
Abstract
Replication timing (RT), the temporal order in which genomic regions replicate, is considered a functional feature of multiple cellular processes and chromatin organization. Two approaches to measure RT are the Repli-seq and DNA copy number (also called S/G1) methods. We previously adapted Repli-seq using 5-ethynyl-2’- deoxyuridine (EdU) pulse-labeling and bivariate flow sorting, and while the approach offers high resolution and exposes heterogeneity in timing, the S/G1 method is a simpler, faster and less resource-intensive assessment. Here we modified the S/G1 technique by using EdU labeling (EdU-S/G1) to facilitate better separation of replicating from non-replicating nuclei during flow sorting, which enables the collection of a more pure sample of G1-phase nuclei. When comparing the three methods we found that profiles from the S/G1 and EdU-S/G1 methods are highly correlated with each other and with Repli-seq profiles for early replication. We also found that the EdU-S/G1 approach offers a better representation of replication in early and late S phase than the conventional S/G1 method. However, the high reproducibility of RT profiles among all three methods indicates that considerations of cost and sample availability can drive the decision of which method to choose.
Similar content being viewed by others

Introduction
DNA replication is fundamental for maintaining genome stability and orchestrating developmental processes vital for plant growth and development. DNA replication is a highly ordered process in which various segments of the genome are copied at different times during S phase1,2,3,4. The DNA replication timing program is highly conserved across cell cycle divisions, and some commonalities, such as euchromatin replicating before heterochromatin, are conserved among eukaryotes including most fungi, mammals and plants5. The maize (Zea mays L.) root tip offers an ideal system to study replication timing (RT) due to its dense population of actively dividing cells6 and accessibility for in vivo labeling with thymidine analogs7. The many cultivars of maize (Z. mays, subsp. mays) also offer a unique opportunity to exploit natural genomic diversity8 to develop and test hypotheses about the variability, regulation, and thus the functional implications of RT. Moreover, by using preparative flow sorting to separate nuclei at different stages of S phase, replication timing can be assigned and assessed within asynchronous mitotic cell populations characteristic of growing root tips.
In previous work, our group developed a genome-wide replication timing profile for maize B73 root tips using the Repli-seq method with an EdU-labeling step9. Repli-seq, as initially developed, involves the metabolic labeling of replicating DNA with bromodeoxyuridine (BrdU), followed by immunoprecipitation to isolate BrdU-labeled DNA from a variable number of S-phase fractions obtained via flow sorting on DNA content only10. This technique has proven successful across diverse model systems, including humans10,11,12, Drosophila13, and mice14,15. When EdU is used as the DNA precursor in place of BrdU, as in our protocol, visualization of EdU using Click chemistry16 preserves nuclear integrity17 and permits flow cytometric separation of replicating and non-replicating nuclei7. As we demonstrated in maize and Arabidopsis9,18,19, the Repli-seq protocol provides high resolution profiles of replication timing across whole genomes.
While Repli-seq with EdU labeling and flow sorting based on both EdU incorporation and DNA content offers high resolution, it is also resource-intensive, requiring substantial starting material before immunoprecipitation of EdU-labeled DNA. This is a challenge when working with limited starting material, species with smaller genomes, or tissue that is not amenable to metabolic labeling. Consequently, the S/G1 method—a simpler, faster, and more cost-effective approach—remains widely used (reviewed in5). The conventional S/G1 method uses flow sorting on DNA content only to separate mostly mid-S phase cells or nuclei from their counterparts in G1 phase to assess the relative copy number of each genomic locus in a replicating population. Higher S/G1 ratios in a given locus correspond to more complete replication, and hence to earlier replication time at that locus. The result is a continuous representation of replication timing across the genome. This method has been applied to multiple species, including yeast20, zebrafish21, fly22, humans23,24 and Arabidopsis25.
In this study, we combined EdU labeling and bivariate flow sorting with the conventional S/G1 method to collect a more inclusive sample of replicating nuclei while also more precisely separating them from non-replicating G1 and G2 nuclei. This adaptation (from here on called EdU-S/G1) provides increased resolution in the early and late portions of the replication timing landscape. Together with the relative simplicity of the S/G1 technique, this increased resolution may be important in future genetic studies of replication timing control. We then compared the strengths and limitations of these three methods, Repli-seq, S/G1 and EdU-S/G1, for measuring replication timing across the genome and at various genomic elements.
Methods
Plant material and growth conditions
Seeds of maize cultivar B73 were surface sterilized in a solution of 1% sodium hypochlorite, 0.05% Tween-20 and then rinsed 4X with water. For the Repli-Seq experiment, seeds were also treated with absolute ethanol for 5 min immediately prior to the hypochlorite step. After rinsing, seeds were imbibed overnight at room temperature in constantly stirred, sterile water that was aerated with an aquarium air pump. After imbibition, seeds were again surface sterilized with 0.5% sodium hypochlorite; 0.05% Tween-20 solution and then rinsed 4X with water. Seeds were placed in 6.5 by 10-inch glass dishes on six layers of paper towels moistened with sterile water. The dishes were covered with a transparent lid, and the seeds were germinated at 28 °C for 3 days under continuous light (Feit Electric OneSync LED light system) adjusted to a 3000 K soft white color temperature and 300–400 lx.
Plant root tip tissue collection and nuclei isolation
Three independently grown biological replicates were used for each experiment. After the 3-day growth period, seedlings were processed differently for each replication timing protocol, as illustrated in (Fig. 1a). For the Repli-seq and EdU-S/G1 methods, seedling roots were labeled in vivo for 20 min in a water solution of 25 µM EdU using previously described conditions9,26, except that after the labeling period, the seedlings were transferred to a 100 µM thymidine chase solution prior root tip isolation. Root tips (the terminal 1 mm) were excised from the primary and first two emerging seminal roots of each seedling, fixed in formaldehyde, and snap-frozen9,26. For the S/G1 method, no EdU labeling was performed, and the terminal 1-mm root tips from the primary and seminal roots were kept separate and processed in parallel, using the same fixation and freezing methods as for the Repli-seq and EdU-S/G1 samples.

