
Abstract
Bone defects are common orthopedic conditions, and due to the limited regenerative capacity of bone tissue, their repair remains a challenge in orthopedic surgery. Mesenchymal stem cells (MSCs) have demonstrated strong potential for osteogenic differentiation; however, their efficiency in vivo remains restricted, particularly in terms of differentiation and migration. Photobiomodulation (PBM), a non-invasive therapeutic technique, has shown great promise in promoting stem cell differentiation. In this study, we cultured human umbilical cord mesenchymal stem cells (hUCMSCs) in vitro and treated them with 635/808 nm laser light. We measured alkaline phosphatase (ALP) activity, mineralized nodule formation, and the expression of osteogenesis-related genes and factors after 7, 14, and 21 days. The results showed that PBM treatment significantly enhanced hUCMSC proliferation and osteogenic differentiation. The mechanism behind this effect involves PBM activating the upstream Akt signaling pathway, increasing P-Akt expression, and elevating reactive oxygen species (ROS) levels to induce mild oxidative stress. This process enhances ALP activity, mineralized nodule formation, and the expression of osteogenesis-related genes and factors, thus promoting the osteogenic differentiation of hUCMSCs.
Similar content being viewed by others

Introduction
Bone tissue defects are common complications resulting from trauma and diseases1. Despite the remarkable self-repair and regenerative capabilities of bone tissue, nearly 10% of fractures do not heal properly2,3. Due to the limitations of current treatments, such as bone grafting, innovative bone tissue engineering approaches, including cell inoculation, scaffold utilization, and cell activation, have emerged as promising strategies for treating bone defects4.
Mesenchymal stem cells (MSCs), multipotent cells derived from mesodermal layers and nerve sheaths5, possess the ability to migrate to injury sites, a crucial step in bone defect repair6. MSCs demonstrate multipotent differentiation abilities, particularly in osteogenesis and chondrogenesis7. Osteoblasts are critical for bone formation and progress through four stages: cell proliferation, extracellular matrix maturation, mineralization, and apoptosis. MSCs secrete paracrine factors that regulate the tissue microenvironment and modulate osteoclast activity, thereby maintaining a dynamic balance during bone formation8. Although there are currently a number of techniques to enhance MSC differentiation into osteoblasts, there continues to be interest in developing simple and noninvasive methods to further enhance osteoblastic differentiation and expedite the bone repair process, which is crucial in the clinical management of bone tissue defects .
Photobiomodulation (PBM) is a non-invasive therapeutic approach that employs red or near-infrared light within the range of 600-1,100 nm to exert an effect on tissues or cells, aiming to expedite and improve bone healing9. The 650-1,100 nm wavelength range is widely used in PBM because it is absorbed by cellular chromophores10. PBM promotes mitochondrial respiration, adenosine triphosphate (ATP) synthesis, and the regulation of reactive oxygen species (ROS) levels, enhancing osteoblast proliferation and differentiation, leading to bone healing. In addition, PBM stimulates inflammatory cells, promotes angiogenesis, and facilitates new bone formation11,12. Lee et al.. demonstrated that photobiomodulation therapy (PBMT) using 660 nm light significantly enhances the healing of cranial defects in a diabetic rat model, promoting osteogenic differentiation and bone regeneration13. In our previous study, we utilized human umbilical cord mesenchymal stem cells (hUCMSCs) as the seeding cells, seeded them onto a filipin/polycaprolactone scaffold, and subjected them to PBM treatment. The results indicated that PBM intervention significantly enhanced the expression of genes associated with mineralization and osteogenesis in hUCMSCs, thereby promoting osteogenic differentiation14. However, the absence of standardized guidelines for the use of PBM in bone regeneration has led to diverse treatment protocols with varying wavelengths, types, and parameters, resulting in inconsistent and sometimes contradictory results15,16,17,18. Furthermore, the specific substances modulated by PBM during bone healing and underlying intrinsic mechanisms remain unclear. Therefore, experimental studies are warranted to elucidate the effects of PBM on MSCs and to understand the regulatory mechanisms of osteoinduction, which will aid in the design of effective therapeutic programs.
MSCs can be isolated from various sources including bone marrow, umbilical cord, adipose tissue, and blood. Among these, hUCMSCs offer distinct advantages due to their ease of extraction and expansion, non-invasive collection, cost-effectiveness, low risk of infection, high proliferation capacity, and minimal immunogenicity19. These characteristics make hUCMSCs an ideal candidate for therapeutic applications.In this study, we investigated the effects of near-infrared light (635 nm and 808 nm) on the osteogenic differentiation of hUCMSCs. We evaluated the impact of near-infrared light on osteogenic differentiation using Alizarin Red staining, alkaline phosphatase (ALP) staining, and the measurement of osteogenesis-related genes and factors. To elucidate the molecular mechanisms underlying the effects of PBM on hUCMSCs, we examined the relationship between PBM-induced mitochondrial ROS production, phosphorylation of Akt (P-Akt), and osteogenic differentiation of hUCMSCs.
Methods
Culture of hUCMSCs
HUCMSCs were obtained from Onsai Cell Company (Tianjin, China), provided by Dr. Han Zhibo. Under specific conditions, these cells differentiate into osteoblasts, chondrocytes, and adipocytes. The hUCMSCs were cultured in an incubator at 37 °C with 5% CO2 using α-MEM medium supplemented (Gibco, USA) with 100 U/mL penicillin-streptomycin (PS, Gibco, USA) and 10% fetal bovine serum (FBS: Gibco, USA). The cells were digested with trypsin (HyClone, USA) and passaged upon reaching 80% confluence. For osteogenic differentiation, once the cells reached 80% confluence the proliferation medium was replaced with an osteogenic induction medium. This osteogenic medium was prepared by further supplementing α-MEM with 10 nM dexamethasone (Sigma, USA), 10 mM β-glycerophosphate (Sigma, USA), and 50 µM L-ascorbic acid (Sigma, USA). In the experiments, 3rd to 5th generation cells were used. The experimental groups included a Control group, a 635 nm laser group, and an 808 nm laser group.
Laser irradiation
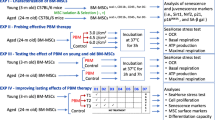
Cells were collected by digestion and centrifugation, then seeded at a density of 2 × 103 cells/well in 96-well plates and 1 × 105 cells/well in 6-well plates. After replacing the complete culture medium(90% α-MEM medium + 10% FBS + 1% PS) with an osteogenic induction medium (Sigma, USA), the cells were incubated overnight at 37 °C in a 5% CO2 environment. Laser irradiation was performed using continuous-output lasers at wavelengths of 635 and 808 nm. The power density was set to 20 mW/cm2 and the energy density was set to 4 J/cm2. Laser irradiation was conducted 1 cm away from the cell culture plates. Control cultures were subjected to the same conditions without laser activation. The output energy was measured and verified using a power meter to ensure accuracy. Each group received irradiation twice daily for three consecutive days (Fig. 1).

