
Abstract
The study of aquatic ecosystems as hydrologic networks requires a metacommunity framework, in which local bacterioplankton communities are interconnected via water movement across the landscape. While evidence suggests that a balance between selection (deterministic) and dispersal (stochastic) processes drives bacterioplankton community composition, little attention has been given to examining the impact of the surrounding terrestrial landscape on these aquatic communities. In this study, we assessed the relative importance of terrestrial landscape composition, local aquatic conditions, and dendritic distance for explaining the variation in aquatic bacterial communities along a small Amazonian hydrological network. Land cover composition had more explanatory power than local aquatic conditions and dendritic distance in explaining bacterial community dissimilarity, particularly in headwaters. Additionally, we observed significant differences in bacterioplankton composition between oxbow lakes and streams, which substantially differed according to local aquatic conditions. These findings underscore the critical role of both local aquatic conditions and terrestrial heterogeneity along the aquatic network in determining bacterioplankton communities. Finally, we identified shifts in the abundance of some bacterial classes associated with terrestrial heterogeneity and aquatic conditions. Our results highlight the crucial role of the terrestrial surrounding landscape as an element that interacts with hydrology and local aquatic conditions in shaping bacterioplankton community structure. Our results contribute to improve our understanding of the processes that drive the dynamics of freshwater bacterioplankton metacommunities and establish clear links between freshwater and surrounding terrestrial environments. These findings advance our knowledge of processes at the land–water interface and may inspire innovative approaches to ecosystem conservation and biomonitoring strategies that include bacterioplankton.
Similar content being viewed by others

Introduction
Freshwater ecosystems are biogeochemical reactors that drive carbon processing and serve as central elements for the flux of dissolved organic matter (DOM) from land to the ocean1,2,3. Within this framework, bacterioplankton-mediated biogeochemical transformations play critical roles in aquatic food webs. Planktonic bacteria process DOM, a larger pool of organic carbon in aquatic ecosystems, transferring the carbon contained within their biomass to higher aquatic trophic levels4. Additionally, carbon can be transferred from DOM to the CO2 pool through bacterial respiration5,6,7. Despite their vital role in biogeochemical cycles and their rapid response to changes in environmental conditions, bacterioplankton are rarely included in monitoring and conservation strategies8,9. This exclusion is partly due to the unpredictable nature of bacterial communities responses to environmental conditions, which makes it difficult to empirically link their taxonomic composition to ecosystem function10. Although the biogeographical patterns of bacterioplankton communities in lakes and rivers imply that their distribution is not random11, we are only beginning to unravel how different factors and ecological processes shape the assembly of these communities.
Selection associated with changes in local aquatic conditions (e.g. pH, pollutants, and biotic interactions), is often attributed as the main factor determining variation in the composition of aquatic bacterial communities12,13,14. The species sorting hypothesis invokes the environment as a filter that selects taxa that grow under specific conditions15. Consequently, sites with similar environments are expected to have similar communities dominated by taxa that are well adapted to such conditions. Other ecological processes, such as dispersal, may increase similarity among communities through the flow of taxa or cells from sites where they are thriving (i.e. high rates of dispersal), which may overwhelm the effect of selection (i.e. mass effects). Given the unidirectional flow of water within draining basins, aquatic ecosystems are increasingly studied within the context of their aquatic networks rather than as a collection of isolated systems16,17. This perspective has expanded the recognition of bacterial metacommunities, which are a group of local communities connected by dispersal mediated by the directional flow of water within aquatic networks15,18,19. However, the observed decrease in bacterioplankton richness along the aquatic network and the presence of dominant soil taxa in the water have highlighted the importance of incorporating terrestrial ecosystems in understanding freshwater bacterioplankton dynamics14,20,21,22,23. This inclusion leads to a more complex conceptual framework, which includes aquatic and terrestrial factors that modulate dispersal and selection in the bacterioplankton metacommunity assembly (Fig. 1).

