
Abstract
This study investigates the selective precipitation of aluminum, iron, and thorium from a rare earth element (REE)-containing pregnant leach solution (PLS) using magnesium carbonate (MgCO3) as a precipitant. The goal is to efficiently remove impurities while minimizing valuable REE losses. A combination of experimental methods and aqueous thermodynamic modeling (OLI software) was used to understand the precipitation behavior of these elements under varying pH, temperature, and hydrogen peroxide (H2O2) conditions. Kinetic experiments confirmed equilibrium is reached within 30 min. A central composite design (CCD) and response surface methodology (RSM) revealed that iron is nearly completely removed at pH 3.5, with thorium and aluminum precipitation occurring at higher pH values. Optimal conditions, 81 °C, pH 3.6, and 0.52 mL H2O2, enabled complete removal of iron, ~ 95% removal of thorium, and ~ 65% of aluminum, with TREE losses under 3%. Solid precipitates were characterized via X-ray diffraction, Raman spectroscopy, inductively coupled plasma mass spectroscopy, and scanning electron microscopy energy dispersive spectroscopy, identifying ferrihydrite, aluminum sulfate, and magnesium carbonate phases. Thermodynamic models supported experimental findings, qualitatively predicting solubility trends. A technoeconomic analysis for a 1000 m3/day PLS treatment plant in Ontario, Canada, estimated monthly operational costs at ~$2.65 million and capital costs at ~$7.85 million. This work advances impurity removal strategies in REE processing, offering scalable, cost-effective, and environmentally responsible solutions for enhancing REE recovery efficiency.
Similar content being viewed by others

Introduction
In the twenty-first century, rare earth elements (REEs) have gained prominence as vital materials crucial to the development and sustenance of industrialized economies. During this era, a select few mines and commercial entities were the primary suppliers of REEs, producing them in substantial quantities. This setup led to a REE crisis that commenced in 2010, prompting a fervent exploration and the development of new sources for REEs. Consequently, the landscape of the REE industry has experienced profound transformations in recent years. Many industrialized nations have initiated efforts to establish circular REE economies by investigating new mining opportunities and tapping into end-of-life secondary sources. However, the transition from promising mineral deposits or secondary sources to marketable products necessitates research and development expertise to effectively address the complicated technological challenges inherent to REE production, separation, and processing1.
The chemistry of these novel ores and secondary sources can significantly deviate from that of conventional ores, necessitating the establishment of new hydrometallurgical practices2. While extensive research has focused on ore or secondary source decomposition, less emphasis has been placed on purging impurities from the resultant processing streams. The challenge lies in the fact that the composition and concentration of these impurity elements can be distinct and unconventional when compared with traditional REE leach solutions. This calls for the development of new procedures or the modification of existing ones to effectively eliminate these impurities. The presence of impurities in solutions containing REEs has a profound impact, not only on the final REE products but also on the overall processing efficiency.
Impurities such as aluminum (Al) and iron (Fe)(III) pose significant challenges in REE processing streams, particularly in solvent extraction circuits. In naphthenic acid circuits, for instance, Al and Fe(III) are preferentially extracted ahead of REEs. During the scrubbing stage, they are the first to be stripped and reintroduced to the feed stage3. As they progressively accumulate in the solvent extraction circuit, they undergo hydrolysis, forming gel-like hydroxides. These hydroxides lead to emulsions during operation, reducing contact area and severely hindering the extraction process4. Silicon is another element that forms gelatinous precipitates and co-extracts in certain newly explored REE deposits, particularly those based on eudialyte5. Calcium, mainly in the form of gypsum, is another major component of the undesirable substances, known as “crud,” that forms in solvent extraction4. The refractory metals, including titanium, zirconium, hafnium, vanadium, niobium, tantalum, chromium, molybdenum, and tungsten, also tend to hydrolyze and contribute to the formation of crud in solvent extraction circuits4. Often, the creation of crud is a direct consequence of the composition of the feed liquor introduced into solvent extraction circuits4. Other elements like thorium and uranium may not form significant crud but are absorbed into the solvent, thereby reducing the recovery of REEs.
Impurities in the REE processing stream have notable impacts on both REE recovery and precipitation efficiency. When sulfate is present, excess calcium in the solution can precipitate as gypsum, leading to some co-precipitation of REEs, which subsequently reduces REE recovery6,7. Likewise, the presence of thorium in the solution interferes with the selective precipitation process and results in the co-precipitation of thorium with REE double sulfates or oxalates8. While Al, Fe, copper (Cu), and lead (Pb) do not co-precipitate with REE oxalates, they hinder precipitation efficiency by consuming oxalic acid through complexation in the solution before REEs can precipitate9. In the case of REE carbonate precipitation, Al is known to hydrolyze, forming a colloidal hydroxide that disrupts the formation of REE carbonate crystals10. Some impurities with relatively positive reduction potentials, such as Cu, can electrodeposit on equipment, piping, valves, or in regeneration circuits, potentially leading to galvanic corrosion. Impurities also impact downstream REE metal or alloy production by molten salt electrolysis in chloride or fluoride melts11. Alkaline earth elements and lead gradually accumulate in the bath, changing their properties and degrading the electrolyte over time. Additionally, phosphorus impurities introduced into molten salt cells become incorporated into REE metals or alloys and subsequently release phosphine, a toxic and flammable gas.
During downstream REE metal or alloy production, many of the impurities present in the feed material are typically transferred in quantifiable amounts to the metallic phase12. Consequently, impurities found in solutions or concentrates often end up in finished REE magnets, where they can have a particularly significant impact. Even small quantities of elements like Al, silicon, refractory metals (such as titanium, zirconium, hafnium, vanadium, niobium, tantalum, chromium, molybdenum, and tungsten), or first-row transition metals (like manganese, cobalt, nickel, and copper) can measurably affect the properties of neodymium iron boron (NdFeB) magnets. These effects extend to characteristics like the Curie temperature, anisotropy field, and coercivity13,14. Heavy metals such as tin, lead, and bismuth are especially problematic in NdFeB magnets because they interact with neodymium in intermetallic compounds and significantly reduce coercivity15. In some cases, the adverse effects of certain elements on the bulk properties of magnetic phases are acknowledged to bring about a better microstructure overall. However, this should be a decision left to magnet manufacturers rather than being dictated by REE supply.
Impurity removal constitutes a pivotal operation that can be integrated at various stages within the REE processing workflow. In certain scenarios, it initiates with the selective leaching of ore or secondary resources, strategically designed to limit the co-extraction of unwanted impurity elements. In contrast, other situations necessitate impurity removal after REE leaching, often preceding the generation of a mixed REE oxide (REO). Alternatively, impurity removal can be strategically positioned after the dissolution of a mixed REE oxide or before the individual REE separation via methods such as solvent extraction or ion exchange. There are instances where impurity removal becomes a post-processing step after REE metal or alloy production, aiming to eliminate residual interstitial elements through methods like vacuum melting, distillation, or electrochemical techniques16. In essence, impurity removal can occur at different points during these stages or even throughout various stages within the REE processing procedure.