Laser exposure. (a) Schematic diagram of the laser irradiation process. The schematic diagram in this figure was created using Adobe Illustrator (https://www.adobe.com/products/illustrator). (b) Osteogenesis test schedule. (c) Light parameter setting.
Stem cell differentiation capacity testing
A 0.1% gelatin (Sigma, USA) solution was prepared to coat the wells for 20 min, after which the cells were seeded. On the second day, the growth medium (90% α-MEM medium + 10% FBS + 1% PS) was replaced with the differentiation medium (Gibco, USA), which was changed every three days for 21 days of osteogenic induction. During lipid induction, cells were cultured in lipid-forming solution A (Procell, China) for three days, followed by culture in lipid-forming solution B (Procell, China) for one day. This alternating solution change was repeated for 20 days. Osteogenic and lipogenic staining was performed at the end of the induction period. Cells were fixed with 500 µL of fixative for 30 min at room temperature, then gently rinsed three times with PBS. The plates were stained with either Alizarin Red (Solarbio, China) or Oil Red O (Beyotime, China) for 15 min, followed by gentle rinsing twice with double distilled water. The plates were observed and photographed under a microscope.
Cell proliferation assay
To detect the growth curves of 5th generation hUCMSCs, the cells were seeded in 96-well plates and incubated at 37 °C with 5% CO2 for seven consecutive days. The effect of PBM on the proliferation of hUCMSCs was assessed using the Cell Counting Kit-8 (CCK-8, Beyotime, China) assay, a widely employed method for evaluating cell viability. The CCK-8 solution was prepared by mixing 90% complete medium with 10% CCK-8 reagent under light/dark conditions. The culture medium in the 96-well plates was discarded and replaced with 100 µL of the CCK-8 solution per well. The cells were then incubated at 37 °C for 1 h. Absorbance was measured at 450 nm using a multifunctional microplate reader (Thermo, USA).
ALP assay and staining
Cells were lysed using Cell Lysis Solution (Beyotime, China) and collected in EP tubes. The samples were centrifuged at 12,000 g for 10 min in a pre-cooled centrifuge at 4 °C. Blank control, sample, and standard wells were prepared according to the ALP Assay protocol (Beyotime, China). Each sample was tested in triplicate and incubated in a thermostat for 10 min. To terminate the reaction, 100 µL of reaction termination solution was added to each well. Absorbance was measured at 405 nm using a multifunctional microplate (Thermo, USA) reader.
The NBT/BCIP (Beyotime, China) ALP staining solution was prepared in a lightproof environment. A mix of 6 mL of color development buffer, 20 µL of BCIP solution, and 40 µL of NBT solution was used. The cell culture medium was discarded and the cells were gently rinsed three times with PBS. Fixation was done with 500 µL of fixative at room temperature for 30 min, followed by rinsing three times with PBS. The BCIP/NBT staining working solution (1 mL/well) was added and incubated at room temperature for 1 h in the dark. The staining solution was discarded, and the wells were rinsed twice with double-distilled water to stop the ALP staining, after which the cells were imaged using a microscope (Olympus, Japan).
Alizarin red staining assay
After 21 days of culture, stromal mineralization was evaluated using alizarin red staining (Solarbio, China). Cells were gently rinsed three times with PBS, then fixed with 500 µL of fixative at room temperature for 30 min. After discarding the fixative, the cells were rinsed three times with PBS. The cells were stained with 1 mL alizarin red dye per well for 15 min, followed by gentle rinsing twice with double-distilled water. The stained cells were observed and photographed under a microscope.
After imaging, the calcium content was quantified by staining with alizarin red. The wells were emptied, and 200 µL of 0.5% cetylpyridinium chloride solution was added to each well to dissolve the calcium nodules. The absorbance was measured at 562 nm using a multifunctional microplate reader (Thermo, USA) after complete dissolution.
Real-time polymerase chain reaction analysis
The expression levels of runt-related transcription factor 2 (RUNX2), collagen type I alpha 1 chain (COL1A1), osteocalcin (OCN), ALP, osteopontin (OPN), and osterix (SP7) mRNA were quantified using RT-PCR. The reaction volume was set to 10 µL, with three replicates per sample. The methods and steps followed the TB Green® Premix Ex Taq™ (Tli RNaseH Plus) protocol. The primer sequences are listed in Table 1.
ELISA
Secretion levels of bone morphogenetic protein type 2 (BMP-2), OCN and OPN in the cells were measured using ELISA kits (Shanghai Tongwei, China). Except for the blank wells, 50 µL of enzyme reagent was added to each well. The wells were covered with a sealing membrane and incubated at 37 °C for 60 min under 5% CO2. After removing the membrane and discarding the waste solution, a wash solution was added, the membrane was allowed to stand for 30 s, and then discarded. The washing process was repeated five times. In the next step, 50 µL of Color Developer A and 50 µL of Color Developer B were added to each well, followed by a 15 min incubation period, protected from light. Lastly, to stop the reaction, 50 µL of a termination solution was added to each well, followed by measuring the absorbance at 450 nm using a multifunctional microplate reader (Thermo, USA).
ROS level detection
A 1:1,000 dilution of DCFH-DA (Beyotime, China) was prepared in a serum-free medium. After removing the medium, each well was treated with 200 µL of diluted DCFH-DA and incubated for 20 min. Subsequently, the cells were washed thrice with serum-free medium to eliminate any unpermeated excess DCFH-DA before imaging. Excess DCFH-DA was avoided to minimize background fluorescence and anti-fluorescence quenching was added to reduce fluorescence fading. Confocal microscopy (Olympus, Japan) was employed to perform the measurements at excitation and emission wavelengths of 488 and 525 nm, respectively.
Immunofluorescence staining
After removing the culture medium, the cells were fixed in 4% paraformaldehyde (Solarbio, China) for 15 min. Subsequently, the fixative was aspirated and the cells were rinsed three times with PBS. The cells were permeabilized on ice with 0.2% Triton X-100 (Solarbio, China) for 10 min and blocked with 5% goat serum (Solarbio, China) at room temperature for 1 h. A working solution of primary antibody targeting P-Akt (1:200, Beyotime, China) was incubated overnight at 4 °C. The next day, the cells were incubated with a fluorescent secondary antibody (Solarbio, China) at room temperature for 2 h. After rinsing with PBS, a DAPI solution (10 µg/mL) was added and incubated for 5 min, followed by three additional rinses with PBS and gentle shaking. Lastly, images were obtained using a confocal microscope (Olympus, Japan).