Conceptual diagram illustrating different perspectives on the relative importance of ecological processes in bacterioplankton metacommunity assembly along hydrological network. The colored circles represent local aquatic conditions. The arrows represent dispersal (width reflects relative importance and, color represents ecosystem connectivity: aquatic in white and soil-aquatic in black). The balance between ecological processes (selection and dispersal) reflects species sorting or mass effect change along the hydrological network. Font size corresponds to relative importance of each process along the network, and color is related to the potential source (water in blue and soil in red). The first perspective (A) predicts that (i) headwater communities are regulated by selection because they are isolated, thus community assembly is described by species sorting, whereas (ii) downstream communities are described by mass effects due to a surplus of dispersal. Therefore, β diversity is expected to decrease with Strahler order. The second (B) and third (C) perspectives add the terrestrial connections of aquatic network with the surrounding soil (dotted rectangle). However, B is a special case of C. B proposes that i) community assemblages in headwaters are predominantly shaped by mass effects, driven by the high dispersal of soil taxa into the water channel (this perspective overlooks terrestrial heterogeneity, assuming that dispersed soil bacteria are uniform across different headwaters), and ii) downstream communities are increasingly influenced by selection exerted by similar local aquatic conditions due to higher water retention time. Hence, β diversity is expected to be more similar in communities from low- and high-order Strahler sites. C proposes an increase in headwater selection due to local environmental changes resulting from landscape heterogeneity (colored patches in background). Therefore, β diversity patterns along the network are expected to be modulated by the degree of landscape heterogeneity (boxplot boxes size).
The network position hypothesis (NPH), which is frequently used in freshwater ecology, highlights the importance of position within the network, considering headwaters as isolated sites and downstream reaches as sites more connected (Fig. 1A)17,24,25. From this perspective, the strength of dispersal increases progressively toward downstream sites; thus, selection is expected to dominate headwater ecosystems and decrease its influence downstream25. On the other hand, empirical evidence, particularly for bacterioplankton (Fig. 1B), recognizes the role of hydrology (i.e., water residence time) and soil-water connectivity as factors that modulate the relative importance of local selection and soil-derived dispersal along aquatic networks20,22. Unlike NPH, mass effects (from soil bacteria communities) are expected to shape community structure in upstream sites, whereas species sorting should increase downstream. Both perspectives recognize variations in aquatic conditions across the network, and the second identifies a contribution from terrestrial taxa, but neither explicitly states the role of the surrounding terrestrial conditions, which may affect the structure of aquatic bacterial communities (Fig. 1C). Although aquatic microbial ecologists currently recognize the landscape as the physical template occupied by the metacommunity, the influence of terrestrial conditions in shaping bacterioplankton metacommunity remains poorly understood26,27.
An approach that integrates terrestrial landscape elements into freshwater microbial community ecology requires the measurement of landscape structure. To evaluate the relative importance of local aquatic conditions, terrestrial landscape composition and connectivity in structuring the bacterioplankton metacommunity along the network, we sampled bacterial communities across a small aquatic network in the upper part of the Guaviare River watershed and immersed in the natural reserve “Serranía de la Lindosa”. This area, located within the Colombian Amazon region, is characterized by several rocky outcrops that generate high levels of geological and floristic heterogeneity within a small spatial extent28,29, whereas other regionally variable factors (i.e. climate) are similar. This unique configuration allowed us to examine how landscape, local aquatic conditions, and within-network-connectivity change along aquatic networks.
Materials and methods

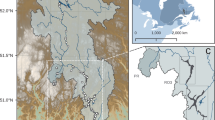
Study site – Serranía de La Lindosa, each point corresponds to a sampling site (A) Hydrological network (intense blue lines), with main caños, as part of Guaviare River basin. The color of points is related to the four sub-basins studied (AB: Agua-Bonita, AY: Arenales-Yamús, CL: Conuco-Lagunas, DLN: Delicias-LagunaNegra). (B) Land cover associated with hydrological network (colors in background) and, type of systems included in the network (shape of points: circles - oxbow lakes and triangles - streams).
Study sites and sampling
The study site was Serranía de La Lindosa in Guaviare, Colombia. This region is characterized by a few rocky outcrops of the Colombian Amazon covering an area of ~ 120 km2. Due to its ecological importance, this region is protected as part of Distrito de Manejo Integrado Ariari-Guayabero29. Its territorial extension houses some small streams that are part of the Guaviare river basin such as caño Negro, caño El Retiro, caño La María, caño Agua Bonita and caño Yamús and oxbow lakes (“madreviejas”) called Laguna Negra, Laguna Chica, and Laguna Grande29. According to stream drainage, we identified four sub-basins (based on an elevation map of the region – https://asf.alaska.edu/datasets/daac/alos-palsar/, resolution: 12.5 m): Delicias-Laguna Negra (DLN), Conuco-Lagunas (CL), Arenales-Yamús (AY), and Agua Bonita (AB). Sampling sites were chosen based on the observed differences in the surrounding land cover, composition and structure, as well as on its accessibility. In total, 40 sites were selected: 13 within DLN, 15 within the CL, and 6 within the AY and AB (Fig. 2).