This study primarily centres on the elimination of impurities stemming from gangue minerals found in ores or secondary resources. These gangue minerals are the predominant source of impurities within the realm of REE processing. Consequently, most impurity removal procedures are implemented downstream of the initial stages, which often encompass physical and chemical beneficiation processes and alkaline cracking, or acid baking followed by leaching. These procedures predominantly involve the hydrometallurgical treatment of REE-containing aqueous solutions. Notably, the core methods of impurity removal can be broadly categorized into the following techniques: (1) Purification techniques using solvent extraction, (2) Purification techniques relying on ion exchange or adsorption, (3) Purification techniques centered on selective precipitation, and (4) Innovative purification techniques, which may not conform to the categories listed above.
These techniques share a fundamental characteristic in that they are designed to be selective. Impurity removal processes must exhibit selectivity either towards extracting REEs or towards extracting specific impurities – achieving both at once is an impractical objective. The choice of technique and selectivity type is contingent upon factors such as the relative concentration of impurities in relation to REEs.
Selective precipitation is an essential process in all REE flowsheets. It involves the conversion of aqueous REE species into solid products, which can be used for separation, impurity removal, sale, transport, or for further manufacturing into high-value products. Various precipitants for REEs are available, and their selection depends on the objectives of the precipitation process. In REE processing, it is often necessary to produce intermediate REE products that can be sold or transported to separation plants. Examples of precipitants include NaOH, Na2CO3, NH4HCO3, H2C2O4, NH4OH, and NaHCO3. Typically, carbonate precipitates exhibit lower crystallinity and may require the use of seed crystals or surfactants. This highlights the significance of precipitation in the REE processing and its potential application at different stages within the REE flowsheet. A previous review article provided a summary of the literature using various precipitants and discussed the advantages and disadvantages of their application1.
Table S1 in Supplementary Materials presents a literature review on the behavior of different precipitation schemes for impurity removal during REE processing. Original research articles on impurity removal by selective precipitation were compiled and systematically categorized based on: (1) Targeted elements in solution (usually REEs), (2) Other elements present in solution (e.g., Fe, Al, Ca), (3) Feed material (chemistry, synthetic or ore, etc.), (4) Precipitation scheme (compounds, pH adjustments, etc.), and (5) Summary and comments (separation efficiencies, recoveries, challenges, etc.).
This study concentrates on the removal of Al, Fe, and Th impurities whose impact on subsequent REE processing and final REE products are presented in Table 1. The primary aim of this research was to achieve the highest possible separation of Al, Fe, and Th through selective precipitation while minimizing the co-precipitation of REEs. Magnesium carbonate (MgCO3) was employed as the precipitating agent, and H2O2 (30 wt%) was added to elevate the oxidation-reduction potential (ORP) of the solution to ensure that all Fe existed as Fe(III) to achieve complete precipitation. Notably, the utilization of MgCO3 in selective precipitation is not extensively documented. Only one previous study by Search Minerals Inc., engaged in the exploration and development of REE recovery deposits in Labrador, Canada17, used MgCO3 for removing Fe from the PLS after acid baking and water leaching of their ore concentrate using H2SO4. Therefore, the application of MgCO3 as a precipitant in a sulfate medium is relatively rare. Furthermore, comprehensive data on process optimization and precipitation mechanisms are lacking, and this study strives to address this knowledge gap, highlighting its novelty.
A response surface methodology was adopted to examine the influence of three key operational parameters: temperature, pH, and added H2O2 amount, on the precipitation efficiency of Al, Fe, Th, and REEs. Through empirical modeling of the gathered data, optimal operating conditions were determined, which maximized impurity removal while minimizing REE losses during selective precipitation. The precipitates obtained at the centre points of the DOE matrix were subject to analysis using Raman spectroscopy and scanning electron microscopy energy dispersive X-ray spectroscopy (SEM-EDX) to identify the mineral phases present and to ascertain their chemical composition; thus, reinforcing the identification of mineral phases.
Results and discussion
Compositional characterization of PLS
In this research, a Canadian ore sample extracted from a deposit situated within peralkaline volcanic rocks was utilized. Table 2 presents the mean elemental composition of the PLS from three measurements, along with the relative standard deviation. The total content of REEs in this sample amounts to 750.55 mg/L, with Ce being the most abundant REE at a concentration of 290.25 mg/L, followed by La and Nd. Furthermore, this sample contains several other elements, including a high amount of Fe at 1624.00 mg/L, Al at 358.40 mg/L, and Th at 12.51 mg/L. The pH of the PLS ranged from 1.6 to 1.7, and its density was measured at 1.013 g/mL.
Kinetic experiment results
At a temperature of 25 °C, kinetic tests were conducted with the addition of 1 mL of 30 wt% H2O2 to ensure all Fe is present as Fe(III) to achieve complete precipitation. The pH of the solution was varied in increments of 0.5, ranging from 3.0 to 4.5, by introducing 20 wt% MgCO3 and maintaining the solution at each pH level for a duration of 30 min. All procedures were repeated in triplicate to ensure consistency. Figure 1a-d illustrate the mean percentage of precipitation for Fe, Al, TREE, and Th respectively. These figures reveal that, with the exception of Fe at pH 3, equilibrium was achieved within 30 min in all systems.
Figure 1e and f depict the pH and ORP measured with a silver reference electrode (Ag/AgCl (3 M KCl)) as functions of time. These figures show that both pH and ORP also reached equilibrium within 30 min. In Fig. 1g, the concentrations of Fe, Al, Th, and TREE at each pH level are presented at the 30-min timepoint. Notably, the concentration of Fe decreased significantly with increasing pH beyond 3. The Al concentration exhibited a slower decrease with increasing pH. The alteration in TREE concentration was not substantial, consistent with the findings in Fig. 1c which show a TREE precipitation percentage ranging from 5 to 10% at pH levels between 3.0 and 4.5. Supplementary Figure S1a–g illustrates the concentration profiles of REEs over time across varying pH levels. The data reveal that light REEs (LREEs) exhibit greater precipitation efficiency compared with heavy REEs (HREEs). At pH values below 4, precipitation efficiency remains below 10%. However, a marked increase is observed at pH 4.5, reaching 16–20%. Based on the outcomes of this experiment, the operating time for the DOE matrix experiments was set at 30 min to ensure that the system had reached equilibrium.

Kinetic experimental results for percentage precipitation of (a) Fe, (b) Al, (c) TREEs and (d) Th at 25 °C, pH 3.0–4.5 up to 30 min residence time. (e) The pH profile over time, and (f) the ORP profile over time. (g) Concentration of Al, Fe, Th, and TREE as a function of pH at a 30-min timepoint.