Introduction to inhibitors
The Akt inhibitor MK-2206 (Beyotime, China) was used at a final concentration of 5 µM. To attain this concentration, MK-2206 (initial concentration: 10 mM) was diluted to 500 µM with DMSO in a 1:20 ratio. Subsequently, the 500 µM MK-2206 was further diluted to 5 µM in osteogenic induction medium using a 1:100 ratio. Cells undergoing Akt pathway inhibition were cultured with 5 µM MK-2206.
To minimize DMSO’s (Sigma, USA) potential impact of DMSO on the results, DMSO was added to the osteogenic induction medium at a 1:100 ratio. This medium was used to culture the cells in the non-inhibited Akt pathway group. After a three-day incubation, the medium was replaced with a regular osteogenic differentiation medium. Following Akt pathway inhibition by MK-2206, the cells were irradiated with 635 and 808 nm lasers. The power density was set to 20 mW/cm2 with an energy density of 4 J/cm2. The experimental groups included the Control, MK-2206, MK-2206 + 635, and MK-2206 + 808 nm group.
Statistical analysis
All experiments were conducted in triplicates. Mean differences were analyzed using SPSS through one-way analysis ANOVA and chi-square test for variance. Image plotting was performed using GraphPad Prism 8. Statistical significance was set at p < 0.05.
Results
HUCMSCs culture and characterization
The cell growth curve exhibited an “S” shape, indicating robust proliferation ability (Fig. 2a). By day 5, the cells were evenly distributed on the surface of the culture flask, with minimal gaps (Fig. 2b). Osteogenic–lipogenic induction, considered the standard for assessing the differentiation potential of MSCs, was performed. After 21 days of osteogenic induction, multilayer growth was observed, characterized by the presence of red-stained calcium nodules (Fig. 2c). Similarly, after 20 days of lipid formation induction, numerous lipid droplets formed in the cytoplasm, exhibiting a dark purple color when stained with Oil Red O (Fig. 2d). These experimental results confirmed that P5 hUCMSCs met the requirements for subsequent experiments.

Impact of PBM intervention on osteogenic differentiation of hUCMSCs. (a) Growth curves of cultured P5 hUCMSCs. (b) Day 5 growth status of P5. (c) Osteogenic staining. (d) Lipogenic staining. (e) Effect of PBM on cell proliferation. (f) Effect of PBM on the level of ALP. (g) Measurement of ALP activity. (h) Effect of PBM on calcium nodules. (i) Quantification of calcium nodule content in hUCMSCs. Data are expressed as mean ± standard deviation. Compared with the Control group, *p < 0.05; Compared with the 635 nm group, #p < 0.05, ns indicates no statistically significant difference.
PBM enhances hUCMSCs activity
We assessed the proliferative activity of each group of cells over three consecutive days to investigate the effects of 635/808 nm laser irradiation on the activity of hUCMSCs. The results indicated that exposure to 635 nm laser significantly enhanced the proliferative activity of hUCMSCs compared to the Control group, while irradiation at 808 nm did not significantly affect cell proliferation (Fig. 2e).
PBM promoted hUCMSCs ALP activity and calcium nodule formation
To evaluate the effects of PBM on the osteogenic differentiation of hUCMSCs, we measured the activity of ALP, a key marker of osteogenic differentiation, after 7 days of induction, as illustrated in Fig. 2f. Compared to the Control group, hUCMSCs exposed to 635/808 nm laser irradiation exhibited a deeper purple color in ALP staining, indicating that PBM induced higher ALP levels. Further validation was provided by measuring ALP activity, as shown in Fig. 2g. The results revealed a significant increase in ALP activity in hUCMSCs following exposure to 635/808 nm laser irradiation compared to the Control group. Notably, the effect of 808 nm laser irradiation on ALP levels was more pronounced than that of the 635 nm laser irradiation.
The formation of mineralization nodules is a crucial marker of the osteogenic process, reflecting the capacity and effectiveness of bone formation. The deposition of extracellular matrix during mineralization provides a foundation for the growth and differentiation of osteoblasts, thereby promoting the regeneration and repair of bone tissue. Therefore, after 21 days of osteogenic induction, we performed Alizarin Red staining and calcium content assays to investigate the impact of 635/808 nm laser irradiation on mineralization nodules. As shown in Fig. 2h, there was a significant increase in the formation of mineralization nodules following treatment with 635/808 nm laser irradiation compared to the Control group. The results of the Alizarin Red calcium content assay in Fig. 2i further confirmed the staining results, indicating that exposure to 635/808 nm laser irradiation induced a greater number of mineralization nodules compared to the Control group.
PBM promotes differentiation of hUCMSCs in the osteogenic direction
After seven days of osteogenesis induction, the mRNA expression levels of osteogenesis-related genes (ALP, COL1A1, OCN, OPN, Runx2, and SP7) were evaluated using RT-PCR. Compared with the Controls, 635 and 808 nm laser treatment resulted in increased expression of osteogenic differentiation marker genes (Fig. 3a). Furthermore, seven days post-osteogenic induction, the levels of osteogenic factors (BMP-2, OCN, and OPN) were measured using ELISA. Irradiation with either the 635–808 nm laser elevated the expression levels of osteogenesis-related elements in comparison with the corresponding values in the Control group (as shown in Fig. 3b).
Low levels of ROS can act as secondary messengers in various signaling pathways involved in cell differentiation. To further explore the mechanism of PBM intervention in the osteogenic differentiation of hUCMSCs, we analyzed ROS production levels in each cell group by irradiating hUCMSCs with 635 nm and 808 nm lasers. Representative confocal microscopy images are shown in Fig. 3c, and quantification of ROS fluorescence is provided in Fig. 3d. The results indicated a minor increase in the production of ROS in both the 635 and 808 nm treatment groups, in comparison to the Control group. Notably, irradiation with the 635 nm laser induced a marginally stronger increase in ROS levels than did irradiation at 808 nm.

Study of the mechanism of PBM intervention in osteogenic differentiation of hUCMSCs. (a) RT-PCR was performed to detect relevant gene expression. (b) ELISA was performed to detect cytokines. (c) Effect of PBM on the level of reactive ROS in hUCMSCs. Scale bar: 100 μm. (d) Relative fluorescent intensities of ROS levels analysed using Image J software. Data are expressed as mean ± standard deviation. Compared with the Control group, *p < 0.05; Compared with the 635 nm group, #p < 0.05, ns indicates no statistically significant difference.