The survey was conducted in June 2021. In order to describe the local aquatic conditions, we sampled 1 L of the surface water and stored it at ~ 4 °C for water chemistry analysis and measured water in situ parameters in each site. To analyze bacterial community composition, 1 L of the surface water was concentrated through a polycarbonate filter (0.22 μm pore size). In order to identify soil-derived bacterial taxa (i.e. taxa shared between soil and water bacterial communities), we collected 250 g of composite topsoil sample from the surrounding area at each sampling site. The polycarbonate filters and the soil samples were stored at -80 °C until DNA extraction. Finally, to characterize the landscape composition, we registered the geographic location of each sample site.
Landscape and local aquatic data
Landscape information was provided by the Instituto Amazónico de Investigaciones Científicas – SINCHI, which generated a land cover classification for the Amazon region (including Serranía de La Lindosa) in 2020, following the Corine Land Cover (CLC) classification (https://datos.siatac.co/pages/coberturas). For each site, we delimited circular buffers with a 30-m radius to calculate the percentage of each land cover class. This radius was chosen by considering the digital elevation model and selecting only the region draining into the stream of interest. Consequently, the samples closest to each other (~ 30 m) had the same buffer. Land cover proportion was calculated using QGIS 3.28 with the LecoS plugin (Landscape Ecology Statistics). We selected level 3 of the CLC classification for the analyses (categories included within the study region can be observed in Fig. 2 and Supplementary file 1-Table S1). Additionally, we measured dendritic distance (i.e. distance between sampling points considering the water flow) in QGIS.
The physical and chemical analysis of water samples included the quantification of total acidity (TAc), organic matter percentage (perOM), biochemical oxygen demand (BOD - mg/L), chemical oxygen demand (COD – mg/L), total suspended solids (TSS - mg/L), total solids (TS – mg/L), and concentrations of total organic carbon (TOC - mg/L), calcium (Ca – mg/L), chlorine (Cl – mg/L), total phosphorus (TP – mg/L), total iron (TFe – mg/L), magnesium (Mg – mg/L), nitrate (NO3 – mg/L), nitrite (NO2 – mg/L), sulfate (SO4 – mg/L), and sulfur (Sul – mg/L). In addition to these variables, environmental characterization incorporated the variables measured in situ: temperature (Temp - °C), pH, percentage of oxygen saturation (perDO), conductivity (Cond - µS/cm), total dissolved solids (TDS – mg/L), redox potential (roP - mV), and turbidity (Turb - NTU) using a multi-parameter sonde. Although we quantified 23 physicochemical variables, only 14 were used to describe the local aquatic environment. We excluded some variables from the study because their measurements were below the detection limit (NO3, NO2, and Sul) or because they were highly correlated with other variables. We considered this set of variables to refer to the local aquatic environment.
Aquatic bacterial community composition
The bacterial community structure was generated by sequencing of the 16 S rRNA gene of the aquatic and soil DNA samples. First, bacterial community DNA was extracted from the stored filter using the QIAGEN DNeasyPowerWater kit and from the stored soil samples using QIAGEN DNeasyPowerSoil kit, both according to the manufacturer’s instructions. The quality and quantity of extracted DNA were measured using a NanoDrop OneC spectrophotometer and a Qubit fluorometer. DNA integrity was evaluated via electrophoresis (data not shown). Then, the hypervariable region V3–V4 of the 16 S rRNA gene were amplified and sequenced in Macrogen, Korea. Amplification was achieved using the primers 341 F and 805R30 and, the sequencing was performed using an Illumina MiSeq platform with a paired-end protocol (300 × 300pb). Finally, the raw sequences were processed using the QIIME2 suite (version 2019.4)31. To obtain high-quality sequences, we used the DADA2 method32, and the deblur plugin was used for OTU aggregation with a 99% similarity threshold. Taxonomic assignment of each OTU was performed using the SILVA132 database33. We filtered and removed the sequences from the Archaea kingdom and we rarefied the aquatic bacterial community dataset to 10.032 reads per sample. We used the raw soil table in order to increase the probability of detecting any OTU shared between aquatic and soil samples.
Statistical analysis
Terrestrial landscape analysis was based on the land cover composition obtained from a 30-m circular buffer at each site, and heterogeneity was measured using the Shannon index. We also described the variation in local aquatic environmental conditions using principal component analysis (PCA) and used a redundancy analysis (RDA) to correlate the aquatic conditions with the land cover composition. As the number of response variables (i.e. local aquatic conditions)14 exceeded the number of explanatory variables (i.e. land cover proportions)8, we reduced the local aquatic conditions to 8 based on collinearity among the variables observed in PCA. We used non-metric multidimensional scaling analysis (NMDS) based on compositional differences among sites, using the Bray-Curtis index, to determine bacterial taxonomic variation across sites. For subsequent analysis, we standardized the datasets as follows: (i) landscape composition was transformed with the “Hellinger” option from the decostand function from the vegan v. 2.6-4 package, (ii) local aquatic conditions were standardized with the scale function, and (iii) OTU abundances were log – transformed.