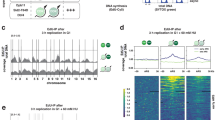
Replication timing (RT) protocols and flow cytometry nuclei sorting strategies. (a) Flow chart of Repli-seq, EdU-S/G1 and S/G1 RT protocols. Replication timing protocol steps are indicated in the black boxes. Steps in the three protocols that were compared are indicated below the text boxes, with a solid right arrow (→) indicating an included step and a dotted line indicating an omitted step. (b-d) Flow cytometry bivariate dot plots and histogram plots showing flow sorting gates used in each RT protocol. Nuclei populations for Repli-seq (b), and EdU-S/G1 (c) are displayed as bivariate dot plots of EdU incorporation (replication, y-axis) vs. DAPI (DNA content, x-axis). The nuclei population for S/G1 (d) is displayed as a DAPI (DNA content) univariate histogram, because no EdU was used. The Repli-seq plot shows the gates (rectangles) used to sort nuclei from early (E), middle (M), and late (L) S phase and G1 fractions. The EdU-S/G1 plot shows the gates used to sort nuclei from S-phase (S) and G1. The gates used to sort nuclei in S-phase (S) or the left half of the G1 peak (G1-left) in the S/G1 experiment are indicated as interval bars. Companion histograms for each RT protocol (e,f,g) show relative DNA content (DAPI) for the total nuclei population overlaid with the position and relative frequency of nuclei that fall in the respective sorting gates.
Nuclei were isolated from frozen root tips ground in cell lysis buffer (CLB) supplemented with a Complete protease inhibitor tablet (Roche # 04693116001) using a small food processor (Cuisinart)7,26. For the Repli-seq and EdU-S/G1 methods, EdU in labeled nuclei was conjugated to Alexa Fluor 488 (AF-488) using a Click-iT EdU Alexa Fluor 488 imaging kit (Invitrogen # C10337)9,26. Clicked nuclei were resuspended in CLB containing 2 µg/mL 4’,6-diamidino-2-phenylindole (DAPI) with 40 µg/mL ribonuclease A and filtered through a 20-µm nylon mesh filter (Partec) prior to flow sorting9,26. For the S/G1 method, nuclei were isolated and prepared for sorting as described above except that the click reaction step was omitted.
Flow sorting of S-phase nuclei
Nuclei were sorted using a FACS Aria III flow cytometer equipped with UV (355 nm) and blue (488 nm) lasers and a 70-micron nozzle. Data was acquired with the BD FACSDiva v9.0.1 software and subsequently analyzed in FlowJo v10.8.2. 1X STE (100 mM sodium chloride; 10 mM Tris, pH 7.5; 1 mM EDTA) was used as sheath buffer and catch tube collection buffer. Nuclei were sorted into 2-mL tubes using the four-way sort purity mode. A sequential gating strategy was used to remove debris and nuclei doublets from the mitotic population prior to sorting (Supplementary Fig. S1). First, side scatter light (SSC) was plotted as a function of DAPI fluorescence, using PMT voltages adjusted to center the G1 (2C) mitotic populations in the bottom third of the plots. A gate drawn in the SSC vs. DAPI plot was used to exclude cellular debris and endocycling nuclei from the mitotic population (for details see7) (Supplementary Fig. S1a). After debris were excluded, plots of FSC-A (forward scatter area) vs FSC-H (forward scatter height) and of SSC-A (side scatter area) vs SSC-H (side scatter height) were used sequentially to gate out nuclei doublets and other aggregates27 (Supplementary Fig. S1b, c). This strategy ensured that comparable populations of nuclei were used as the input for sorting in the three types of RT experiments, after which additional gates were used to define the nuclei of interest, as shown in (Fig. 1b–g).
For both the Repli-seq and EdU-S/G1 methods, the AF-488 signal representing incorporated EdU was used to distinguish replicating, EdU-labeled nuclei from non-replicating, unlabeled nuclei on the y-axis, with the DAPI signal indicating DNA content on the x-axis. For the Repli-seq method, gates were drawn around the G1 nuclei and three separate S-phase fractions with increasing DNA contents. Nuclei representing early, middle, and late stages of S phase9 were collected separately (Fig. 1b,e). For the EdU-S/G1 method, an S-phase gate was drawn to include most of the labeled, replicating nuclei in the S-phase arc and a G1 gate was drawn to include the unlabeled, non-replicating G1 nuclei (Fig. 1c,f). For the S/G1 method, nuclei were sorted on the basis of DNA content only, and a histogram plot of DAPI fluorescence vs number of nuclei was used to draw sorting gates (Fig. 1d,g). To minimize contamination of G1 nuclei with nuclei in early S phase, the G1 gate included only the left half of the G1 peak and the S-phase gate was placed between the G1 and G2 peaks, as indicated in (Fig. 1g). For this method, we increased the DAPI PMT voltage on the flow sorter to expand the relative width of the DAPI histogram along the x-axis and then drew the S-phase sorting gate to minimize contamination from nuclei with 2C and 4C DNA contents (compare x-axis of histogram in Fig. 1e–g). After sorting, all nuclei were snap frozen in liquid nitrogen and stored at -80ºC until DNA isolation.
DNA isolation, shearing, and immunoprecipitation
Reversal of formaldehyde cross links and isolation of DNA were carried out as described in26. Isolated DNA was diluted to a total volume of 120–125 uL in 1X or 0.1X TE and sheared for 100 s in a Covaris S220 Ultrasonicator using the following settings: Peak Incident Power, 140; Duty Cycle, 10%; and Cycles/burst, 200. These conditions resulted in an average sheared size of 325–425 bp. For the Repli-seq method, EdU-labeled DNA clicked to AF-488 was immunoprecipitated from each S-phase fraction. DNA samples were pre-cleared using magnetic Dynabeads protein G beads (Invitrogen # 10004D), and labeled DNA was immunoprecipitated using a 1:200 dilution of Alexa Fluor 488 polyclonal antibody (Invitrogen # A-11094, lot 2,551,344) as described in26. DNA from the G1 fraction was not immunoprecipitated and directly used for library preparation. For the EdU-S/G1 and S/G1 methods, sheared DNA preparations from S and G1 nuclear populations were used for library preparation without further processing.