Akt activation regulates numerous downstream protein functions including cell proliferation, migration, survival, metabolism, and angiogenesis. To investigate the effects of PBM on Akt signaling, P-Akt protein expression was evaluated using immunofluorescence staining. PBM treatment was administered continuously for three days, and Akt expression was measured one hour after the completion of the three-day intervention. Representative confocal microscopy images are shown in Fig. 4a, and quantitative fluorescence measurements are shown in Fig. 4b. The results revealed a significant increase in intracellular red fluorescence intensity in cells treated with either the 635–808 nm lasers compared to the Control group, indicating an upregulation of P-Akt protein expression.

The effect of PBM intervention on P-Akt expression in hUCMSCs. (a) Immunofluorescence assay to detect the expression of P-Akt in hUCMSCs. Data are expressed as mean ± standard deviation. P-Akt (red), DAPI (blue), Scale bar: 100 μm. (b) Relative fluorescent intensities of P-Akt analysed using Image J software. Compared with the Control group, *p < 0.05. Compared with the 635 nm group, #p < 0.05, ns indicates no statistically significant difference.
ALP activity and calcium nodulation in hUCMSCs decreased when Akt was inhibited
Previous experimental results demonstrated that PBM intervention can promote osteogenic differentiation of hUCMSCs, enhancing ALP activity, mineralized nodule formation, and the expression of osteogenesis-related genes and factors. Additionally, ROS and P-Akt levels were also altered. However, it remains unclear whether there is a regulatory relationship between these factors. To explore this further, we used the inhibitor MK-2206 to suppress the Akt signaling pathway and examined the effects of 635/808 nm laser intervention on ALP activity in hUCMSCs (as shown in Fig. 5a). Compared to the Control group, 635/808 nm laser treatment significantly increased the area of purple staining, reflecting elevated ALP activity. However, this increase was markedly reduced when the Akt pathway was inhibited. Furthermore, the use of 635/808 nm laser irradiation after Akt inhibition did not restore the decreased ALP activity.
Similarly, we evaluated the impact of 635/808 nm laser treatment on the formation of mineralized nodules in hUCMSCs after Akt pathway inhibition. As shown in Fig. 5b, compared to the Control group, laser irradiation significantly increased mineralized nodule formation and calcium content. However, inhibition of the Akt pathway significantly diminished these effects. After suppression of the Akt pathway, 635/808 nm laser treatment no longer promoted mineralized nodule formation or calcium accumulation in hUCMSCs.

Impact of Akt pathway inhibition on hUCMSCs osteogenic differentiation. (a) Effect of inhibiting Akt pathway on ALP activity of hUCMSCs. Data are expressed as mean ± standard deviation. (b) The effect of inhibiting the Akt pathway on calcium nodules of hUCMSCs osteogenic differentiation, *p < 0.05 compared with the Control group; #p < 0.05 compared with 635–808 nm irradiation group.
Osteogenic differentiation of hUCMSCs decreased by Akt pathway inhibition
To further evaluate the role of the Akt signaling pathway in PBM-induced osteogenic differentiation of hUCMSCs, we inhibited the Akt pathway using MK-2206 and examined the effects of 635 nm laser irradiation on the mRNA expression levels of osteogenic genes ALP, COL1A1, OCN, OPN, RUNX2, and SP7, as shown in Fig. 6a. Compared to the Control group, 635 nm laser treatment significantly increased the mRNA expression of these osteogenic genes. However, inhibition of the Akt signaling pathway led to a marked decrease in the expression of these osteogenic markers. Notably, following the inhibition of the Akt pathway, PBM intervention did not further enhance osteogenic gene expression compared to the group treated solely with 635 nm laser.
Figure 6b illustrates the impact of 808 nm laser irradiation on the expression of osteogenesis-related genes in hUCMSCs. The 808 nm laser significantly increased the expression of these genes compared to the Control group. However, when PBM was applied following Akt pathway inhibition, there was no further significant increase in osteogenic gene expression. These findings suggest that when the Akt signaling pathway is inhibited, PBM is unable to further enhance the expression of osteogenesis-related genes, indicating that the Akt pathway plays a critical role in PBM-mediated osteogenesis of hUCMSCs.

Effect of inhibition of the Akt pathway on osteogenesis-related gene expression. (a) Influence of 635 nm laser exposure on osteogenesis-associated gene expression in hUCMSCs following Akt pathway inhibition. (b) Influence of 808 nm laser exposure on osteogenesis-associated gene expression of hUCMSCs after Akt pathway inhibition. Data are expressed as mean ± standard deviation, *p < 0.05 compared to the Control group and #p < 0.05 compared with 635–808 nm irradiation group.
Figure 7a and b demonstrate the effects of 635 nm and 808 nm laser irradiation on the osteogenesis-related factors BMP-2, OCN and OPN in hUCMSCs after inhibition of the Akt pathway. The results showed that laser irradiation at 635–808 nm significantly increased the levels of osteogenesis-related factors compared with the Control group. In contrast, inhibition of the Akt pathway significantly reduced the expression of osteogenic factors. Notably, the application of laser intervention after inhibition of the Akt pathway did not reverse the decrease in osteogenesis-related factor levels compared with the group irradiated with 635–808 nm laser only.
Low level of ROS generation can promote the differentiation of hUCMSCs in the osteogenic direction, in order to further investigate the relationship between Akt on mitochondrial ROS level generation in this study. Figure 7c, d and e demonstrates the effect of laser irradiation on ROS levels in hUCMSCs after inhibiting the Akt pathway using MK-2206. Compared with the Control group, hUCMSCs receiving 635–808 nm laser irradiation showed stronger green fluorescence, indicating elevated ROS levels. However, when the Akt pathway was inhibited, the intensity of green fluorescence diminished, resulting in a significant decrease in ROS levels. Importantly, application of PBM after Akt pathway inhibition failed to restore ROS levels compared to the groups receiving 635–808 nm laser irradiation alone. These results suggest that ROS production is closely related to the Akt pathway and that the Akt pathway plays an upstream regulatory role in this process.