To evaluate the relative importance of environmental factors (i.e. land cover composition and local aquatic conditions) and connectivity (i.e. dendritic distance) in explaining variations in bacterial community composition, we applied a Generalized Dissimilarity Model (GDM) using the gdm v. 1.5.0-9.1 package34. We used (i) dendritic distance and differences of NMDS scores between sites from (ii) land cover composition and (iii) local aquatic conditions ordinations, as predictors of bacterial community dissimilarity within the GDM model. The weight of each predictor was assessed using the gdm.partition.deviance() function. In addition, we quantified the number of shared OTUs between sites, also as proxy to connectivity between communities within network. Finally, we explored the influence of the terrestrial landscape heterogeneity on bacterial community composition via differential abundance analysis using the ANCOM-BC method35 - ancombc v.1.4.0 package. For this analysis, we considered landscape heterogeneity clustering to probe the differential abundance of bacterial classes and used a canonical analysis of principal coordinates (CAP) to demonstrate the relationship between the abundance of selected classes and the dissimilarity of bacterial communities. For all analyses, we used R software (version 4.0.2) and the statistical significance at a p-value < 0.05.
Results
Terrestrial landscape heterogeneity
The terrestrial landscape surrounding the aquatic sites was variable and heterogeneous although grassland and pastureland usually dominated the landscape (Fig. 3a). In addition, we identified three site clusters with different levels of landscape heterogeneity: High, Medium and Low (Supplementary file 1 – Figure S1).

Terrestrial landscape heterogeneity surrounding aquatic sites in the Serranía de La Lindosa. (a) Comparison of land cover composition across sites by sub-basin (AB, AY, CL and DLN); colors represent different types of land cover. (b) PCA bi-plot of sites according to land cover proportions. All variables (represented by arrows) are expressed as percentages (per).
Variation in local aquatic conditions
Variation in local aquatic conditions was more related to the type of system and land cover composition than to sub-basin membership (Fig. 4). PC1 clearly separated oxbow lakes and streams, with the former having higher conductivity (17 µS/cm), temperatures (30.87 °C), and pHs (pH 6) and lower oxygen levels than streams. Meanwhile, PC2 separated some sites of the AB and YL sub-basins associated with high BOD and turbidity or sulfates and acidity (Fig. 4a). Other conditions, such as Fe, P, and Ca concentration showed low loading and explained little variation along PC1 and PC2 components (Supplementary file 1 - Table S2). Finally, we observed correlations between: (i) the proportion of grassland and secondary vegetation cover and the concentrations of SO4, Mg, and OD; (ii) the proportion of fragmented forest and TFe concentration; and (iii) the proportion of water surface area and conductivity (Fig. 4b).

Description of the local aquatic conditions in the hydrological network of Serranía de La Lindosa. (a) PCA bi-plot of sites colored by sub-basin (AB, AY, CL, DLN). (b) RDA model (p < 0.05) explaining changes in environmental variables (red arrows) according to land cover proportions (black arrows).
Variation in the bacterial community composition (BCC) within hydrological networks
In total, we obtained 8688 OTUs with their taxonomic assignments after rarefaction. We observed differences in the taxonomic composition of the bacterial communities among the aquatic sites (Fig. 5). There was a clear segregation between oxbow lakes and streams (ANOSIMType: p < 0.01, R = 0.82) although, the bacterial community composition tended to be more similar between lakes than between streams. In contrast, we found no evidence of separation in BCC between sub-basins (ANOSIMsubBasin: p = 0.23, R = 0.36) (Fig. 5a). Additionally, we identified a declining pattern of bacterial community dissimilarity with increasing hydrological - Strahler - order (from upstream to downstream sites in the network) (Fig. 5b).
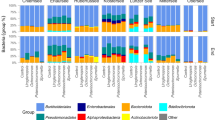
In general, Gammaproteobacteria was the most dominant class with > 25% relative abundance across sites (Supplementary file 1 – Figure S2). Taxonomic differences in bacterial communities between oxbow lakes and streams were related to an increase in the relative abundance of: Parcubacteria and Acidobacteriia toward lower river orders and Actinobacteria, Oxyphotobacteria, and Acidimicrobiia toward lakes.

Beta diversity of aquatic bacterial communities in the hydrological network of Serranía de La Lindosa. (a) NMDS of BCC using the Bray-Cutis index. (b) Bacterial community dissimilarity among sites with same hydrological Strahler order. OL denotes the oxbow lakes.