Effect of operating parameters and empirical model Building
The removal of impurities from the PLS was examined under three distinct operating conditions: temperature (A), H2O2 quantity (B), and pH (C). An empirical model was employed to quantify the impact of these three operating parameters on the removal process. The impurity removal experiments were structured based on a 23 full-factorial matrix, and the results concerning the target REE (Nd, Ce) and total REE (TREE), along with Al, Fe, and Th at the corresponding operating conditions, are presented in Table 3.
The effect of each factor (A, B, and C) and their interactions are calculated using Eq. 1:
where n represents the number of data points and y corresponds to the associated responses, with the high level indicated by a plus sign (+) and the low level by a minus sign (–).
The developed empirical model describes the removal efficiency (\(\:{\widehat{y}}_{i}\)) of the target elements as a function of the experimental parameters (A to C) (Eq. 2). The experimental data were fitted to this empirical model using multiple linear least squares regression (mLLSR) (Eq. 3). The vector \(\:\widehat{\beta\:}\) contains each of the model parameters (\(\:{\widehat{\beta\:}}_{0}\), \(\:{\widehat{\beta\:}}_{1}\), \(\:{\widehat{\beta\:}}_{2}\), …), corresponding to each parameter (A to C) and their second-order interaction terms. The design matrix X (comprising A, B, C) represents the experimental setup, while \(\:{\text{y}}_{i}\) is the response vector encompassing each of the measured experimental extraction efficiencies.
The removal efficiency \(\:{\widehat{y}}_{i}\) was calculated using the mass of element i in the solid phase divided by the total mass of element i in the output which included the solid phase (residue), the liquid phase, and the wash solution (Eq. 4):
where msolid is the mass of precipitate, Ci, s is the concentration of i in the precipitate, Vfiltrate is the volume of the filtrate, Ci, filtrate is the concentration of i in the filtrate, Vwash is the volume of wash solution, and Ci, wash is the concentration of i in the wash solution.
Upon fitting the initial empirical model to the data points, it was observed that the model achieved statistical significance, and the lack of fit (LOF) was deemed significant as well. When a model attains significance and the lack of fit is also significant within a design of experiments, it implies that the model is effective for predicting the response variable. However, it is important to note that the model lacks precision in accurately predicting outcomes across all cases. Significant curvature was observed, prompting the augmentation of the study with a Response Surface Methodology (RSM) design. This augmentation facilitated a thorough exploration of the factors contributing to the observed curvature or whether the curvature aligned with the desired direction for the objectives of the study. Therefore, a second block of experiments was conducted which entails six runs, represented as block 2 summarized in Table 4 as standard order experiment numbers 14 to 19. These experiments were conducted to provide sufficient information to adequately model the true surface. The central composite design (CCD) contains five levels of each factor: low axial, low factorial, centre, high factorial, and high axial. With this many levels, it generates enough information to fit the quadratic model shown in Eq. (5) to (10) in terms of coded factors.
The intercept value (β₀) signifies the mean of all actual responses. When models are expressed in a coded format, the units of measure for the predictors (A, B, and C in this context) become irrelevant. Consequently, the model coefficients (β) can be directly compared, serving as a gauge of their respective impact.
The reactions that take place in the system are shown in reactions (11)–(16). The REEs form carbonate precipitates while Al, Fe, and Th form hydroxides. The Ksp at 25 °C for all REE carbonates and Al, Fe, and Th hydroxide are presented in Table 5.
To elucidate the impact of various parameters on the precipitation efficiency of REEs, Fe, Th, and Al, thermodynamic calculations in aqueous solutions were performed using two distinct models: the Mixed Solvent Electrolyte (MSE) model and the Aqueous (AQ) model, both integrated into OLI Studio 11.0.1.9 (OLI Systems, Inc.). A detailed explanation of the models and their associated parameters in the context of REE sulfate systems can be found elsewhere18.
For the purpose of these calculations, Nd was selected as a representative of the REEs due to the limited availability of databases for other REE carbonates. In Fig. 2a, the solubility profile of Nd2(CO3)3 as a function of temperature in an aqueous medium, as modeled by the MSE software from OLI is presented. It is evident that the solubility of Nd2(CO3)3 decreases as temperature rises, consistent with the exothermic nature of the dissolution reaction. Worth noting is that the pH of this salt in water at 25 °C is computed to be 8.1, and this value decreases as the temperature increases, reaching 6.8 at 90 °C.
In Fig. 2b, the solubility of Nd2(CO3)3 is displayed as a function of pH within the range of 2.5 to 7 at 25 °C. Notably, it is observed that the salt exhibits high solubility up to a pH of 4.5. Beyond this point, as the pH exceeds 4.5, the solubility of the salt starts to decline, reaching a concentration of 2.9 × 10− 5 mol/L at pH 5.5. These findings are consistent with the observed trend of increased REE losses with increasing pH from 3 to 5.5. It should be noted that experimental data for the solubility of Nd2(CO3)3 at different temperatures or pH levels was unavailable. Consequently, we were unable to validate the OLI results. It is important to acknowledge that the OLI database may necessitate some parameter adjustments; thus, the modeling results elucidate the observed trends only qualitatively.
In Fig. 2c, the solubility of Al(OH)3, Fe(OH)3, and Th(OH)4 is depicted as a function of temperature, as calculated using the OLI software MSE model for Fe and Al and the Aqueous model for Th. The trend shows that the solubility of all three salts increases with rising temperature, attributed to the endothermic nature of the dissolution reactions. It is also evident that Th(OH)4 exhibits the lowest solubility, followed by Fe(OH)3, and then Al(OH)3. Consequently, it is anticipated that near-complete removal of Fe and Th can be accomplished, while Al may remain in the solution due to its relatively higher solubility. The pH for all three salts starts at around 6.9 at 25 °C and decreases to approximately 6.2 at 90 °C. This information is important for understanding the behavior of these salts during the removal process.
In Fig. 2d, the solubility of Al(OH)3, Fe(OH)3, and Th(OH)4 is illustrated as a function of pH at 25 °C, as calculated using the OLI software. The composition of the solution was determined based on the PLS assay, as indicated in Table 2, with the exception of Th, which was selected at a higher concentration to make the changes in concentration visible on the graph. The graph shows that Th(OH)4 initiates precipitation at a pH of 2.5. For Fe(OH)3, precipitation begins at a pH of 3, and beyond pH 3.5, it completely precipitates. The Al(OH)3, on the other hand, starts precipitating at a pH of 3.5, and above pH 4, it fully precipitates. This data is valuable for understanding the conditions under which these compounds will form solid precipitates in the solution.