Effect of inhibition of the Akt pathway on osteogenesis-related factors and ROS. (a) Influence of 635 nm laser exposure on hUCMSCs expression of osteogenic correlated factors after Akt pathway inhibition. (b) Influence of 808 nm laser exposure on hUCMSCs expression of osteogenic correlated factors after Akt pathway inhibition. (c) Impact of Akt pathway inhibition on the level of ROS in hUCMSCs. (d) Fluorescence quantification was performed using ImageJ to evaluate the effect of 635 nm laser irradiation on ROS levels in hUCMSCs following Akt pathway inhibition. (e) Fluorescence quantification was performed using ImageJ to assess the impact of 808 nm laser irradiation on ROS levels in hUCMSCs after Akt pathway inhibition. Scale bar: 100 μm. Data are expressed as mean ± standard deviation, *p < 0.05 compared to the Control group and #p < 0.05 compared with 635–808 nm irradiation group.
Discussion
In this study, we examined how PBM using dual-wavelength near-infrared light (635 nm and 808 nm) affects the osteogenic differentiation of hUCMSCs. We also explored the mechanisms by which PBM impacts these osteogenic processes. First, our results showed that 635 nm laser irradiation significantly enhanced the viability of hUCMSCs, while 808 nm laser irradiation had no significant effect on cell viability. PBM interacts with key intracellular chromophores to activate multiple signaling pathways, thereby promoting cell proliferation, migration, and differentiation20. Moreover, the biological effects of PBM typically follow a biphasic dose-response: an energy density that is too low may fail to activate the necessary signaling pathways, whereas an energy density that exceeds the optimal range can lead to excessive ROS generation and local thermal damage, ultimately impairing cell function or inducing apoptosis21,22. Consistent with our observations, de Andrade et al.. evaluated the effect of PBM at different energy levels on the proliferation of MSCs of adipose tissue origin. Low-energy lasers (660 nm, 40 mW, 0.56/1.96 J/cm2) promoted MSCs proliferation, whereas irradiation at higher energy (5.04 J/cm2) resulted in cellular damage23. Similarly, Tani et al.. investigated the effects of PBM on human osteoblasts and mesenchymal stromal cells, concluding that 808 nm wavelength laser irradiation did not significantly affect cell viability16. Apart from this, Zare et al.. investigated the impact of 630 and 810 nm single-wavelength irradiation and combined interventions on the survival and recovery of MSCs derived from human bone marrow and stem cells derived from human adipose tissue, the authors reported that the combined 630 and 810 nm PBM treatment was more effective in promoting cell viability and significantly reducing apoptosis24. In summary, these findings indicate that PBM induced by low-energy lasers, particularly at a wavelength of 635 nm, can significantly enhance the viability of MSCs. Our study highlights the varying effects of different wavelengths and energy levels on cell viability, underscoring the importance of selecting appropriate laser parameters to achieve optimal therapeutic outcomes.
Osteoblastic differentiation is crucial for bone development, fracture repair, and tissue maintenance3. Elevated ALP activity and mineralized nodule formation are early markers of osteoblast differentiation and maturation. Our study shows that laser irradiation at 635 and 808 nm significantly increased ALP activity and mineralized nodule formation in hUCMSCs. This aligns with the previous findings. For instance, Bueno et al.. isolated bone marrow MSCs from diabetic rats and found that irradiation at 660 nm increased the viability of stem cells and the activity of the osteogenic marker ALP, and the level of mineralization ultimately restored the osteogenic differentiation potential of MSCs from diabetic rats25. Furthermore, Amaroli et al.. evaluated the efficacy of 808-nm laser therapy in vitro by irradiating bone marrow mesenchymal cells with an 808-nm laser26. Their findings showed that PBM increased ALP expression, elevated anti-inflammatory factor levels, and decreased pro-inflammatory factor levels, thereby effectively influencing bone marrow mesenchymal osteogenic differentiation of BMSCs. However, our study has certain limitations. We observed that PBM promoted cell proliferation (as shown in Fig. 2), but we did not normalize the mineralization levels to cell number or total protein. Therefore, we cannot completely rule out the possibility that the observed increases in ALP activity and mineralization levels are partly attributable to increased cell numbers, rather than a direct effect of PBM on single-cell osteogenic activity. To more accurately assess the impact of PBM on single-cell osteogenic activity, in future studies, we will actively explore single-cell level detection methods to gain a deeper understanding of the effects of PBM on osteogenic differentiation.
Osteogenic differentiation is a complex process regulated by various transcription factors. In this study, we assessed the expression of osteogenesis-related genes, including RUNX2, COL1A1, OCN, ALP, OPN, and SP7, as well as other factors such as BMP-2, OCN, and OPN. These genes and factors play critical roles in the osteogenic differentiation process. For instance, RUNX2 is a primary regulatory factor in osteogenic differentiation, promoting the maturation and mineralization of osteoblasts27. COL1A1 is one of the main components of bone matrix, essential for the strength and stability of bone structure. OCN, a protein secreted by mature osteoblasts, facilitates mineralization and serves as a marker for bone metabolism. ALP acts as an early marker of osteogenic differentiation, participating in the mineralization of inorganic phosphate, while OPN promotes the adhesion and mineralization of osteoblasts during the bone repair process. Additionally, SP7 plays a significant role in the transcriptional regulation of osteoblast differentiation and is involved in bone development and mineralization. BMP-2 is a crucial factor in bone tissue formation, stimulating the proliferation and differentiation of osteoblasts while promoting bone matrix formation. In our study, these results demonstrate that PBM significantly enhanced the expression of these genes and factors in hUCMSCs, which is consistent with previous studies. De Farias et al.. assessed the impact of 660 nm on the migration of osteoblasts, ALP, COL-1, and RUNX2 in dentin, and study showed that laser intervention at 660 nm and 3.2 J/cm2 can significantly enhance the migration of osteoblasts and promote the expression of osteogenic genes, this could potentially help form a new layer of tooth bone28. In addition, Abdelgawad et al.. investigated human dental ligament stem cells using a combination of an 808-nm laser and vitamin D. They discovered that PBM combined with vitamin D significantly increased hUCMSCs proliferation as well as the expression of osteogenic genes (ALP, RUNX2, and COL-1), further promoting osteogenic differentiation of hUCMSCs29. These studies indicate that PBM can enhance the osteogenic differentiation of hUCMSCs and accelerate bone defect healing.