Sequencing
DNA-seq libraries were made using the NEBNext Ultra II DNA Library Prep Kit for Illumina (# E7645). For S/G1 and EdU-S/G1 libraries, we used 100 ng DNA for library input, and following the library kit instructions, performed a SPRIselect (Beckman Coulter # B23317) bead-based size selection targeting 300–400 bp (not including adaptors) sequenceable DNA inserts after adaptor ligation, and used 7 PCR cycles to amplify the libraries. Average library sizes (adaptors + DNA insert) ranged between 503–540 bp for the S/G1 samples and 472–492 bp for EdU-S/G1 samples. Repli-seq libraries were made with 0.5 ng DNA input for each fraction, no size selection after adaptor ligation, and 11 PCR cycles of amplification. Average library sizes (adaptors + DNA insert) ranged from 390–436 bp.
For each type of replication timing experiment, libraries from the flow sorted fractions were barcoded using unique dual indexes prior to sequencing. For Repli-seq, barcoded libraries of each sorted fraction (G1, early, mid, late) corresponding to three independent biological replicates were pooled, sequenced using 150-bp paired-end reads on one lane of an Illumina NovaSeq 6000 S4 flow cell, and demultiplexed. For the EdU-S/G1 and S/G1 experiments, barcoded libraries from three independent biological replicates were similarly pooled, sequenced, and demultiplexed. In the case of S/G1, samples from primary and seminal roots were processed separately through the sequencing and demultiplexing. A preliminary analysis found no differences in replication timing profiles between primary and seminal roots from the same seedlings, and the data from matching sort gates and biological replicates were merged (i.e., primary root S-phase biological replicate 1 was merged with seminal root S-phase biological replicate 1) prior to processing and analysis. Data from all sequenced libraries were deposited in NCBI SRA under the accession numbers listed in Supplementary Table 4.
Comparing input material and cost of Repli-seq vs EdU-S/G1 and S/G1 experiments
While the source tissue in this study was maize root tips, any tissue or cell culture amenable to EdU labeling could be used for a DNA replication timing experiment if the number of available flow sorted nuclei is sufficient for library production, which in turn depends on genome size and the final DNA yield/nucleus. In maize, with a diploid genome size of ~ 2.4 Gb, about a million nuclei would be needed for each S-phase fraction in the Repli-seq protocol to yield a target library input of 100 ng DNA. The high number of nuclei in the Repli-seq protocol is necessary because immunoprecipitation of EdU-AF488 labeled DNA, the unique step that gives the protocol its increased specificity in measuring replication activity across different fractions of S phase, greatly reduces DNA yield. However, we have successfully used much less than 100 ng of input DNA for Repli-seq library construction. In contrast, the low DNA yield after immunoprecipitation is not a constraint in the S/G1 methods, for which 100,000 nuclei from each sorting gate are sufficient to generate well over 100 ng of DNA for a sequencing library.
The immunopreciptiation step in Repli-seq requires the purchase of Alexa Fluor 488 antibody and associated costs of magnetic beads, buffers, and personnel time at the bench. Repli-seq uses a G1 gate and three S-phase gates while both S/G1 methods use a G1 gate and one S-phase gate, so the biggest cost difference is a twofold increase in the number of libraries to prepare and sequence for Repli-seq. In addition, if the input material is expensive or rare, the need for additional starting material could also contribute significantly to the cost of Repli-seq.
Creating an in silico low mappability droplist
We developed an in silico strategy to identify segments of reference genomes with inherent low mappability. These segments are enriched in repeats and low-complexity regions that preclude the unambiguous alignment of short reads to unique locations in the genome. Hard to map regions in the reference genome is a concern in the S/G1 methods because late replicating regions are defined by their lower read coverage relative to G1. It is therefore important to exclude low mappability regions from the analysis to avoid misclassifying some of them as late replicating. We located these regions and created a low mappability drop list for masking the genome during downstream analysis (Supplementary Fig. S2a, Github link: https://github.com/ewheeler7/genome_mappability/).
To develop a “low mappability drop list” for the maize B73 assembly (AGPv5), artificial 150-bp reads were created by taking partially overlapping 150-bp windows, with a step size of 10 bp, across the whole B73 (AGPv5) genomic reference assembly. These artificial reads were then mapped back to the reference genome with bowtie2 v2.5.1. Mapped artificial reads were filtered by MAPQ score, keeping only reads with scores ≥ 6 to focus on uniquely mapping reads (see Supplementary Fig. S2b for the distribution of MAPQ values). The coverage of uniquely mapping reads was determined at the single base pair level (see Supplementary Fig. S2c–e for chromosome 1 and an example of expanded genomic regions). Then the genome was divided into non-overlapping 10-kb bins and the percentage of each bin with zero coverage by the MAPQ filtered artificial reads was calculated. We used non-overlapping 10-kb bins to align with the binning strategy we used with the experimental data. If 60% or more of a bin was void of artificial reads, that 10-kb bin was included in the “low mappability” drop list (see Supplementary Fig. S2e for an example of a 10-kb drop listed bin). Using 60% zero read coverage as a cutoff removes 2% of the genomic 10-kb bins and 1% of the genes from the maize reference genome. While several software packages are available to measure reference genome mappability28,29, using the same read mapper as used with experimental data is a simple and quick method to identify regions of inherently poor mappability.
Read pre-processing
Commands used for data processing can be found on Github (link: https://github.com/ewheeler7/replication_timing_methods_comparison). Sequenced reads were trimmed using Trimmomatic v0.3930 with default parameters. The two sequencing runs (from the same libraries) for the EdU-S/G1 method were merged and the sequencing runs for the primary and seminal roots for the S/G1 method were merged. See Supplementary Tables 1–3 for the number of reads processed at each step per sample. Trimmed, merged reads from each replication timing method were mapped using bowtie2 v2.5.131 to the B73 NAM version 5 reference genome, including scaffold sequences8. Duplicate mapped reads were removed using sambamba v1.0.032 markup, and only properly paired reads with a bowtie2 MAPQ value of 6 or greater were extracted using samtools v1.9–433 with -bf 0 × 2 and -q 6 parameters. Finally, reads from the scaffolds and the low mappability drop list were removed.
The coverages of the final mapped reads from all three replication timing experiments were calculated in non-overlapping 10-kb windows using DeepTools v3.5.434. To remove spikes in read coverage, which are likely due to collapsed repeats, 0.25% of 10-kb bins windows with the highest coverage in each G1 sample (for EdU-S/G1 and S/G1 independently) were combined into a pooled high coverage drop list and removed from all S and G1 samples. While spikes in read coverage are rare, they accumulate a disproportionate number of reads which distort the 1X normalization step, so these regions are removed prior to normalization.