It should be noted that experimental data for the solubility of these three salts at different temperatures or pH levels was also unavailable. Consequently, we were unable to validate the OLI results. It is important to acknowledge that the OLI database may require parameter adjustments. Thus, the modeling results elucidate the observed trends only qualitatively Therefore, comparisons between the modeling results and the experimental data for precipitation pH of the three salts should also be made qualitatively to highlight the observed trends.

Thermodynamic modeling of the system using OLI software. (a) Solubility of Nd2(CO3)3 in water as a function of temperature in the range of 25–90 °C. (b) Solubility of Nd2(CO3)3 in water as a function of pH in the range of 2.5–7 at 25 °C. (c) Solubility of Al(OH)3, Fe(OH)3, and Th(OH)4 in water as a function of temperature in the range of 25–90 °C. (d) Solubility of mixed Al(OH)3, Fe(OH)3, and Th(OH)4 in water as a function of pH in the range of 1–7 at 25 °C. The HCl and MgCO3 were used as the pH adjusters.
To elucidate the impact of H2O2 addition, the Eh–pH diagram for Fe was generated using the MSE model in OLI software, as shown in Fig. 3. The diagram illustrates the stability regions of various Fe species as a function of pH and oxidation-reduction potential (Eh), referenced against the standard hydrogen electrode (SHE). Notably, the precipitation of Fe as FeO(OH)(s) occurs at ORP values exceeding approximately 0.59 V vs. SHE and at pH levels above ~ 2.5. Given that the reference potential of a silver/silver chloride electrode in 3 M KCl is 0.22 V vs. SHE19, the solution ORP must exceed 0.37 V (i.e., 0.59 V–0.22 V) to enter the FeO(OH)(s) stability domain. The initial PLS had an ORP of 311 mV (0.311 V), which is insufficient to achieve Fe(III) precipitation. Therefore, H2O2 was introduced to oxidize Fe2+ to Fe3+, effectively increasing the ORP and shifting the system into the desired region of the Eh–pH space. This oxidation step is crucial, as Fe(III) readily precipitates as FeO(OH)(s), while Fe(II) remains soluble across a wider pH range. By elevating the ORP, the addition of H2O2 ensures complete transformation of soluble Fe species into insoluble hydroxides, thereby enabling efficient and selective removal of Fe from the solution without significant co-precipitation of REEs.

The Eh-pH diagram of Fe using the MSE model of OLI software (Fe concentration = 0.01 mol/L, 25 °C).
Optimization of operating parameters
Having characterized the system and identified the crucial factors along with their respective ranges, the subsequent focus shifts to optimization. Specifically, the objective was to determine the optimal settings for these influential factors that yield desirable values for the response variables. The optimization strategy employed in this study aims to address two antagonistic responses: minimizing TREE, Ce, and Nd loss through precipitation while maximizing the removal of impurities such as Fe, Al, and Th from the PLS.
In this study, the desirability optimization methodology (DOM) is employed to augment RSM. The DOM is a methodology that complements RSM by providing a systematic approach to balance and optimize multiple properties of a product. The DOM involves mapping each property onto a zero-to-one desirability function and then combining these individual desirabilities into a composite desirability scale. This allows for the evaluation and optimization of multiple properties simultaneously, providing a comprehensive view of the trade-offs involved in the optimization process20. The objective of an optimization exercise is to find the right set of conditions that will meet al.l the goals; this may not always translate to a desirability value of 1.0. In Supplementary Figure S2, ramps views show the desirability for temperature, H2O2 addition, and pH for minimum REE losses and maximum impurity removal from the PLS. The highlighted points show both the exact values and how well the goal was satisfied. With a desirability factor value of 0.74, the optimum conditions for the laboratory-scale impurity removal were determined. These optimum conditions, limited by the chosen range of factor levels, are the following: 81 °C temperature, 0.52 mL H2O2 addition, and pH 3.6. Figure 4 is crafted by overlaying contour plots for all three responses, ensuring tight control around their respective targeted values. Within Fig. 4, a discernible sweet spot emerges, where all specifications can be successfully attained.

Overlay plot reveals a window of operability.
To validate conclusions drawn from the DOE, confirmation runs were executed under optimal conditions. Confirmation involved predicting responses using the model equation, comparing them with observed values, and assessing agreement. Results from three confirmation runs (standard run orders 20, 21, and 22) are detailed in Table 4. Removal and loss levels experimentally observed and predicted values are closely aligned, confirming a correct DOE interpretation. In instances of discrepancies between predicted and observed values, determining whether deviations are reasonable is essential. Constructing a prediction interval (PI) for future observations aids in this assessment. Further detailed explanations can be found in literature21. Statistical analysis of confirmation runs compared with the developed model is presented in Table 6. Some responses fell slightly outside the PI but yielded favourable outcomes—lower losses of Nd and Ce and higher removal levels of Al than predicted.
Mechanistic investigation of the precipitation process
As described in the Experimental section, in addition to the DOE matrix, two more replications of the central points were conducted at a pH of 3.5. This entailed the use of 20 wt% MgCO3, a temperature of 57.5 °C, and the addition of 0.5 mL of H2O2 (30 wt%) with a residence time of 30 min. The resulting filtrate from this step was then transferred to the reactor. The temperature was subsequently set to 57.5 °C. Following this, 20 wt% MgCO3 was introduced to raise the pH to 4.5, and a 30-min interval was allowed for the precipitation process to conclude. This entire process was iterated at pH levels of 5 and 5.5, with each step maintaining a 30-min residence time. The solid precipitate obtained at a pH of 5.5 underwent characterization using Raman spectroscopy, SEM-EDX, and ICP-MS/OES.
Figure 5 presents the percentage precipitation of Fe, Al, Th, and TREEs calculated based on the concentration of these elements in the aqueous phase, plotted against pH. At pH 3.5, 100% of Fe precipitated and was subsequently removed from the system through filtration. Following the removal of solid material at pH 3.5, the solution’s pH was systematically increased up to 5.5. This figure also shows that Th achieved 100% precipitation at pH 4.5. However, due to its very low concentration, it may not be detectable through standard characterization techniques. The Al precipitation increased with rising pH and reached 100% at pH 5.5. As for TREEs, their precipitation reached 34% at pH 5.5.
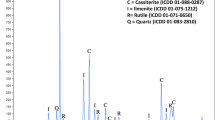
The solid phase at pH 3 from another test was characterized using X-ray diffraction and the results are presented in Fig. 6. As shown, the solid phase is ferrihydrite, Fe9.78O14(OH)2 with ICDD 00-058-0899. The broad nature of the peaks indicates that the ferrihydrite is poorly crystalline.

The percentage precipitation of Al, Fe, Th, and TREEs as a function of pH at 57.5 °C and 0.5 mL 30 wt% H2O2.