Research highlights that PBM’s primary mechanism is the modulation of mitochondrial function, which subsequently influences various cellular activities30. High levels of ROS are cytotoxic, causing damage by oxidizing and nitrating essential macromolecules such as DNA, RNA, proteins, and lipids. Conversely, controlled ROS production plays a pivotal role in cellular signaling, acting as secondary messengers in pathways that regulate metabolism, proliferation, differentiation, survival, and apoptosis. This redox regulation reflects a delicate balance between ROS generation and their neutralization by the cell’s antioxidant systems31,32. PBM reacts photochemically with target cells by absorbing photons via the mitochondrial chromophore cytochrome C oxidase, leading to changes in the activity of ATP, ROS, and Ca[2+ 33. Another key finding of our study was the slight increase in ROS production in hUCMSCs after PBM. This finding is consistent with those of the previous studies. For instance, Yaralı Çevik et al.. studied the impact of 655 and 808 nm wavelength laser irradiation on osteogenic differentiation in human umbilical vein endothelial cells and human bone marrow stem cells co-cultured on 3D scaffold-free microtissues. The findings indicated that PBM elevated ROS and ATP levels in hUCMSCs, enhancing ALP activity, calcium content, and expression of genes associated with ALP, COL, and OPN. This resulted in an improved osteogenic differentiation capacity within the co-culture34. Ateş et al.. utilized the photosensitizer indocyanine green in conjunction with PBM to generate low ROS doses, which promoted human osteoblast activity, increased ALP activity, and enhanced mineralization35. Therefore, regulation of bone defect repair by PBM is associated with low levels of mitochondrial ROS production. It is noteworthy that our study results indicate that, compared to the 808 nm laser, the 635 nm laser generates higher levels of ROS.
Akt, a 57-kDa serine/threonine kinase commonly known as Protein Kinase B (PKB), plays a crucial role in various cellular processes. Numerous studies have highlighted the role of Akt in the regulation of various cellular processes, such as mitochondria-mediated apoptosis, redox reactions, dynamic homeostasis, autophagy, and metabolism36,37. Our study revealed that PBM activated the Akt signaling pathway in hUCMSCs, leading to elevated expression levels of P-Akt, the phosphorylated form of Akt. The role of the Akt pathway in osteogenic differentiation was also investigated. For example, Liu et al.. found that ammonia promoted the proliferation of bone marrow-derived MSCs by modulating the Akt/Mtor/S6k pathway38. Zhang et al.. indicated that phenytoin significantly increased the expression of the osteogenic factors ALP and RUNX2 by activating the PI3K/Akt pathway, ultimately improving osteogenic differentiation efficiency in human bone marrow stem cells39. Additionally, Wang et al.. detailed how moderate calcium intake could lower apoptosis levels, reverse histopathological damage, and increase ALP activity in osteofluorosis-affected mice by activating the PI3K/Akt pathway40. Thus, the activation of the Akt signaling pathway may promote the osteogenic differentiation of stem cells.
To further investigate the impact of Akt on the generation of low levels of ROS in the mitochondria and the differentiation of hUCMSCs in the osteogenic direction, we employed Akt inhibitors for reverse validation. Our results demonstrate that the addition of an Akt inhibitor reduced ROS levels, ALP activity, mineralized nodule formation, and the expression of genes and factors associated with osteogenesis in hUCMSCs. Notably, PBM intervention did not reverse the declining trend of these osteogenesis-related indices, further confirming the crucial role of PBM in regulating osteogenic differentiation of hUCMSCs through the Akt-ROS signaling pathway. The interplay between the Akt signaling pathway and ROS levels has been extensively studied. Di Sante et al.. found that the mitochondrial flavoenzyme monoamine oxidase A promotes ROS generation and calcium homeostasis, activates the Akt/WNT signaling pathway, and facilitates human pluripotent stem cell differentiation into cardiomyocytes41. Zhang et al.. investigated the effect of formyl peptide receptors on neural stem cell differentiation and observed that the activation of formyl peptide receptors increased ROS production and Akt phosphorylation in neural stem cells42. This process encouraged neural stem cells to differentiate into neurons. Conversely, when the corresponding inhibitors were employed, both ROS levels and Akt phosphorylation decreased, and neural stem cell differentiation into neurons was blocked. Collectively, these results indicate that the activation of P-Akt can promote the generation of ROS, inducing a low level of oxidative stress, which effectively enhances the differentiation capability of stem cells.
Our study yielded noteworthy results, highlighting the distinct effects of 635 and 808 nm laser treatment on hUCMSCs. Specifically, we observed that the 635 nm laser intervention significantly enhanced the activity of hUCMSCs, whereas the 808 nm laser had no appreciable effect on their activity. However, the 808 nm laser outperformed the 635 nm laser in enhancing ALP activity, osteogenic differentiation gene expression, and osteogenic factor production. Previous studies have demonstrated that different wavelengths of light can activate distinct intracellular signaling pathways21,43,44. Based on these findings, we speculate that the low levels of ROS induced by 635 nm laser exposure may function as signaling molecules within an appropriate range, activating pathways related to cell cycle regulation and promoting proliferation, whereas the 808 nm laser may facilitate an earlier exit from the cell cycle and initiate differentiation. Although previous studies have individually explored the therapeutic effects of these wavelengths on osteoblast differentiation and related gene expression, our study offers a side-by-side assessment. Based on these research findings, we believe that dual-wavelength combination therapy may be an effective strategy that can enhance cell activity while promoting osteogenic differentiation and bone repair. Therefore, our future research will further explore the potential of combined use of 635 nm and 808 nm lasers to optimize their application in bone defect repair and regenerative medicine.
In summary, our findings indicate that PBM can significantly enhance the osteogenic differentiation of hUCMSCs. PBM can upregulate P-Akt expression and increase ROS levels to induce low levels of oxidative stress, thereby promoting the osteogenic differentiation of hUCMSCs (Fig. 8). Future research will focus on further elucidating the complex mechanisms underlying the PBM effect. While our results highlight the involvement of the Akt signaling pathway and the role of low levels of ROS as important signaling molecules promoting osteogenic differentiation, PBM may also regulate other potential cellular pathways and biological processes. For instance, studies have indicated that PBM can influence mitochondrial function, potentially increasing the production of ATP, thereby providing the necessary metabolic support for osteogenic differentiation45. Therefore, systematically investigating the interplay between these signaling pathways and specific PBM parameters, such as wavelength, power density, and irradiation time, is crucial for a comprehensive understanding of its therapeutic potential and for optimizing its clinical applications. These findings not only deepen our understanding of the regulatory role of PBM in hUCMSCs but also establish a robust foundation for the prospective clinical application of PBM in the treatment of bone defects. The results of this study introduce novel possibilities for future clinical applications.

Schematic diagram summarizing the interaction between PI3K/Akt pathway and ROS and enhancement of osteogenic differentiation during PBM intervention in hUCMSCs osteogenic differentiation. The schematic diagram in this figure was created using Adobe Illustrator (https://www.adobe.com/products/illustrator).
Data availability
All data in support of the findings of this paper are available within the article. Raw data are available from corresponding author on reasonable request.
