cells with an inflammatory phenotype sensitizing tumors to immune checkpoint blockade. Nat. Commun. 14, 7490. https://doi.org/10.1038/s41467-023-43274-3 (2023).
De Bono, J. et al. Olaparib for metastatic castration-resistant prostate cancer. N. Engl. J. Med. 382, 2091–2102. https://doi.org/10.1056/nejmoa1911440 (2020).
Yamamoto, G. et al. WEE1 confers resistance to KRAS(G12C) inhibitors in non-small cell lung cancer. Cancer Lett. 611, 217414. https://doi.org/10.1016/j.canlet.2024.217414 (2024).
Fujimoto, Y. et al. Combination treatment with a PI3K/Akt/mTOR pathway inhibitor overcomes resistance to anti-HER2 therapy in PIK3CA-mutant HER2-positive breast cancer cells. Sci. Rep. 10, 21762. https://doi.org/10.1038/s41598-020-78646-y (2020).
Iwai, K. et al. A CDC7 inhibitor sensitizes DNA-damaging chemotherapies by suppressing homologous recombination repair to delay DNA damage recovery. Sci. Adv. https://doi.org/10.1126/sciadv.abf0197 (2021).
Yonemori, K. et al. Safety and tolerability of the olaparib tablet formulation in Japanese patients with advanced solid tumours. Cancer Chemother. Pharmacol. 78, 525–531. https://doi.org/10.1007/s00280-016-3106-7 (2016).
Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677. https://doi.org/10.1093/nar/gkae909 (2025).
Coppé, J.-P. et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, e301. https://doi.org/10.1371/journal.pbio.0060301 (2008).
Huang, W., Hickson, L. J., Eirin, A., Kirkland, J. L. & Lerman, L. O. Cellular senescence: The good, the bad and the unknown. Nat. Rev. Nephrol. 18, 611–627. https://doi.org/10.1038/s41581-022-00601-z (2022).
Hwang, E. S., Yoon, G. & Kang, H. T. A comparative analysis of the cell biology of senescence and aging. Cell. Mol. Life Sci. 66, 2503–2524. https://doi.org/10.1007/s00018-009-0034-2 (2009).
Frank, T. et al. Cell cycle arrest in mitosis promotes interferon-induced necroptosis. Cell Death Differ. 26, 2046–2060. https://doi.org/10.1038/s41418-019-0298-5 (2019).
Tanzer, M. C. A proteomic perspective on TNF-mediated signalling and cell death. Biochem. Soc. Trans. 50, 13–20. https://doi.org/10.1042/bst20211114 (2022).
Kim, C., Wang, X. D. & Yu, Y. PARP1 inhibitors trigger innate immunity via PARP1 trapping-induced DNA damage response. Elife 9, e60637. https://doi.org/10.7554/elife.60637 (2020).
Wang, T. et al. Combination of <scp>PARP</scp> inhibitor and <scp>CDK4</scp>/6 inhibitor modulates <scp>cGAS</scp>/<scp>STING-</scp>dependent therapy-induced senescence and provides “one-two punch” opportunity with <scp>anti-PD-L1</scp> therapy in colorectal cancer. Cancer Sci. 114, 4184–4201. https://doi.org/10.1111/cas.15961 (2023).
Pantelidou, C. et al. STING agonism enhances anti-tumor immune responses and therapeutic efficacy of PARP inhibition in BRCA-associated breast cancer. NPJ Breast Cancer https://doi.org/10.1038/s41523-022-00471-5 (2022).
Fleury, H. et al. Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell reversible senescence. Nat. Commun. https://doi.org/10.1038/s41467-019-10460-1 (2019).
Lombard, A. P. et al. Olaparib-Induced senescence is bypassed through G2–M checkpoint override in olaparib-resistant prostate cancer. Mol. Cancer Ther. 21, 677–685. https://doi.org/10.1158/1535-7163.mct-21-0604 (2022).
Dong, Z. et al. Cellular senescence and SASP in tumor progression and therapeutic opportunities. Mol. Cancer https://doi.org/10.1186/s12943-024-02096-7 (2024).
Muthuswamy, R. et al. NF-κB hyperactivation in tumor tissues allows tumor-selective reprogramming of the chemokine microenvironment to enhance the recruitment of cytolytic T effector cells. Can. Res. 72, 3735–3743. https://doi.org/10.1158/0008-5472.can-11-4136 (2012).
Acknowledgements
We would like to thank members of the Aki Lab: Kumi Kinoshita, Emi Ito, Yasushi Kawagishi, Takuya Nakatsuru, and Keita Hirouchi. We also thank Katsuya Tsuchihara, Tohru Kiyono, Riu Yamashita for their valuable comments on the manuscript and technical support.
Funding
This research was supported by the Grants-in-Aid for Scientific Research (B) (20H03541) to A.O., the Grants-in-Aid for Fostering Joint International Research (B) (20KK0187) to A.O., Takeda Science Foundation (2019, 2022) to A.O., the Grants-in-Aid for Scientific Research (C) (23K06708) for R.K., and the Grant-in-Aid for Research Activity Start-up (24K23370) for K.N.
Author information
Authors and Affiliations
Contributions
M.K.: Data curation, Formal analysis, Investigation, Visualization, Writing¬–original draft, R.K.: Conceptualization, Formal analysis, Investigation, Writing¬–review & editing H.S.: Formal analysis, Investigation, G.Y.: Resources, C.M.: Resources, T.Y.: Resources, T.N.: Resources, Y.S.: Resources, T.Y.M.: Resources, K.N.: Resources, Y.H.: Resources, M.T.: Supervision, A.O.: Supervision, A.Oh.: Conceptualization, Funding acquisition, Project administration, Supervision, Writing¬–original draft, Writing¬–review & editing.
Corresponding authors
Ethics declarations
Competing interests
T.Y. was an employee of Axcelead Drug Discovery Partners, Inc (2023-present). A.Oh. was an employee of Takeda Pharmaceutical Company, Ltd (2006–2018). A.Oh. is a part-time employee of Astellas Pharma Inc. as a cross-appointment system (2022-present). A.Oh. reported paid consulting or advisory roles for Ono Pharmaceutical Company Ltd., Craif Inc., and GEXVal Inc. out of this study. A.Oh. has received research funding from Takeda Pharmaceutical, Daiichi-Sankyo, and Astellas Pharma companies out of this study. M.K., R.K., H.S., G.Y., C.M., T.N., Y.S., T.Y.M, K.N., Y.H., M.T., and A.O. declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kamii, M., Kamata, R., Saito, H. et al. PARP inhibitors elicit a cellular senescence mediated inflammatory response in homologous recombination proficient cancer cells. Sci Rep 15, 15458 (2025). https://doi.org/10.1038/s41598-025-00336-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-00336-4
Keywords
This article is cited by
-
Cellular senescence in cancer: from mechanism paradoxes to precision therapeutics
Molecular Cancer (2025)