Land cover as a driver of bacterioplankton community composition
Our approach allowed us to determine the relative importance of environmental factors (i.e. land cover and local aquatic conditions) and connectivity (i.e. dendritic distance) in the explanation of bacterial community dissimilarity. Considering that we identified a clear separation of BCC between lakes and streams (Fig. 5a), we present both GDM results for lakes and streams (i.e. all sites) and those that excluded lakes (i.e. only streams). The two GDM models were statistically significant and explained 49.02% (all sites – Fig. 6a) and 52.62% (only stream sites – Fig. 6b) of the deviance. In the model including all sites, the interaction between aquatic conditions and land cover composition explained the largest deviance (10.65%), followed by land cover composition alone (10.05%). In this case, the dendritic distance was the least important factor (2.64% of the deviance). In contrast, in the model that only included stream sites, land cover composition alone explained most of the deviance (14.34%), followed by the intersection between land cover composition and dendritic distance (12.96%), and the least important factor was aquatic conditions alone.

Deviance partition in GDM model for BCC from Serranía de La Lindosa. The values correspond to the percentage of deviance explained. (a) All sites (b) Only stream sites.
Discussion
We analyzed the changes in bacterioplankton composition in relation to dendritic distance, local aquatic conditions, and terrestrial landscape across different sub-basins located on the upper part of the Guaviare River in the Colombian Amazon. Our results support the notion that environmental factors (i.e. land cover composition and local aquatic conditions) rather than connectivity-related factors (i.e. dendritic distance) shape the bacterioplankton community composition within this aquatic network. Although the relative influence of local aquatic conditions and land cover composition on bacterioplankton community structure changed along the network, the magnitude of their deviance was always higher than that of connectivity (Fig. 6). In the lotic portion of the aquatic network, the terrestrial landscape was the main driver of the bacterioplankton community structure, whereas in complete network, aquatic conditions and their interaction with the terrestrial landscape explained the differences in bacterioplankton communities. This work highlights the role of terrestrial landscape heterogeneity on bacterioplankton dissimilarity through the modulation of local aquatic conditions (Figs. 4b and 6)36. Also, it adds landscape heterogeneity (Fig. 1C) to previous evidence of the importance of hydrology (i.e. water residence time – WRT) and water chemistry (Scenario Fig. 1B) in the structuring of freshwater bacterioplankton communities14,22,37.
We show a decreasing pattern in community dissimilarities from upstream to downstream sites (Fig. 5b), a pattern that fits the expectation of scenarios A and C of our perspectives in Fig. 1. However, GDM revealed that the beta diversity pattern was better predicted by the landscape and local aquatic environment than by dendritic distance (Fig. 6). The last implies that the lower compositional dissimilarities observed downstream cannot be explained only by stronger dispersal, as suggested by NPH (Fig. 1A); in fact, we found that only between 46 and 257 OTUs persisted along each sub-basin (Supplementary file 1 - Table S4 and Figures S3 and S4). Instead of that, this decreasing pattern may be explained by the combination of longer WRTs with more similar local environments at downstream sites14 and larger differences in the aquatic environmental conditions toward the upper parts of the network16, both of which favor selection processes. The analysis of aquatic environmental conditions also indicated that these conditions are related to changes in land cover composition (Fig. 4b). Therefore, our analysis of the GDM model revealed that land cover composition seems to capture environmental or spatial changes in unmeasured environmental gradients associated with differences in bacterial community composition between upstream and downstream sites. This finding highlights the importance of including landscape variables and opens a new avenue for exploring the hierarchical relationships between regional (e.g. climate and land cover) and local environmental variables to better understand bacterioplankton dynamics in the aquatic networks38.