Abbreviations
- MSCs:
-
Mesenchymal stem cells
- PBM:
-
Photobiomodulation
- HUCMSCs:
-
Human umbilical cord mesenchymal stem cells
- ROS:
-
Reactive oxygen species
- P-Akt:
-
Phosphorylated Akt
- ATP:
-
Adenosine triphosphate
- PBMT:
-
Photobiomodulation therapy
- ALP:
-
Alkaline phosphatase
- RUNX2:
-
Runt-related transcription factor 2
- COL1A1:
-
Collagen type I alpha 1 chain
- OCN:
-
Osteocalcin
- OPN:
-
Osteopontin
- SP7:
-
Osterix
- BMP-2:
-
Bone morphogenetic protein type 2
References
Wang, W. & Yeung, K. W. K. Bone grafts and biomaterials substitutes for bone defect repair: A review. Bioact Mater. 2, 224–247. https://doi.org/10.1016/j.bioactmat.2017.05.007 (2017).
Duda, G. N. et al. The decisive early phase of bone regeneration. Nat. Rev. Rheumatol. 19, 78–95. https://doi.org/10.1038/s41584-022-00887-0 (2023).
Wang, C. et al. NOTCH signaling in skeletal progenitors is critical for fracture repair. J. Clin. Invest. 126, 1471–1481. https://doi.org/10.1172/jci80672 (2016).
Huang, S., Jin, M., Su, N. & Chen, L. New insights on the reparative cells in bone regeneration and repair. Biol. Rev. Camb. Philos. Soc. 96, 357–375. https://doi.org/10.1111/brv.12659 (2021).
Li, P., Gong, Z., Shultz, L. D. & Ren, G. Mesenchymal stem cells: From regeneration to cancer. Pharmacol. Ther. 200, 42–54. https://doi.org/10.1016/j.pharmthera.2019.04.005 (2019).
Guo, Y. et al. HtrA3 paves the way for MSC migration and promotes osteogenesis. Bioact Mater. 38, 399–410. https://doi.org/10.1016/j.bioactmat.2024.05.016 (2024).
Guérit, D. et al. Inhibitory effect of miR-29a on the chondrogenic differentiation of mesenchymal stem cells. Ann. Rheum. Dis. 71, A65–A66. https://doi.org/10.1136/annrheumdis-2011-201237.12 (2012).
Han, Y., You, X., Xing, W., Zhang, Z. & Zou, W. Paracrine and endocrine actions of bone-the functions of secretory proteins from osteoblasts, osteocytes, and osteoclasts. Bone Res. 6, 16. https://doi.org/10.1038/s41413-018-0019-6 (2018).
Selestin Raja, I. et al. Tailoring photobiomodulation to enhance tissue regeneration. Biomaterials 309, 122623. https://doi.org/10.1016/j.biomaterials.2024.122623 (2024).
Maghfour, J. et al. Photobiomodulation CME part I: overview and mechanism of action. J. Am. Acad. Dermatol. https://doi.org/10.1016/j.jaad.2023.10.073 (2024).
Hamblin, M. R. Mechanisms and mitochondrial redox signaling in photobiomodulation. Photochem. Photobiol. 94, 199–212. https://doi.org/10.1111/php.12864 (2018).
Karu, T. I. Cellular and molecular mechanisms of photobiomodulation (low-power laser therapy). IEEE J. Sel. Top. Quantum Electron. 20, 143–148. https://doi.org/10.1109/JSTQE.2013.2273411 (2014).
Lee, J. H. et al. The effects of photobiomodulation on bone defect repairing in a diabetic rat model. Int. J. Mol. Sci. 22 https://doi.org/10.3390/ijms222011026 (2021).
Chen, H. et al. Study on the osteogenic differentiation of hUCMSCs in silk fibroin/polycaprolactone membrane under the intervention of photobiomodulation. IEEE J. Sel. Top. Quantum Electron. 29, 1–8. https://doi.org/10.1109/JSTQE.2022.3200367 (2023).
Lopes, C. C. A. et al. Effectiveness of photobiomodulation therapy on human bone healing in dentistry: A systematic review. Photobiomodul Photomed. Laser Surg. 40, 440–453. https://doi.org/10.1089/photob.2021.0092 (2022).
Tani, A. et al. Red (635 nm), near-infrared (808 nm) and violet-blue (405 nm) photobiomodulation potentiality on human osteoblasts and mesenchymal stromal cells: A morphological and molecular in vitro study. Int. J. Mol. Sci. 19 https://doi.org/10.3390/ijms19071946 (2018).
Cubas-Mogollón, J., Jiménez-Sánchez, S., Ramirez, E. & Erazo-Paredes, C. Effect of energy level of photobiomodulation therapy on bone repair in rats. Odovtos - Int. J. Dent. Sci. 25, 324–335. https://doi.org/10.15517/ijds.2023.54077 (2023).
Hosseinpour, S., Fekrazad, R., Arany, P. R. & Ye, Q. Molecular impacts of photobiomodulation on bone regeneration: A systematic review. Prog Biophys. Mol. Biol. 149, 147–159. https://doi.org/10.1016/j.pbiomolbio.2019.04.005 (2019).
Shang, F. et al. Human umbilical cord MSCs as new cell sources for promoting periodontal regeneration in inflammatory periodontal defect. Theranostics 7, 4370–4382 (2017).
Zein, R., Selting, W. & Hamblin, M. R. Review of light parameters and photobiomodulation efficacy: Dive into complexity. J. Biomed. Opt. 23, 1–17. https://doi.org/10.1117/1.Jbo.23.12.120901 (2018).
Huang, Y. Y., Sharma, S. K., Carroll, J. & Hamblin, M. R. Biphasic dose response in low level light therapy—an update. Dose Response 9, 602–618. https://doi.org/10.2203/dose-response.11-009.Hamblin (2011).
Coskun, M. E., Coskun, K. A. & Tutar, Y. Determination of optimum operation parameters for low-intensity pulsed ultrasound and low-level laser based treatment to induce proliferation of osteoblast and fibroblast cells. Photomed. Laser Surg. 36, 246–252. https://doi.org/10.1089/pho.2017.4354 (2018).
de Andrade, A. L. M. et al. Photobiomodulation effect on the proliferation of adipose tissue mesenchymal stem cells. Lasers Med. Sci. 34, 677–683. https://doi.org/10.1007/s10103-018-2642-2 (2019).
Zare, F. et al. Photobiomodulation with 630 plus 810 Nm wavelengths induce more in vitro cell viability of human adipose stem cells than human bone marrow-derived stem cells. J. Photochem. Photobiol B. 201, 111658. https://doi.org/10.1016/j.jphotobiol.2019.111658 (2019).