X-ray diffraction spectra of the solid precipitate at pH 3. The mineral phase is ferrihydrite, Fe9.78O14(OH)2 with ICDD 00-058-0899.
Numerous regions of the mounted powder obtained at pH 5.5 underwent Raman Spectroscopy analysis, and a spectral database search was conducted to identify the components within the material. Despite analyzing over 50 areas, the spectra acquired from different parts of the sample exhibited remarkable consistency. This suggests that the mineral composition of the analyzed regions is quite similar. The outcomes reveal that the majority of the powder primarily comprises aluminum sulfate and magnesium carbonate. In Fig. 7a, selected Raman spectra are presented alongside reference spectra for aluminum sulfate hexadecahydrate (Al2(SO4)3•16H2O) and rostite (Al(SO4)(OH)•5H2O), and Fig. 7b showcases dypingite, which is a hydrated magnesium carbonate (formula: Mg5(CO3)4(OH)2•5H2O). Regrettably, the Raman analysis did not reveal the presence of REEs. The absence of REEs in the Raman analysis results could indicate that these components are Raman-inactive or that their concentration is below the detection limit of the instrument.

Raman spectrum of the precipitate at pH 5.5 (the bottom spectra in all panels). (a) Aluminum sulfate hexadecahydrate (Al2(SO4)3•16H2O) and Rostite (Al(SO4)(OH)•5H2O). (b) Dypingite (Mg5(CO3)4(OH)2•5H2O).
The backscattered electron (BSE) images and accompanying EDX semi-quantitative elemental data are depicted in Figs. 8 and 9. The BSE images serve to illustrate the diverse particulates within the powder, while the EDX data provides insight into their composition. In certain fragments (for example, spectra 5 and 7 in Fig. 15a), notable levels of O, Mg, and C are present, consistent with the dypingite findings from the Raman analysis. Figure 9b indicates that the bright, spherical particles primarily consist of C, O, F, and Al. However, some of these bright particles contain C, O, and Pb (specifically, spectrum 12 in Fig. 9b). The EDX data validates the presence of C, O, Mg, Al, and S. It is worth noting that while Raman analysis did not identify REEs, the EDX data does reveal the presence of various REEs, including Ce, La, Y, Pr, Nd, Sm, Gd, and Dy. However, these elements are present in relatively smaller quantities compared with the other mentioned elements.
The composition of the solid precipitate was also determined using alkali fusion followed by ICP-MS for REEs and ICP-OES for bulk elements. The composition results are presented in Table 7.

BSE images of the solid precipitate at pH 5.5.

(a,b) BSE images and EDX semi-quantitative data (wt%) collected from the precipitate solid at pH 5.5.
Technoeconomic analysis of the process
In this section, a preliminary technoeconomic analysis was performed to estimate the operating and capital costs of the proposed process. The plant capacity is set at 1000 m3/day of PLS, and it is assumed to operate in Ontario, Canada. The necessary chemicals are MgCO3 and H2O2, with their quantities determined based on the optimal operating conditions of the process. To handle the proposed PLS capacity, two jacketed and agitated continuous stirred tank reactors (CSTRs) with a volume of 50 m3 each were selected. Figure S3 shows the process flow diagram of the proposed plant.
The process requires five single-stage centrifugal pumps for the feed, MgCO3, H2O2, and product, with one spare pump for each. Assuming a 30-min residence time for the CSTR and 15 min each for filling and emptying the reactors, the feed and product pumps need a capacity of 200 m3/h. The capacities of the reagent pumps were calculated based on the ratio of the required reagent volume to the feed volume.
Filtration is also needed, for which a vacuum drum filter was chosen. It was assumed that 3 m3 of feed could be processed with 2 m2 of filter surface, requiring an approximate filter surface area of 20 m2. Additionally, two 1000 m3 cone roof storage tanks were selected for feed and product storage, and two 100 m3 tanks for reagents. The details of mass and energy balance are presented in Supplementary Note 1.
Table 8 presents the materials and utility inputs to the plant, their capacity, and unit cost. Since this processing plant will be part of a larger facility that produces REE oxides and processes PLS, no cost for PLS was included in the calculations.
Table 9 presents the monthly operating costs of the plant. It is hypothesized that the optimal operation and maintenance of the plant require 7 skilled operators, as indicated by Eq. 17:
where NOL is the number of operators per shift, P is the number of processing steps of particle solids (e.g., transportation and distribution, particle size control, and particle removal), Nnp is the number of non-particle processing steps including compression, heating, cooling, mixing, and reaction22.
Each operator is assumed to work 49 weeks per year, five days a week, with one shift per day. The salary rate is set at 26 USD per hour, reflecting the average hourly wage for chemical plant operators in Canada (Payscale.com, 2022; Economic Research Institute, 2022). An additional 5% of direct labor costs is included for benefits and overhead. Moreover, direct supervisory and clerical labor costs are estimated at 18% of the operating labor costs22.
Electricity costs are calculated based on rates for a consumer with similar consumption levels as the plant, including delivery and regulatory charges, converted to USD (“Ontario Energy Board,” 2024). Similarly, natural gas costs for heating are based on rates for a comparable consumer, including supply, delivery, carbon, and other regulatory charges (“Ontario Energy Board,” 2024). Both electricity and natural gas prices are projected to increase by 2% per year. Material (reagent) and utility costs are summarized in Table 9.
The capital cost of the plant is presented in Table 10. The costs of the equipment were taken from the Capital Equipment Cost Database23. The prices were provided in the year 2007. The Chemical Engineering Plant Cost Index (CEPCI)24 was used to adjust equipment costs for time effects. The equipment costs were calculated using the following formula:
To calculate the total EPC (engineering, procurement, and construction management) costs, the following items are considered: equipment cost, spare parts (4% of equipment cost), and installation, piping, and electrical (30% of equipment cost) which result in the installed equipment cost. The next items are engineering, procurement, and construction management costs that are 20% of the installed equipment cost, construction costs that are 8% of the installed equipment cost, and the freight cost for equipment and construction materials which is 10% of the installed equipment cost. Finally, a contingency cost which is 10% of the total mentioned costs is added to obtain the total EPC costs. In calculating the total Capex, development costs (including environmental studies and acquiring licences), land purchase price, owner’s project management, owner’s engineer, and construction insurance were not considered since this process will be parts of the plant producing REE oxide. Therefore, these costs should be considered in the Capex of the overall plant.
While significant improvements in the selective precipitation of impurities have been demonstrated by these findings, it is important to consider the broader environmental implications of these processes. In the following section, the potential ecological and human health impacts associated with the chemicals and methods used in the REE impurity removal process are addressed.
Environmental implications of impurity removal from PLS
This study focuses on the selective precipitation of impurities, specifically Al, Fe, and Th, from a PLS containing REEs. The primary objective is to improve the efficiency and sustainability of REE recovery while acknowledging the potential environmental impacts of the chemicals and methods used and the associated waste generated.