Regarding the role of hydrology and soil-derived bacteria in structuring aquatic metacommunities (Fig. 1B), previous studies have indicated that water residence time interacts with local environmental conditions to drive bacterial community composition14,37. Although we did not explicitly measure water residence time, we observed the largest divergence in bacterial community composition between streams and oxbow lakes (Fig. 5a; Supplementary file 1- Figure S2). This pattern was related to a lower prediction power of local aquatic conditions on bacterial community composition in the GDM model that excludes oxbow lakes (Fig. 6b). We argue that this observation is related to an increasing relative importance of environmental selection toward downstream sites10,22, associated with the largest differences in local aquatic conditions between stream and lakes (Fig. 4a). These differences may impose spatially variable selection pressures resulting in distinct bacterial communities toward downstream sites21,37. On the other hand, we expected an increasing relative importance of dispersal of soil-derived taxa (i.e. mass effects) toward upstream sites (Fig. 1B) but measuring this process may be a challenging task. However, we found a low number of shared OTUs between aquatic and surrounding terrestrial samples along the whole network (329 OTUs; ~5% of total soil community – Supplementary file 1-Figure S5). The last indicates that dispersal of soil-derived bacteria into aquatic environments is occurring, although our data seem not enough to match the expected numbers to support the mass effects previously proposed10,20,22,23. We acknowledge that our sampling design and sequencing depth (about 10.000/sample) may not be optimal for capturing the rarest portion of the bacterial communities. Nonetheless, soil-water interface is a strong environmental filter that prevents most taxa from reaching the aquatic environment22,23, which may reconcile the expected large flow of bacteria from soil to water with the small proportion of shared OTUs shown here. Regardless of these limitations, the larger deviance explained by dendritic distance in the only-streams GDM model (Fig. 6) suggests that dispersal with the network may be relatively more important upstream than in downstream sites. This is partially consistent with the expectations of our second scenario (Fig. 1B), which proposes a gradual change from upstream sites dominated by mass effects10,22 to downstream sites dominated by species sorting.
The influence of terrestrial landscape on bacterioplankton community composition has been largely neglected39 but recently started to be examined by a few authors. The conclusions of these studies have not always been consistent40,41,42, because some of them still emphasize the dominance of local aquatic conditions whereas others highlight the role of the terrestrial landscape. We attribute these contrasting results to differences in the spatial extent (i.e. draining areas) and to the position of the sampling sites within the networks (e.g. upstream or downstream), which represent partial views of the processes driving bacterial communities in aquatic networks and therefore may influence the relative role of landscape. In this regard, we decided to study a network that includes aquatic systems with low and high WRT (i.e. streams and lakes), with a known variable geological and floristic heterogeneity29, which is unusual for such a small area (120 km2). The increase in the deviance explained by land cover composition in the only-stream sites compared to the all-sites GDM (Fig. 6) suggests that the relative role of the landscape decreases toward downstream sites, regardless of the heterogeneity of the landscape. The last, probably due to changes in the water: soil ratio along the network, which is related to the interchange and the dilution of whatever is moving from soil to water. However, we acknowledge that how the bacterial community responds to the influence of landscape along the network may be modulated by how heterogeneous the landscape is. In fact, the landscape heterogeneity did not increase or decrease with the flow of water in our dataset. Instead, we found that a small portion of bacterial taxa (22 out 87 Classes) were actually changing in abundance, as a response to our groups of landscape heterogeneity (High, Medium and Low in Fig. 3b) (Supplementary file 1 – Figure S6 and Table S3), which contained sites from different sub-basins (Supplementary file 1- Figure S1). Thus, although we added evidence of how hydrological and environmental gradients influence bacterioplankton community changes along the aquatic networks, we also showed that they seem to be responding to landscape heterogeneity and, in general, to how the landscape is structured across space.
Conclusion
In this study, we highlight the role of landscape in the structuring of aquatic bacterial communities, and how this role may change along aquatic networks. We propose that landscape composition captures unmeasured aquatic environmental gradients that drive changes in bacterial community composition in aquatic networks. These aquatic environmental gradients may result from the flow of water across the landscape, which gather and convey several soil properties that promote selection of bacterial communities within aquatic ecosystems. The relative importance of these landscape-mediated properties vary depending on the position within the network, in a close relationship with changes in hydrology. This landscape perspective may improve our understanding of bacterial community patterns and microbes-mediated biogeochemical processes in aquatic ecosystems. This approach may also promote the use of this biological component in monitoring processes that may extend beyond the boundaries of aquatic ecosystems.
Data availability
Sequence data that support the findings of this study have been deposited in the National Center for Biotechnology Information -NCBI- with the code PRJNA1146926.
Abbreviations
- SINCHI:
-
Instituto Amazónico de Investigaciones Científicas
- OTU:
-
Operational taxonomic unit
- GDM:
-
Generalized dissimilarity model
- ANCOMB-BC:
-
Analysis of composition of microbiomes with bias correction
- BCC:
-
Bacterial community composition
- DOM:
-
Dissolved organic matter
- NPH:
-
Network position hypothesis
References
Battin, T. J. et al. River ecosystem metabolism and carbon biogeochemistry in a changing world. Nature 613(7944), 449–459. https://doi.org/10.1038/s41586-022-05500-8 (2023).
Bernhardt, E. S. et al. The metabolic regimes of flowing waters. Limnol. Oceanogr. 63, S99–S118 (2018).
Cole, J. J. et al. Plumbing the global carbon cycle: Integrating inland waters into the terrestrial carbon budget. Ecosystems 10(1), 171–184 (2007).