After removal of both the high coverage and low mappability drop list reads, each EdU-S/G1 and S/G1 sample was 1X normalized using DeepTools v3.5.4 bamCoverage with parameters of a bin size of 10 kb, RPGC mode (Reads Per Genomic Content, or 1X normalization), ignoring duplicates and extending reads. The dropped regions from the high and low coverage drop lists were subtracted from the effective genome size used to normalize the data.
For Repli-seq, after removing both the high coverage and low mappability drop lists the mapped reads are processed using the Repliscan application35, which includes the 1X normalization step and classifies genomic regions into discrete replication signatures (see below). The theoretical coverage obtained by the number of filtered mapped reads is reported in the last column of (Supplementary Tables 1–3).
Repli-seq RT classification
After DNA from all Repli-seq fractions was sequenced, trimmed, mapped, and filtered, the Repliscan pipeline35 (link to Repliscan container: https://zenodo.org/records/13937103, DOI: https://doi.org/10.5281/zenodo.13937103) was run to calculate the strength of signal from the early, mid, and late S-phase gates and to segment the genome into one of five classes: Early (E), Early-Mid (EM), Mid (M), Mid-Late (ML), or Late (L). Although only three S-phase gates are sorted, Repliscan can computationally define regions that replicate in intermediate classes like EM or ML. The following parameter modifications were used for the Repliscan pipeline: (1) read densities were aggregated in 10-kb non-overlapping windows across the genome ( –window 10,000) and (2) a segmentation threshold of 1.0 (–threshold value, –value 1.0).
Data from the new Repli-seq experiment was compared with Repli-seq data published in Wear, et al.9 2017, which was originally mapped to B73 RefGen_v3 (AGPv3) (NCBI SRA # PRJNA327875). The earlier Repli-seq data was re-mapped to the B73 NAM version 5 genome assembly (Zm-B73-NAM-5.08) using the same filtering parameters, high coverage drop list and low mappability removal as described for the EdU-S/G1 and S/G1 data processing. The re-mapped data was run through Repliscan with the same parameters described above to create the RT signal profiles from each S-phase nuclei population and to assign RT classes (see Supplementary Table 5 for GEO accessions for these files, with both low mappability and high coverage drop lists removed).
The early, mid, and late replication timing signal outputs from Repliscan from the 20179 and current experiments have correlation coefficients of r = 0.98, 0.93, and 0.96 respectively (Supplementary Fig. S3a). However, there is a slight early shift in the 2025 coverage of each RT class defined by Repli-seq, such that some of the Early-Mid class regions are re-assigned into Early and some Late regions are re-assigned to Mid (see Supplementary Fig. S3b and Supplementary Table S6).
Comparing experiments with different sequencing depths
Sequencing depths differed considerably among the experiments for the different replication timing methods (see Supplementary Tables S1–S3 last column). We down-sampled the EdU-S/G1 data to 6X for more appropriate comparisons with S/G1 data. However, there are no apparent differences between the different coverages in the EdU-S/G1 down-sampled data, indicating that the EdU-S/G1 method can be useful even at moderate coverages (Supplementary Fig. 4a–c).
Calculating the S/G1 ratio, smoothing, and averaging bioreps
For both S/G1 approaches, replication is inferred from the ratio of normalized S-phase reads to normalized G1 reads, with higher ratios corresponding to earlier replication. The S/G1 ratios were calculated in sequential, non-overlapping 10-kb bins across the genome. In each bin, the S-phase signal for a given biological replicate was divided by the average signal of all the G1 replicates for that bin. All 10-kb bins with a ratio value of zero in any replicate were removed from all replicates. We also removed “stand-alone” single bins of data flanked on both sides by a drop listed bin. Haar wavelet smoothing36 (https://staff.washington.edu/dbp/WMTSA/NEPH/wavelets.html) was performed at level 2 on each replicate. After replicate data was processed through the smoothing step, an average profile of all bioreps was made for EdU-S/G1 and S/G1.
Because replication duplicates the DNA at each locus, it is intuitive for S/G1 ratios to vary between 1 for unreplicated regions and 2 for replicated regions. Both the S and G1 sequencing data are normalized to 1X coverage, which sets the mean ratio of the two datasets to 1. In principle, with this normalization—and assuming pure sorted populations of S phase and G1 nuclei—the earliest replicating regions of the genome should have S/G1 = 2 and the latest regions should have S/G1 = 0. However, in reality, there is enough heterogeneity in populations of nuclei that very few loci exhibit these theoretical extreme ratios.
Pearson’s correlation coefficients between and among the EdU-S/G1 and S/G1 bioreps were calculated and, as shown in Supplementary Fig. S4a, the minimum correlation coefficient was r = 0.93. A visual comparison of the S/G1 and EdU-S/G1 profiles at all coverages show consistent signal structure between all biological replicates (Supplementary Fig. S4b,c). The biorep average for the EdU-S/G1 or S/G1 profiles were also compared to the Repli-seq early, mid, and late profiles (Supplementary Fig. S5). The EdU-S/G1 6X coverage profile and Repli-seq E profile had a correlation coefficient of r = 0.89, while the S/G1 6X coverage profile and Repli-seq E profile had a correlation coefficient of r = 0.85. Similarly strong negative correlations with the Repli-seq L profile were observed. A negative correlation between S/G1 data and Repli-seq L data is expected because in S/G1 type data, late replication is identified by a depletion of reads, whereas in Repli-seq, late replicating DNA is identified by elevated read coverage from EdU-labeled DNA regions in nuclei from late S phase. A two-tailed paired Wilcoxon rank sum test using the R package rstatix v0.7.2 was performed to compare the EdU-S/G1 and S/G1 methods for the genome and for each individual RT class (Supplementary Table 7). The Wilcoxon effect size (r) was also calculated. Replication time at genes and TE families was calculated by averaging the genomic 10-kb bin data at each gene and TE element, using annotation files from the NAM project annotation8.