The mitigation strategies to address environmental concerns during PLS processing are well-established in the industry. The process would begin with a thorough characterization of precipitation residue (tailings) content including Al, Fe, and Th as well as leaching rates using kinetics studies25. These leaching rates are incorporated into an environmental risk assessment model that predicts the levels of Al, Fe, and Th released into the environment, based on the selected mitigation measures.
Using these predictions, mitigation measures for tailings containing Fe, Al, and Th include disposal in above-ground tailings management facilities with an engineered cover placed on top at the end of operations to limit releases. Alternatively, tailings can be used as backfill material in underground workings or open mined-out pits, if this approach also limits leaching.
In REE mining, Th and U-containing leaching wastes (residues) are typically neutralized with limestone before disposal. Solid radioactive wastes are kept wet to minimize dust formation. Impoundment facilities use waterproof materials like high density polyethylene (HDPE) to prevent soil and groundwater contamination, and barium hydroxide is used to precipitate radioactive elements. The Australian company Lynas stores Th waste in HDPE-lined ponds. To meet the maximum allowed concentration limit of 500 ppm for safe disposal, radioactive waste is mixed with other residues to dilute Th and U concentrations. This procedure is also followed at the Mountain Pass mine, where regular tailings are co-disposed with radioactive wastes26,27,28. For detailed regulations on the disposal of radioactive materials and maximum authorized concentrations of prescribed deleterious substances29.
Depending on the risk assessment model’s predictions, water quality management may involve additional measures such as water collection ponds and groundwater pumping wells around the tailings management facilities. Collected water would be treated by an advanced water treatment plant before being discharged into the environment.
Ensuring environmental compliance and sustainability in the REE extraction process is therefore achievable and aligns with current industry practices, promoting efficiency and safety29,30.
Conclusion
This study presents a detailed and systematic approach to the selective removal of Al, Fe, and Th from a REE-bearing PLS using MgCO3. The objective was to maximize impurity removal while minimizing the co-precipitation of valuable REEs—a critical challenge in REE hydrometallurgy. Kinetic experiments confirmed that a 30-min residence time is sufficient to reach equilibrium across all studied systems, forming the basis for process optimization through experimental design.
A central composite design (CCD) combined with response surface methodology (RSM) was employed to evaluate the influence of temperature, pH, and H2O2 addition on the precipitation efficiency of target impurities and the retention of REEs. The use of H2O2 was essential to maintain Fe in the Fe(III) oxidation state, promoting its complete removal at low pH. Optimal conditions—81 °C, 0.52 mL H2O2, and pH 3.6—resulted in complete Fe removal, 95% Th removal, and 65% Al removal, with TREE losses limited to under 3%. A stepwise pH adjustment strategy further demonstrated that impurity removal can be staged to progressively extract Fe, Th, and Al while preserving REEs.
Solid characterization using X-ray diffraction (XRD), Raman spectroscopy, and scanning electron microscopy with energy-dispersive X-ray spectroscopy (SEM-EDX) revealed that ferrihydrite was the dominant Fe phase at low pH, while aluminum sulfate and magnesium carbonate were prevalent at higher pH levels. EDX analysis also confirmed the presence of REEs in minor quantities within the precipitates, although Raman analysis did not detect them, likely due to their low concentrations.
To evaluate scalability, a technoeconomic analysis was conducted for a plant treating 1000 m3/day of PLS. The capital expenditure was estimated at ~$7.85 million, and monthly operational costs were ~$2.65 million, with MgCO3 comprising the bulk of material costs. These findings demonstrate the process’s feasibility for industrial deployment, particularly in regions like Canada where REE development is a strategic priority.
In summary, this study provides a robust framework for impurity removal from REE leach solutions using MgCO3. The findings offer valuable insights into both the fundamental chemistry and applied process design, with direct relevance to industrial REE processing. The developed methodology enhances process efficiency, supports environmental compliance, and lays the groundwork for broader implementation in REE value chains.
Methodology
Materials
In this research, a Canadian ore sample extracted from a deposit situated within peralkaline volcanic rocks was utilized. The chosen ore for this study was determined to contain a total of 1.25% REE oxides (TREO + Y2O3), which encompasses the combined oxides of lanthanum through lutetium, in addition to yttrium oxide. Magnesium carbonate (MgCO3, FCC/USP grade) was obtained from Fisher Chemical by Thermo Fisher Scientific. Hydrogen peroxide (H2O2, ACS Reagent grade, 30 wt%) was obtained from VWR. All solutions were prepared using deionized water (> 18 MΩ cm) produced by the Milli-Q Integral water purification system of MilliporeSigma (Merck KGaA, Darmstadt, Germany). Nitric acid (HNO3, ACS Reagent Grade, 68.0–70.0 wt% Assay, VWR) was used for sample dilution for chemical analysis.
Apparatus
The impurity removal experimental setup featured a 2 L jacketed glass reactor equipped with a Liebig condenser, a receiving flask, and a mechanical stirrer operating at 150 rpm, controlled via a JGR-2 L, YH CHEM control panel. Vapor condensation and distilled water collection were achieved using a Liebig condenser and a receiving flask. To maintain a constant temperature within the reactor, a Fisher Scientific, Inc. water bath circulated hot water through the jacket layer. Additionally, a chiller from Fisher Scientific, Inc. circulated coolant through the condenser. The use of a condenser was essential to prevent the loss of evaporated liquid, ensuring it condensed and returned to the system. This integrated array of equipment ensured precise control and execution of each experimental procedure.
Procedures
The ore sample was ground and subjected to a magnetic-gravity hybrid separation, achieving a 92% recovery of total REE oxides (TREO). The beneficiated product then underwent thermal treatment with sulfuric acid at 250 °C, followed by water leaching at 80 °C, resulting in a 92% REE extraction. The pregnant leach solution (PLS) obtained was filtered and prepared for subsequent impurity removal investigations. Detailed descriptions of the beneficiation and leaching processes are beyond the scope of this paper, which focuses on the impurity removal from the PLS. The experimental procedure for the DOE matrix experiments began with the reactor being filled with 800 mL of PLS. The process started by elevating the bath temperature from room temperature to the desired set temperature, maintaining this temperature for 30 min. Subsequently, a predetermined quantity of H2O2 was introduced to elevate the ORP of the solution. Following this, 20 wt% MgCO3 was added to raise the pH to the specified value. Initial kinetic experiments, performed at 25 °C, were used to assess Al, Fe, Th, and total REEs (TREEs) concentrations in the solutions. The results, determined via inductively coupled plasma optical emission spectroscopy (ICP-OES) and inductively coupled plasma mass spectroscopy (ICP-MS), indicated that equilibrium was reached in approximately 30 min. Consequently, in subsequent experiments, systems were held for 30 min after reaching the target pH.