Bueno, N. P. et al. Recovering the osteoblastic differentiation potential of mesenchymal stem cells derived from diabetic rats by photobiomodulation therapy. J. Biophotonics. 14, e202000393. https://doi.org/10.1002/jbio.202000393 (2021).
Amaroli, A. et al. The effects of photobiomodulation of 808 Nm diode laser therapy at higher fluence on the in vitro osteogenic differentiation of bone marrow stromal cells. Front. Physiol. 9, 123. https://doi.org/10.3389/fphys.2018.00123 (2018).
Liu, Z. et al. Mediator MED23 cooperates with RUNX2 to drive osteoblast differentiation and bone development. Nat. Commun. 7, 11149. https://doi.org/10.1038/ncomms11149 (2016).
de Farias, C. S., Garcez, A. S., Teixeira, L. N. & Suzuki, S. S. In vitro effects of photobiomodulation on cell migration and gene expression of ALP, COL-1, RUNX-2, and osterix in cementoblasts. Lasers Med. Sci. 38, 121. https://doi.org/10.1007/s10103-023-03775-5 (2023).
Abdelgawad, L. M., Abdelaziz, A. M., Sabry, D. & Abdelgwad, M. Influence of photobiomodulation and vitamin D on osteoblastic differentiation of human periodontal ligament stem cells and bone-like tissue formation through enzymatic activity and gene expression. Biomol. Concepts 11, 172–181. https://doi.org/10.1515/bmc-2020-0016 (2020).
Yang, L., Youngblood, H., Wu, C. & Zhang, Q. Mitochondria as a target for neuroprotection: role of methylene blue and photobiomodulation. Transl Neurodegener. 9, 19. https://doi.org/10.1186/s40035-020-00197-z (2020).
Atashi, F., Modarressi, A. & Pepper, M. S. The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: A review. Stem Cells Dev. 24, 1150–1163. https://doi.org/10.1089/scd.2014.0484 (2015).
Tyagi, A., Haq, S. & Ramakrishna, S. Redox regulation of dubs and its therapeutic implications in cancer. Redox Biol. 48 (2021).
Chen, H. et al. Photobiomodulation therapy mitigates depressive-like behaviors by remodeling synaptic links and mitochondrial function. J. Photochem. Photobiol B. 258, 112998. https://doi.org/10.1016/j.jphotobiol.2024.112998 (2024).
Yaralı Çevik, Z. B., Karaman, O. & Topaloğlu, N. Photobiomodulation therapy at red and near-infrared wavelengths for osteogenic differentiation in the scaffold-free microtissues. J. Photochem. Photobiol B 238, 112615. https://doi.org/10.1016/j.jphotobiol.2022.112615 (2023).
Ates, G. B., Ak, A., Garipcan, B. & Gulsoy, M. Indocyanine green-mediated photobiomodulation on human osteoblast cells. Lasers Med. Sci. 33, 1591–1599. https://doi.org/10.1007/s10103-018-2530-9 (2018).
Chae, Y. C. et al. Mitochondrial Akt regulation of hypoxic tumor reprogramming. Cancer Cell. 30, 257–272. https://doi.org/10.1016/j.ccell.2016.07.004 (2016).
Majewski, M. et al. Activation of mitochondrial Raf-1 is involved in the antiapoptotic effects of Akt. Cancer Res. 59, 2815–2819 (1999).
Liu, Y. et al. Ammonia promotes the proliferation of bone marrow-derived mesenchymal stem cells by regulating the Akt/mTOR/S6k pathway. Bone Res. 10, 57. https://doi.org/10.1038/s41413-022-00215-y (2022).
Zhang, Z., Shang, W., Zhao, X. & Lin, L. Phenytoin regulates osteogenic differentiation of human bone marrow stem cells by PI3K/Akt pathway. Regen Ther. 24, 201–210. https://doi.org/10.1016/j.reth.2023.06.015 (2023).
Wang, J. et al. Calcium relieves fluoride-induced bone damage through the PI3K/AKT pathway. Food Funct. 11, 1155–1164. https://doi.org/10.1039/c9fo02491c (2020).
Di Sante, M. et al. Monoamine oxidase A-dependent ROS formation modulates human cardiomyocyte differentiation through AKT and WNT activation. Basic. Res. Cardiol. 118, 4. https://doi.org/10.1007/s00395-023-00977-4 (2023).
Zhang, L. et al. Formyl peptide receptors promotes neural differentiation in mouse neural stem cells by ROS generation and regulation of PI3K-AKT signaling. Sci. Rep. 7, 206. https://doi.org/10.1038/s41598-017-00314-5 (2017).
Rojas, J. C. & Gonzalez-Lima, F. Low-level light therapy of the eye and brain. Eye Brain. 3, 49–67. https://doi.org/10.2147/eb.S21391 (2011).
Hamblin, M. R. Mechanisms and applications of the anti-inflammatory effects of photobiomodulation. AIMS Biophys. 4, 337–361. https://doi.org/10.3934/biophy.2017.3.337 (2017).
Yaralı Çevik, Z. B., Karaman, O. & Topaloğlu, N. Synergistic effects of integrin binding peptide (RGD) and photobiomodulation therapies on bone-like microtissues to enhance osteogenic differentiation. Biomater. Adv. 149, 213392. https://doi.org/10.1016/j.bioadv.2023.213392 (2023).
Acknowledgements
We would like to thank Adobe Illustrator (https://www.adobe.com/products/illustrator) for the assistance in creating figures.
Funding
This work was supported by the National Key R&D Program of China (No. 2024YFC3505503), the Natural Science Foundation of Hebei Province (No. F2024110001), and the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (No. 2021-I2M-1-058).
Author information
Authors and Affiliations
Contributions
C.Y.M.: Assist to complete the experiment and analyse and interpret the data. Y.T.Y.: Data analysis and curate. X.Y.S.: Data interpretation, visualization, manuscript revision. N.L.: Conception and design, data collection and analysis and manuscript writing. Y.F.C.: Provision of study material and data collection. X.F.S.: Assist to complete the experiment and analysis of data. H.L.C.: Conception and design, data analysis, supervision, and financial support. All the authors have approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, C., Ye, Y., Shi, X. et al. Photobiomodulation promotes osteogenic differentiation of mesenchymal stem cells and increases P-Akt levels in vitro. Sci Rep 15, 17844 (2025). https://doi.org/10.1038/s41598-025-02428-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-02428-7
Keywords
This article is cited by
-
Advances in Photobiomodulation: Effects on Mesenchymal Stem Cells and their Paracrine Factors
Stem Cell Reviews and Reports (2025)