To explore replication timing across the entire maize genome in a meta-chromosome or relative position from the centromere format, we divided each chromosome arm evenly into 10 windows each representing 10% of the chromosome arm. For this analysis, the chromosome arm boundary extends to the edge of the centromere but does not include the centromere. Short arms and long arms both exhibit the trend of earlier replication in the distal end and later replication in the pericentromeric end of the chromosome arm. By dividing each arm into 10% windows we can associate the relative telomeric ends of all chromosome arms together and the relative centromeric regions together, regardless of absolute chromosomal size. The median EdU-S/G1 and S/G1 ratios from all the genomic 10-kb bins contained in each 10% window was calculated, as were the ratios for all genes and TE superfamilies in each window. Processed files for each method were deposited at GEO, see Supplementary Table S5 for accession numbers.
Results and discussion
Flow cytometry considerations for measuring replication time
Separating replicating and non-replicating nuclei is a critical step for determining RT for both the Repli-seq and S/G1 approaches. This is frequently accomplished by using flow sorting to separate nuclei in G1 and S phase. In Repli-seq, nascent DNA is immunoprecipitated from nuclei in different S-phase sorting gates and compared to a G1 DNA sample (Fig. 1b). In the S/G1 methods, total DNA coverage in a gate that includes much of S phase is compared to DNA coverage in a G1 gate (Fig. 1c,d). It is important to obtain a non-replicating G1 population to accurately calculate RT in both approaches.
When sorting for conventional S/G1, only the left side of the G1 peak is collected to minimize contamination by early S-phase nuclei, and a narrow S gate is used to avoid G1 nuclei (Fig. 1d,g). However, using DAPI staining to measure DNA content has limitations, and cannot completely separate nuclei at the very beginning and very end of S phase from G1 and G2 populations, respectively. This can be seen in the EdU-labeled bivariate sorting profiles (Fig. 1b,c), where labeled S-phase nuclei actually span the whole width of the G1 and G2 peaks. The presence of S-phase nuclei in the G1 peak can also be visualized by overlaying the bivariate gates in a DNA content profile (Fig. 1e,f). An in silico overlay of the G1 left peak sorting gate of non-EdU, DAPI-only sorting experiments onto a EdU bivariate plot shows that ~ 10% of the G1 left peak nuclei are early S contaminants (Supplementary Fig. S6b,c). This contamination of the G1 control by early S-phase nuclei results in higher DNA coverage values in the G1 control that then decreases the RT signal ratio, primarily in the earliest-replicating regions. This dampening of early RT ratios in early regions could reduce resolution for researchers interested in early replicating regions that are enriched for genic sequences37.
Even with the ability to sort pure replicating vs non-replicating nuclei when using EdU, we did not extend the S-phase gate to fully include the 4C DNA content region of the EdU-labeled arc because, as is common in plant systems, maize terminal 1-mm root tips contain a small subpopulation of cells that undergo endocycling6. Thus, a population of labeled nuclei with a 4C DNA content containing primarily late mitotic S phase nuclei will also include some early replicating endocycling nuclei. We chose to avoid adding this complication to the EdU-S/G1 data.
Chromosomal distribution of replication time
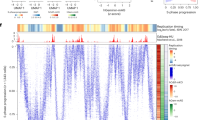
Using chromosome 1 as an example, Fig. 2 shows a comparison of the chromosomal distribution of replication timing data generated from Repli-seq and the two S/G1 methods. In the Repli-seq protocol, newly synthesized DNAs from the early, middle and late S phase nuclei produce replication signals where a high signal from early S phase indicates earlier replication and a high signal from late S phase indicates later replication. For S/G1 methods higher signal represents earlier replication, and earlier replicating sequences will have higher copy numbers than later replicating sequences20,23,38. Because both G1 and S phase reads are normalized, a S/G1 ratio above 1 indicates an earlier replicating locus whereas a ratio below 1 indicates a later replicating locus. Because of variations in sequencing depth (see Methods and Supplementary Tables S1–3) the EdU-S/G1 data were down-sampled to 6X to remove coverage depth effects as a variable when comparing to the S/G1 data.

Chromosome 1 replication timing data from Repli-seq, EdU-S/G1 and S/G1 methods. (a) Repli-seq intensity profiles of 1X normalized signal from each S-phase fraction as a ratio to G1 in 10-kb windows across chromosome 1 (Chr 1) for early (E, blue), middle (M, green), and late (L, red) S phase (scales at 0-5). The RT Class annotation track shows the genome segmentation assigned by Repliscan. The scale at the top of panel a shows the chromosome coordinates in Mbp, and is used for both panels a-b. The locations of the enlarged regions shown in panels c-d are indicated on the chromosome graphic with red boxes, and the centromere is marked with “CEN”. (b) The S/G1 ratio signal for EdU-S/G1 (purple) and conventional S/G1 (black) methods for each 10-kb bin (scale of 0.5-1.5). Higher S/G1 ratios indicate earlier replication and lower S/G1 ratios indicate later replication. (c) Enlargement of a mostly early replicating Chr1 region, with vertical dashed lines aligning Repli-seq early peaks with EdU-S/G1 and S/G1 peaks. (d) Enlargement of a mostly late replicating Chr1 region, with vertical dashed lines aligning Repli-seq late peaks with EdU-S/G1 and S/G1 valleys. (e) An enlargement of a 7-Mbp region of chromosome 1 showing the same profiles as in panels a-b. The dashed rectangles outline typical examples of early (blue dashed box) and late (red dashed box) Repli-seq peaks, and the corresponding ratio data for the S/G1 methods. The black dashed rectangle outlines an example of replication progression that is most clear in the Repli-seq data. A peak of early replication (blue arrowhead) proceeds bidirectionally to two flanking peaks in mid (green arrowheads) and then to two peaks in late (red arrowheads). The S/G1 methods, which only have a single S-phase population, have a peak of signal indicating early replication that transitions to a valley, indicating late replication (black arrow).
The Repli-seq early profile (Fig. 2a, top blue track) shows high replication activity at the gene-rich ends of the chromosome arms while the Mid profile shows a more evenly dispersed activity across the chromosome arms. In contrast, the Repli-seq Late profile exhibits notably high replication activity in the pericentromeric regions. The final track in Fig. 2a displays the replication timing class assigned by Repliscan.