Immediately before sampling, 10 mL volumetric flasks were prepared by carefully adding 5 mL of a 5 wt% HNO3 solution, using the Hamilton Microlab 600 diluter/dispenser system. The saturated solution samples were withdrawn from the flasks through the use of a syringe and plastic tubing. To ensure that any solid particles were removed during the extraction, nylon syringe filters (Basix, 0.45 μm) were attached to the syringes. This process was implemented to prevent any solid materials from re-dissolving within the syringes. Once the samples were extracted, they were promptly transferred to volumetric flasks, being filled up to the precisely calibrated 10 mL mark.
Following the completion of the experiment, the solution underwent filtration using vacuum filtration, employing Whatman Grade 3 filter paper, and the resulting filtrate was gathered. A comparison was conducted between the composition of the filtrate and the average of the three samples taken directly from the reactor to verify their consistency. Subsequently, the filter cake was subjected to a washing process involving 300 mL of deionized water, with the resulting filtrate being collected and analyzed, while also being factored into the mass balance. The filter cake was then carefully dried in an oven at 50 °C for a duration of 24 h. Once fully dried, it was placed into sample bags and stored within a desiccator until the time of analysis.
The solid samples underwent digestion through an alkali fusion process, which was facilitated using a fusion fluxer known as Claisse LeNeo. The resulting compositions were subjected to analysis for REEs, thorium (Th), uranium (U), and bulk elements (Al, Fe, Ti, Mn). The concentrations of REEs, Th, U, Mn, and Ti were determined using ICP-MS while those of Fe and Al were determined using ICP-OES. After conducting solid and solution analyses, a mass balance was executed on individual REE and impurities within the system. The calculation of a metal accountability factor for each REE, along with Fe, Al, and Th as impurities, was based on the net metals entering and exiting the process. The closure of the mass balance was successfully achieved by taking into account the composition of the PLS both before and after the precipitation process, along with the wash solution and the solid precipitate. Utilizing the compositions of both solution and solid samples proved essential in calculating the precipitation percentage during the process.
In addition to the DOE matrix, two further trials at the conditions of central points were executed at a pH of 3.5, employing 20 wt% MgCO3, 57.5 °C, and the addition of 0.5 mL of H2O2 (30 wt%) with a residence time of 30 min. The filtrate generated from this step was transferred to the reactor, and the temperature was set to 57.5 °C. Subsequently, 20 wt% MgCO3 was introduced to elevate the pH to 4.5, and a 30-min interval was allowed for the precipitation process to conclude. This sequence was reiterated at pH levels of 5 and 5.5, with each step maintaining a 30-min residence time. The primary aim of this investigation was to attain nearly complete removal of Fe at the initial pH of 3.5, followed by the monitoring of Al elimination and the assessment of TREE loss as the pH increased incrementally to 5.5. The solid precipitate gathered at a pH of 5.5 underwent characterization using Raman spectroscopy and SEM-EDX, as elaborated in the subsequent section.
Analytical methods
To analyze the PLS, first, it was diluted, and then the concentrations of REEs, Th, U, Mn, and Ti in the PLS were quantified using ICP-MS with a PerkinElmer NexION 2000 instrument. For measuring Fe, and Al concentrations, ICP-OES was employed, utilizing a PerkinElmer Optima 8000 instrument and the wavelengths employed were Al (396.153 nm) and Fe (238.204 nm). The ICP-OES system was calibrated using Fe and Al standards spanning concentrations of 1–50 mg/L, which were prepared from certified standard solutions sourced from Inorganic Ventures. The ICP-MS system was calibrated using REE, Th, U, Ti, and Mn standards spanning concentrations of 1–50 µg/L, which were prepared from certified standard solutions sourced from Inorganic Ventures. The uncertainty of ICP-MS and ICP-OES measurements was determined through repeated calibration and testing with standard solutions, resulting in an average relative standard deviation of 2% for REE, Th, U, Ti, and Mn concentrations and 5% for Fe and Al measurements.
The solid precipitate from the centre point replicate runs but at pH 5.5 (57.5 °C, H2O2 = 0.5 mL, pH 5.5) were characterized using Raman spectroscopy and SEM-EDS. For the Raman analysis, the powder was first homogenized by stirring. A portion of the homogenized powder was then placed onto a glass slide and subjected to analysis using a Renishaw InVia Raman Spectrometer. This spectrometer utilized a 633 nm wavelength laser. During analysis, the sample was exposed to the laser at 50% power, with an exposure time of 20 s. A 50× objective was employed to focus the laser onto the surface, resulting in a spot size of approximately 1 μm in diameter. Raman spectroscopy, a technique that employs lasers to probe the molecular vibrations of materials, was used to gain insights into the structural composition of the particles. Spectra collected through this method were then examined to confirm the mineralogical makeup of the particles.
For the SEM-EDX analysis, the powdered sample underwent the following process: it was affixed to a carbon adhesive, received a thin indium coating to minimize potential sample charging artifacts, and then was subjected to examination using either a Hitachi SU3500 Variable Pressure SEM, in conjunction with an Oxford AZtec X-Max50 SDD X-ray analyzer, or a Hitachi SU3900 Variable Pressure SEM, combined with an Oxford ULTIM Max65 SDD X-ray analyzer.
EDX spectroscopy, a semi-quantitative technique, was employed for this analysis. It has the capability to detect elements spanning from carbon to uranium, probing the sample to a depth of a few microns and offering a detection limit of approximately 0.5 wt%. The SEM images were captured using a back-scattered electron (BSE) detector. These BSE images reveal variations in the distribution of elements with differing atomic numbers. In these images, elements with lower atomic numbers, like carbon, appear darker, while elements with higher atomic numbers, such as Fe, appear brighter. An accelerating voltage of either 15 kV or 25 kV was used for these analyses.
Images and EDX spectra were collected during this process to identify the chemical composition of the powder. It is important to note that the EDX lines for various REEs (e.g., Ce, Pr, Sm, Nd, Gd) overlap with the EDX lines for Fe, potentially affecting the quantification.
The saturating solid phase at pH 3 was subjected to X-ray diffraction (XRD) analysis. This analysis was conducted utilizing a Rigaku D/max 2500 rotating anode powder diffractometer, which employed monochromatic Cu Kα radiation. The instrument’s operational conditions were configured as follows: Two theta angular range: 5–70°, step size: 0.020°, scan speed: 1° per min, voltage (kV) of 40, and current (mA) of 200. To identify the present phases in the sample, the analysis was performed using JADE version 9.3, which was integrated with the ICSD (Inorganic Crystal Structure Database) and ICDD (International Centre for Diffraction Data) diffraction databases.