Cole, J. J., Findlay, S. & Pace, M. L. Bacterial production in fresh and saltwater ecosystems: A cross-system overview. Mar. Ecol. Progr. Ser. 43, 1–10. https://doi.org/10.3354/meps043001 (1988).
Berggren, M., Lapierre, J. F. & Del Giorgio, P. A. Magnitude and regulation of bacterioplankton respiratory quotient across freshwater environmental gradients. ISME J. 6(5), 984–993 (2012).
Graeber, D. et al. Going with the flow: Planktonic processing of dissolved organic carbon in streams. Sci. Total Environ. 625, 519–530. https://doi.org/10.1016/j.scitotenv.2017.12.285 (2018).
Azam, F. et al. The ecological role of water-column microbes in the sea. Mar. Ecol. Prog. Ser. 10, 257–263 (1983).
González, C., Vallarino, A., Pérez, C. & Low Pfeng, A. Bioindicadores: Guardianes de nuestro futuro ambiental. México, D.F. (2014).
Sagova-Mareckova, M. et al. Expanding ecological assessment by integrating microorganisms into routine freshwater biomonitoring. Water Res. 191, 116767. https://doi.org/10.1016/j.watres.2020.116767 (2021).
Ruiz-González, C. Microbial metacommunities: Dispersal and connectivity as key drivers of the diversity and function of aquatic microorganisms. Premi IEC de la Secció de Ciències Biològiques Pius Font i Quer de Ciències de la Vida 2020. 46 (2020).
Brendan Logue, J. & Lindström, E. S. Biogeography of Bacterioplankton in Inland Waters. Freshw. Rev. 1(1), 99–114 (2008).
Gao, F.-Z. et al. Anthropogenic activities and seasonal properties jointly drive the assemblage of bacterial communities in subtropical river basins. Sci. Total Environ. 806, 151476. https://doi.org/10.1016/j.scitotenv.2021.151476 (2022).
Liu, S. et al. Ecological insights into the disturbances in bacterioplankton communities due to emerging organic pollutants from different anthropogenic activities along an urban river. Sci. Total Environ. 796, 148973. https://doi.org/10.1016/j.scitotenv.2021.148973 (2021).
Niño-García, J. P., Ruiz-González, C. & Del Giorgio, P. A. Interactions between hydrology and water chemistry shape bacterioplankton biogeography across boreal freshwater networks. ISME J. 10(7), 1755–1766 (2016).
Leibold, M. A. et al. The metacommunity concept: A framework for multi-scale community ecology. Ecol. Lett. 7(7), 601–613 (2004).
Altermatt, F. Diversity in riverine metacommunities: A network perspective. Aquat. Ecol. 47(3), 365–377 (2013).
Brown, B. L. & Swan, C. M. Dendritic network structure constrains metacommunity properties in riverine ecosystems. J. Anim. Ecol. 79(3), 571–580 (2010).
Heino, J. et al. Metacommunity organisation, spatial extent and dispersal in aquatic systems: Patterns, processes and prospects. Freshw. Biol. 60(5), 845–869 (2015).
Ruiz-González, C., Niño-García, J. P., Kembel, S. W. & Del Giorgio, P. A. Identifying the core seed bank of a complex boreal bacterial metacommunity. ISME J. 11(9), 2012–2021 (2017).
Crump, B. C., Amaral-Zettler, L. A. & Kling, G. W. Microbial diversity in arctic freshwaters is structured by inoculation of microbes from soils. ISME J. 6(9), 1629–1639. https://doi.org/10.1038/ismej.2012.9 (2012).
Read, D. S. et al. Catchment-scale biogeography of riverine bacterioplankton. ISME J. 9(2), 516–526 (2015).
Ruiz-González, C., Niño-García, J. P. & del Giorgio, P. A. Terrestrial origin of bacterial communities in complex boreal freshwater networks. Ecol. Lett. 18(11), 1198–1206 (2015).
Stadler, M. & del Giorgio, P. A. Terrestrial connectivity, upstream aquatic history and seasonality shape bacterial community assembly within a large boreal aquatic network. ISME J. 16(4), 937–947 (2022).
Doretto, A., Piano, E. & Larson, C. E. The River Continuum Concept: Lessons from the past and perspectives for the future. NRC Res. Press 1864(July), 1853–1864 (2020).
Henriques-Silva, R. et al. A comprehensive examination of the network position hypothesis across multiple river metacommunities. Ecography (Cop) 42(2), 284–294 (2018).
Mony, C., Vandenkoornhuyse, P., Bohannan, B. J. M., Peay, K. & Leibold, M. A. A Landscape of opportunities for microbial ecology research. Front. Microbiol. https://doi.org/10.3389/fmicb.2020.561427 (2020).