The replication profiles obtained with the two S/G1 methods (Fig. 2b) look very similar to the early signal of Repli-seq. Peaks of the S/G1 profiles, which indicate local regions of earlier replication, align with Repli-seq early signal peaks, as indicated by the dashed lines in (Fig. 2c). Repli-seq early peaks appear somewhat sharper than their corresponding S/G1 peaks, but the positional alignment of peaks is consistent for all profiles. Peaks in Repli-seq Late correspond to valleys in the S/G1 profiles (Fig. 2d dashed lines, and Fig. 2e red dashed box), as expected because regions of late replication have a high signal in Repli-seq Late, but a low ratio in both S/G1 methods. The concordance of these profiles indicates that all three methods offer a reliable measure of replication timing.
From these replication profiles we can also visualize the chromosomal progression of S phase in both Repli-seq and S/G1 types of experiments. Local S-phase progression in Repli-seq can be seen as chromosomally sequential peaks of signal from the early to mid to late S-phase sorting gates9,10,18,39 (Fig. 2e, triangles in black dashed box), while the S/G1 methods have a peak of signal at early replication that transitions gradually to a valley in late replicating regions (Fig. 2e, see arrow). Using the Repli-seq as the benchmark measure of replication timing, we show that the S/G1 methods faithfully captures RT profiles in our maize root tip system.
Sorting with EdU improves measurement of early and late replication timing regions
The replication timing profiles generated by the EdU-S/G1 and the S/G1 methods look remarkably similar (Fig. 3a), but a closer examination reveals that in distal regions of chromosome arms aligned peaks show earlier values in the EdU-S/G1 method (Fig. 3b). To a lesser degree, the reverse is true in pericentromeric regions, with EdU-S/G1 having later values at valleys in common between the two methods (Fig. 3c). This effect is likely due to the ability to generate a pure G1 population in the EdU-S/G1 protocol. In the S/G1 protocol, ca. 10% of the G1 population are in early S phase (Supplementary Fig. S6) and have already started replicating the genome. The contaminating S phase nuclei will increase the copy number in the G1 control and result in a corresponding reduction in the S/G1 ratio values in early replicating regions.

Comparison of replication time distributions of the EdU-S/G1 and S/G1 ratios. An overlay of the EdU-S/G1 (purple) and S/G1 (black) ratio data along chromosome 1 (a), and representative enlarged regions at the end of the chromosome arm, which is mostly early replicating (b), and near the centromere, which is mostly late replicating (c). (d) The whole genome distribution of S/G1 ratios from EdU-S/G1 and conventional S/G1 (outliers are not shown). (e) The same ratio values in panel d separated by regions of the genome represented in each Repli-seq RT segment class (RT class). EdU-S/G1 ratios are earlier than S/G1 ratios in early Repli-seq segments. In mid segments, the distributions for both methods are similar. EdU-S/G1 has later ratios than S/G1 in late Repli-seq segments. A red line at S/G1 = 1 indicates mid S.
When comparing the EdU-S/G1 or S/G1 ratio values irrespective of genomic location, i.e., by plotting the distribution of all genomic bin S/G1 ratio values (Fig. 3d), the bulk of the data is very similar between the two methods, as seen by the nearly identical median and first and third quartile values. However, in the EdU-S/G1 distribution the first and fourth quartile tail populations are extended toward earlier and later replication ratio values compared to the more compressed S/G1 distribution. When the genomic distributions are separated by Repli-seq segmentation classes (Fig. 3e) this extension of earlier and later data points in the EdU-S/G1 relative to the S/G1 method can be most clearly seen in the Repli-seq early (E) and late (L) segment classes, respectively.
Using the Wilcoxon rank sum test to compare EdU-S/G1 and S/G1 in each RT class, we see that in all classes EdU-S/G1 and S/G1 are statistically different. However, this statistical significance could be due to the large number of genomic bins considered (sample size). Consequently, we measured the effect size to quantify the magnitude of the differences. This analysis found small effects for the Early-Mid, Mid, and Mid-Late classes, but large effects for the Early and Late classes (Supplementary Table 7). Although the direction of the effect is not indicated in the statistical test, genome browsing (such as in Fig. 3b,c) and comparing the distributions by RT class (Fig. 3e) indicated that the EdU-S/G1 method has the effect of producing slightly earlier early values and later late values. Taken together, these observations suggest that the EdU-S/G1 approach can provide a wider range of early and late replicating ratio values at the maxima of early peaks and minima of late valley regions, allowing for more resolution in these regions. The slight improvement in EdU-S/G1 over S/G1 in early and late peaks and valleys could be useful if the genomic area under study is in early or late replicating regions.
Comparing genic and transposable element (TE) replication
The maize genome is made up of features that can be classified as genes, TEs, intergenic regions or repeats. Genes constitute ca. 8% of the maize genome while TE families comprise over 75% of the genome40. Despite their repetitive nature, most maize TEs can be uniquely mapped allowing us to assess their replication timing compared to other genomic features. The TE superfamilies each have different distributions and we wanted to investigate possible differences in replication timing among the TE families, especially given the notable effect of relative chromosomal position on replication timing.
Figure 4 compares the distribution of replication times obtained by the S/G1 and EdU-S/G1 methods for the genome as a whole (“Genomic bins”), genes, class I retrotransposon superfamilies (Copia, Gypsy, and unknown LTR), and class II DNA transposon superfamilies (Helitron, hAT, CACTA, Pif-Harbinger, Mutator, and Tc1-Mariner). Because of their small number of elements, individual superfamilies within the Class I retrotransposon LINE order were analyzed together (Supplemental Table 8).

Distributions of EdU-S/G1 and S/G1 ratios for genes and TE superfamilies. The distributions of EdU-S/G1 (purple) and S/G1 (black) ratio values for all genomic bins, genes, and indicated TE superfamilies. The percent genomic coverage of each gene and TE superfamily is included. A red line at S/G1 = 1 indicates mid S.
The EdU-S/G1 and S/G1 RT distributions are similar to each other whether looking at the genome, genes, or TE superfamilies (Fig. 4). As seen previously in the genomic data (Fig. 3), all gene and TE EdU-S/G1 distributions show extended tail populations in earlier and later values compared to the S/G1 distributions. The median values of the genes and the LINE, Pif-Harbinger, hAT, Tc1-Mariner, Helitron, and Mutator TE superfamilies are earlier to varying degrees than the distribution for the genome as a whole (“Genomic bins”). In contrast, the median values for the unknown LTR and Gypsy superfamilies are somewhat later than the genome as a whole. In all cases, the differences between the spread of the EdU-S/G1 and S/G1 distributions are likely explained by the reduced dynamic range attributable to minor contamination of samples without EdU-based sorting.