Data availability
The data will be made available upon request. Please contact the corresponding author, Dr. Gisele Azimi (g.azimi@utoronto.ca).
References
Judge, W. D. & Azimi, G. Recent progress in impurity removal during rare Earth element processing: A review. Hydrometallurgy 196, 105435 (2020).
Anawati, J. & Azimi, G. Separation of rare Earth elements from a South American ionic clay lixivium by sequential precipitation. Hydrometallurgy 213, 105946 (2022).
Zhang, J., Zhao, B. & Schreiner, B. Seperation Hydrometallurgy of Rare Earth Elements (Springer International Publishing, 2016). https://doi.org/10.1007/978-3-319-28235-0
Ritcey, G. M. Crud in solvent extraction processing — a review of causes and treatment. Hydrometallurgy 5, 97–107 (1980).
Davris, P. et al. Leaching of rare Earth elements from eudialyte concentrate by suppressing silica gel formation. Min. Eng. 108, 115–122 (2017).
Dutrizac, J. E. The behaviour of the rare Earth elements during gypsum (CaSO4·2H2O) precipitation. Hydrometallurgy 174, 38–46 (2017).
Azimi, G., Papangelakis, V. G. & Dutrizac, J. E. Modelling of calcium sulphate solubility in concentrated multi-component sulphate solutions. Fluid Phase Equilib. 260, 300–315 (2007).
Verbaan, N., Bradley, K., Brown, J. & Mackie, S. A review of hydrometallurgical flowsheets considered in current REE projects, in: Symposium on Critical and Strategic Materials 3. British Columbia Geological Survey Paper. 147–162 (2015).
Xia, C. & Griffith, W. Direct oxalate precipitation for rare earth elements recovery. (2018).
Zuo, X., Liu, Q., Deng, X., Liu, K. & Chi, Y. Preparation method of crystalline rare-earth carbonate. (2014).
Eisele, J. A. & Bauer, D. J. Recovery and separation of rare-earth elements, barium, and strontium from bastnäsite with sulfuric acid. U.S. Bureau of Mines Report of Investigations 7990, U.S. Dept. of the Interior: Washington. (1974).
Gschneidner, K. A. & Daane, J. A. H. Physical Metallurgy, Handbook on the Physics and Chemistry of Rare Earths (North-Holland, 1988).
Fidler, J. & Schrefl, T. Overview of Nd–Fe–B magnets and coercivity (invited). J. Appl. Phys. 79, 5029–5034 (1996).
Salazar, D. et al. Effect of Nb and Cu on the crystallization behavior of under-stoichiometric Nd–Fe–B alloys. J. Phys. D Appl. Phys. 50, 015305 (2016).
van Mens, R. Ternary phase studies of Nd-Fe-X where X = C, Si, Pb, Sn. J. Magn. Magn. Mater. 61, 24–28 (1986).
Zhang, Z., Jia, Q. & Liao, W. Chapter 277 - Progress in the Separation Processes for Rare Earth Resources. in Including Actinides (eds. Bünzli, J.-C. & Pecharsky, V. K. B. T.-H. on the P. and C. of R. E.) 48, 287–376 (Elsevier, 2015).
Dreisinger, D., Andrews, G., Verbaan, N., Johnson, M. & Bourricaudy, E. in The Demonstration Pilot Plant Results for the Search Minerals Direction Extraction Process for Rare Earth Recovery BT - Rare Metal Technology 2018. 3–14 (eds Kim, H.) (Springer International Publishing, 2018).
Das, G. et al. Rare Earth sulfates in aqueous systems: thermodynamic modeling of binary and multicomponent systems over wide concentration and temperature ranges. J. Chem. Thermodyn. 131, 49–79 (2019).
Bates, R. G. & MacAskill, J. B. Standard potential of the silver-silver chloride electrode. Pure Appl. Chem. 50, 1701–1706 (1978).
Derringer, G. & Suich, R. Simultaneous optimization of several response variables. J. Qual. Technol. 12, 214–219 (1980).
Montgomery, D. C. Design and Analysis of Experiments (Wiley, 2017).
Turton, R., Bailie, R. C., Whiting, W. B. & Shaeiwitz, J. A. Analysis, synthesis and design of chemical processes (Pearson Education, 2008).
Townsend, W., Faber, G. & AssessCCUS Capital Equipment Cost Database. (2021). Available at: https://assessccus.globalco2initiative.org/.
The Chemical Engineering Plant Cost Index. (2024).
Price, W. A. Prediction manual for drainage chemistry from sulphidic geologic materials, MEND Report 1.20.1. (2009).
Findeiß, M. & Schäffer, A. Fate and environmental impact of thorium residues during rare Earth processing. J. Sustain. Metall. 3, 179–189 (2017).
Findeiß, M. Effects of Radioactive By-products Along with the Extraction of Rare Earth Elements on Aquatic and Terrestrial OrganismsAachen (2016).
Talan, D. & Huang, Q. A review of environmental aspect of rare Earth element extraction processes and solution purification techniques. Min. Eng. 179, 107430 (2022).
Government of Canada. Guidelines for the disposal of radioactive materials. (2002).
Jain, R. K., Cui, Z. & Domen, J. K. Environmental Impact of Mining and Mineral Processing Management, Monitoring, and Auditing Strategies. https://doi.org/10.1016/C2014-0-05174-X (2016).
Acknowledgements
The authors acknowledge the financial support provided by the CanmetMINING of Natural Resource Canada through the Critical Minerals Research, Development & Demonstration program. The authors also acknowledge OLI Systems Inc. for providing access to OLI Studio. The authors acknowledge Mr. Spencer Cunningham for help with ICP-MS and Ms. Baian Almusned for help with Raman Spectroscopy and SEM-EDX analysis. We gratefully acknowledge the invaluable assistance of the environmental specialists at CanmetMINING, Natural Resources Canada, Dr. Richard Goulet, and Albert Stoffers, for their expert review of the environmental implications section and their insightful contributions.
Author information
Authors and Affiliations
Contributions
S.C.: Investigation; Methodology; Data curation; Formal analysis; Writing - review & editing. M. E. S.: Investigation; Methodology; Data curation; Formal analysis; Visualization; Validation; Writing - review & editing. T.S.: Investigation; Writing - review & editing. G.A.: Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Supervision; Validation; Visualization; Writing - original draft; and Writing - review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, S., Sauber, M.E., Sun, T. et al. Iron, aluminum, and thorium impurity removal from a rare earth element pregnant leach solution using magnesium carbonate. Sci Rep 15, 15951 (2025). https://doi.org/10.1038/s41598-025-00847-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-00847-0
Keywords
This article is cited by
-
Selective Cerium Separation from Rare Earth Chloride Solutions Using Calcium Hypochlorite: Thermodynamics and Process Optimization
Journal of Sustainable Metallurgy (2026)