Mony, C., Uroy, L., Khalfallah, F., Haddad, N. & Vandenkoornhuyse, P. Landscape connectivity for the invisibles. Ecography (Cop) 1–14 (2022)
Briceño Vanegas, G. & Gallego Herrera, J. C. Heterogeneidad espacial y oferta de hábitats en tres ecosistemas lóticos de la Serranía de La Lindosa Amazonía colombiana. Geográfica Digit. 20, 20–33 (2023).
Cárdenas López, D., Castaño Arboleda, N., Zubieta Vega, M. & Jaramillo Echeverry, M. Flora de las formaciones rocosas de la Serranía de La Lindosa. 2008. 162 paginas. Available from: http://hdl.handle.net/20.500.12324/18988
Herlemann, D. P. R. et al. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 5(10), 1571–1579. https://doi.org/10.1038/ismej.2011.41 (2011).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37(8), 852–857 (2019).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13(7), 581–583 (2016).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41(D1), 590–596 (2013).
Mokany, K., Ware, C., Woolley, S. N. C., Ferrier, S. & Fitzpatrick, S. A working guide to harnessing generalized dissimilarity modelling for biodiversity analysis and conservation assessment. Global Ecol. Biogeogr. 31(4), 802–821. https://doi.org/10.1111/geb.13459 (2022).
Lin, H. & Peddada, S. D. Analysis of compositions of microbiomes with bias correction. Nat. Commun. https://doi.org/10.1038/s41467-020-17041-7 (2020).
Erős, T. & Campbell Grant, E. H. Unifying research on the fragmentation of terrestrial and aquatic habitats: Patches, connectivity and the matrix in riverscapes. Freshw. Biol. 60(8), 1487–1501. https://doi.org/10.1111/fwb.12596 (2015).
Bambakidis, T. et al. Temperature, water travel time, and dissolved organic matter structure river microbial communities in a large temperate watershed. Limnol. Oceanogr. 69(7), 1618–1635. https://doi.org/10.1002/lno.12591 (2024).
Weigel, B. et al. Local eukaryotic and bacterial stream community assembly is shaped by regional land use effects. ISME Commun. https://doi.org/10.1038/s43705-023-00272-2 (2023).
Johnson, L. B. & Host, G. E. Recent developments in landscape approaches for the study of aquatic ecosystems. J. North Am. Benthol. Soc. 29(1), 41–66 (2010).
Shu, W. et al. Coupled effects of landscape structures and water chemistry on bacterioplankton communities at multi-spatial scales. Sci. Total Environ. 811, 151350. https://doi.org/10.1016/j.scitotenv.2021.151350 (2022).
Zhou, L., Chen, W., Sun, J., Liu, L. & Huang, X. Spatial variation in bacterioplankton communities in the Pearl River, South China: Impacts of land use and physicochemical factors. Microorganisms 8(6), 814. https://doi.org/10.3390/microorganisms8060814 (2020).
Zhao, J., Peng, W., Ding, M., Nie, M. & Huang, G. Effect of water chemistry, land use patterns, and geographic distances on the spatial distribution of bacterioplankton communities in an anthropogenically disturbed riverine ecosystem. Front. Microbiol. 12(May), 1–14 (2021).
Acknowledgements
We thank Clara María Arboleda Baena and Emiliano Pereira, members of the µSudAqua network, for discussion and feedback.
Funding
This study was supported by the Ministry of Environmental and Sustainable Development via a grant for Research on the conservation and sustainable use of biological, socioeconomic, and cultural diversity of the Colombian Amazon [Grant Number: BPIN 2017011000137], University of Antioquia (CODI 2021-41830 and CODI 2022–53597) and the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; Processes:20/03716-0 and 22/14160-9). Additionally, H.S. acknowledges continuous funding through Research Productivity Grants provided by the Conselho Nacional de Desenvolvimento Cientifico e Tecnológico – CNPq (Process: 303906/2021-9).
Author information
Authors and Affiliations
Contributions
Conceptualization: NDR, MCGC, JPNG; Sampling: MCGC, GC, MN, SS, JPGN; Lab work: NDR; Data analysis: NDR, JLP, JPNG; Funding acquisition: GC, MCGC, JPNG, HS; Writing: NDR, MCGC, JLP, JPNG, HS. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Díaz-Ruiz, N., Parra, J.L., García-Chaves, M.C. et al. Terrestrial landscape as the main driver of bacterioplankton communities along a hydrological network located in the Colombian Amazon. Sci Rep 15, 23706 (2025). https://doi.org/10.1038/s41598-025-01692-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-01692-x