Replication timing of genes and TEs can also be visualized in a chromosomal context by plotting RT values in a meta-chromosome-arm format. This approach allows the S/G1 ratios of specific genomic elements to be analyzed as a function of relative chromosome position (Fig. 5a). Using this approach, comparison of both S/G1 methods yielded highly similar results for all genes and TE families. Minor differences were observed near the telomeres and centromeres where the EdU-S/G1 method has earlier or slightly later RT values, respectively (Fig. 5a). Notably, the general pattern of chromosome replication, using either S/G1 method, mirrors the replication profiles illustrated in Fig. 2a and b where the ends of chromosome arms replicate earlier and centromeric regions replicate later. This pattern is suggestive that all genomic elements are subject to a large-scale chromosome position effect on RT.

Meta-chromosome-arm plots of genomic, genic and TE replication time. The median EdU-S/G1 and S/G1 ratios from each 10% window were calculated for the genome, genes, and the indicated TE superfamilies and plotted as a meta-chromosome-arm line graph (a). Comparison of EdU-S/G1 (b) and S/G1 (c) ratio signal at genes, hAT and Gypsy superfamilies (solid line) to the genome (dashed line). The centromere (marked C) is not given an RT value. A red line at S/G1 = 1 indicates mid S.
However, local differences between genomic elements and their local genomic environment can be seen as well by comparing the RT of genes and TE superfamilies to the genome (Fig. 5b,c and Supplemental Fig. S7). For the same relative chromosomal position, genes and some TE superfamilies, like hAT, replicate earlier and some TE superfamilies, like Gypsy, replicate slightly later than the genome as a whole, especially in the distal arms. These observations suggest that RT is affected by both genome-wide scale and by more localized influences.
In pericentromeric regions of the genome, the genes, as well as the LINE, Pif-Harbinger, hAT, and Tc1-Mariner elements have earlier RT compared to the genome as a whole, indicating they replicate earlier than the genomic regions around them. The CACTA, unknown LTR and Gypsy superfamilies replicate similarly to the genome throughout the chromosome arms. However, given that Gypsy elements are highly abundant, constituting up to 60% of some of the 10% relative windows, and are concentrated in pericentromeric and centromeric regions, (Supplementary Fig. S8), it is difficult to distinguish between overall genomic replication and Gypsy replication in those windows. Although their late RT could be due to their chromosomal distribution, the Gypsy and unknown LTRs also show slightly later RT compared to the genome in the distal arms (Supplementary Fig. 7a, b). Further study is needed to determine if these TEs families are active or passive elements in determining replication time in their local environment.
Conclusion
Repli-seq, EdU-S/G1 and S/G1 are three useful and highly correlated approaches for measuring replication times during S phase. A summary of each method’s features is listed in (Table 1). If EdU labeling of source tissue is feasible and enough material is available for bivariate sorting of labeled nuclei into multiple gates within S phase, Repli-seq offers the highest resolution as well as the ability to distinguish timing heterogeneity at each locus. When starting material is limited, or when the need to analyze many species or genotypes makes Repli-seq impractical, the S/G1 method can be applied successfully to characterize average RT at each locus across the genome. Labeling replicating DNA is not required for this method. However, when EdU labeling is feasible, a broader sample of replicating nuclei and better separation from non-replicating nuclei can be obtained by bivariate sorting. Thus, the EdU-S/G1 procedure can further improve the resolution of early and late replication times compared to the conventional S/G1 procedure.
Data availability
Sequencing data is stored at the NCBI SRA database under the accession PRJNA1219493, PRJNA1136904, and PRJNA1137362. Processed files are stored at the NCBI GEO database under the accession GSE288800, GSE287258, GSE287085.
References
Gilbert, D. M. Evaluating genome-scale approaches to eukaryotic DNA replication. Nat. Rev. Genet. 11, 673–684. https://doi.org/10.1038/nrg2830 (2010).
Sequeira-Mendes, J. & Gutierrez, C. Links between genome replication and chromatin landscapes. Plant J. 83, 38–51. https://doi.org/10.1111/tpj.12847 (2015).
Savadel, S. D. & Bass, H. W. Take a look at plant DNA replication: recent insights and new questions. Plant Signal Behav. 12, e1311437. https://doi.org/10.1080/15592324.2017.1311437 (2017).
Vouzas, A. E. & Gilbert, D. M. Mammalian DNA replication timing. Cold Spring Harb. Perspect. Biol. https://doi.org/10.1101/cshperspect.a040162 (2021).
Hulke, M. L., Massey, D. J. & Koren, A. Genomic methods for measuring DNA replication dynamics. Chromosome Res. 28, 49–67. https://doi.org/10.1007/s10577-019-09624-y (2020).
Bass, H. W. et al. A maize root tip system to study DNA replication programmes in somatic and endocycling nuclei during plant development. J. Exp. Bot. 65, 2747–2756. https://doi.org/10.1093/jxb/ert470 (2014).
Wear, E. E. et al. Isolation of plant nuclei at defined cell cycle stages using EdU labeling and flow cytometry. Methods Mol. Biol. 1370, 69–86. https://doi.org/10.1007/978-1-4939-3142-2_6 (2016).
Hufford, M. B. et al. De novo assembly, annotation, and comparative analysis of 26 diverse maize genomes. Science 373, 655–662. https://doi.org/10.1126/science.abg5289 (2021).
Wear, E. E. et al. Genomic analysis of the DNA replication timing program during mitotic S phase in maize (Zea mays) root tips. Plant Cell 29, 2126–2149. https://doi.org/10.1105/tpc.17.00037 (2017).
Hansen, R. S. et al. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl. Acad. Sci. USA 107, 139–144. https://doi.org/10.1073/pnas.0912402107 (2010).
Chen, C. L. et al. Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res. 20, 447–457. https://doi.org/10.1101/gr.098947.109 (2010).
Rivera-Mulia, J. C. et al. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res. 25, 1091–1103. https://doi.org/10.1101/gr.187989.114 (2015).
Schübeler, D. et al. Genome-wide DNA replication profile for Drosophila melanogaster: a link between transcription and replication timing. Nat. Genet. 32, 438–442. https://doi.org/10.1038/ng1005 (2002).
Hiratani, I. et al. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 6, e245. https://doi.org/10.1371/journal.pbio.0060245 (2008).
Rivera-Mulia, J. C. et al. Allele-specific control of replication timing and genome organization during development. Genome Res. 28, 800–811. https://doi.org/10.1101/gr.232561.117 (2018).
Salic, A. & Mitchison, T. J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. USA 105, 2415–2420. https://doi.org/10.1073/pnas.0712168105 (2008).
Kotogany, E., Dudits, D., Horvath, G. V. & Ayaydin, F. A rapid and robust assay for detection of S-phase cell cycle progression in plant cells and tissues by using ethynyl deoxyuridine. Plant Methods https://doi.org/10.1186/1746-4811-6-5 (2010).
Concia, L. et al. Genome-wide analysis of the Arabidopsis replication timing program. Plant Physiol. 176, 2166–2185. https://doi.org/10.1104/pp.17.01537 (2018).
Wear, E. E. et al. Comparing DNA replication programs reveals large timing shifts at centromeres of endocycling cells in maize roots. PLoS Genet. 16, e1008623. https://doi.org/10.1371/journal.pgen.1008623 (2020).
Koren, A., Soifer, I. & Barkai, N. MRC1-dependent scaling of the budding yeast DNA replication timing program. Genome Res. 20, 781–790. https://doi.org/10.1101/gr.102764.109 (2010).
Siefert, J. C., Georgescu, C., Wren, J. D., Koren, A. & Sansam, C. L. DNA replication timing during development anticipates transcriptional programs and parallels enhancer activation. Genome Res. 27, 1406–1416. https://doi.org/10.1101/gr.218602.116 (2017).
Armstrong, R. L. et al. Chromatin conformation and transcriptional activity are permissive regulators of DNA replication initiation in drosophila. Genome Res. 28, 1688–1700. https://doi.org/10.1101/gr.239913.118 (2018).
Woodfine, K. et al. Replication timing of the human genome. Hum. Mol. Genet. 13, 191–202. https://doi.org/10.1093/hmg/ddh016 (2004).
Sasaki, T. et al. Stability of patient-specific features of altered DNA replication timing in xenografts of primary human acute lymphoblastic leukemia. Exp. Hematol. 51, 71-82.e73. https://doi.org/10.1016/j.exphem.2017.04.004 (2017).
Voichek, Y. et al. Cell cycle status of male and female gametes during Arabidopsis reproduction. Plant Physiol. 194, 412–421. https://doi.org/10.1093/plphys/kiad512 (2023).
Mickelson-Young, L. et al. A protocol for genome-wide analysis of DNA replication timing in intact root tips. Methods Mol. Biol. 2382, 29–72. https://doi.org/10.1007/978-1-0716-1744-1_3 (2022).
Wersto, R. P. et al. Doublet discrimination in DNA cell-cycle analysis. Cytometry 46, 296–306. https://doi.org/10.1002/cyto.1171 (2001).
Derrien, T. et al. Fast computation and applications of genome mappability. PLoS ONE 7, e30377. https://doi.org/10.1371/journal.pone.0030377 (2012).
Pockrandt, C., Alzamel, M., Iliopoulos, C. S. & Reinert, K. GenMap: ultra-fast computation of genome mappability. Bioinformatics 36, 3687–3692. https://doi.org/10.1093/bioinformatics/btaa222 (2020).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. https://doi.org/10.1093/bioinformatics/btu170 (2014).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. https://doi.org/10.1038/nmeth.1923 (2012).
Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J. & Prins, P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034. https://doi.org/10.1093/bioinformatics/btv098 (2015).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience https://doi.org/10.1093/gigascience/giab008 (2021).
Ramirez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160-165. https://doi.org/10.1093/nar/gkw257 (2016).
Zynda, G. J. et al. Repliscan: a tool for classifying replication timing regions. BMC Bioinform. 18, 1–14. https://doi.org/10.1186/s12859-017-1774-x (2017).
Percival, D. B. & Walden, A. T. Wavelet Methods for Time Series Analysis (Cambridge University Press, 2000).
Rhind, N. & Gilbert, D. M. DNA replication timing. Cold Spring Harb. Perspect. Biol. 5, a010132. https://doi.org/10.1101/cshperspect.a010132 (2013).
Armstrong, R. L., Das, S., Hill, C. A., Duronio, R. J. & Nordman, J. T. Rif1 functions in a tissue-specific manner to control replication timing through its PP1-binding motif. Genetics 215, 75–87. https://doi.org/10.1534/genetics.120.303155 (2020).
Zhao, P. A., Sasaki, T. & Gilbert, D. M. High-resolution Repli-Seq defines the temporal choreography of initiation, elongation and termination of replication in mammalian cells. Genome Biol. 21, 76. https://doi.org/10.1186/s13059-020-01983-8 (2020).
Stitzer, M. C., Anderson, S. N., Springer, N. M. & Ross-Ibarra, J. The genomic ecosystem of transposable elements in maize. PLoS Genet. 17, e1009768. https://doi.org/10.1371/journal.pgen.1009768 (2021).
Acknowledgements
We thank Larkin C. Hannah, University of Florida, for the B73 seed used for the 2025 Repli-seq, EdU-S/G1, and S/G1 replication timing experiments.
Funding
The funding was supported by National Science Foundation, 2025811.
Author information
Authors and Affiliations
Contributions
HWB, WFT and LH-B funding acquisition, conceptualization. EEW, LMY, MB designed and executed the experiments. EAW, LMY, EEW, and LC analyzed the data. EEW, LMY, and EAW created figures and tables. EAW, LMY, EEW, and WFT drafted the manuscript and all authors critically revised and approved the final manuscript. LC, HB, LH-B reviewed and edited the manuscript. All authors have reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wheeler, E., Mickelson-Young, L., Wear, E.E. et al. A comparison of genomic methods to assess DNA replication timing. Sci Rep 15, 17761 (2025). https://doi.org/10.1038/s41598-025-02699-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-02699-0
