
Abstract
We evaluated the patterns of genetic diversity, structure, and differentiation both within and among populations levels of Sophora koreensis in South Korean populations using the genotyping-by-sequencing method. We obtained 1,463 single nucleotide polymorphisms from 92 individuals from 12 populations. Genetic diversity analysis revealed a significant heterozygote deficiency (P < 0.001) and low levels of genetic diversity (average expected heterozygosity = 0.150, mean observed heterozygosity = 0.015, and nucleotide diversity = 0.163) in S. koreensis. Three peripheral populations (SC, SE, and SF) located at the southeastern margin of S. koreensis distribution showed lower levels of genetic diversity than other populations. Further to the population admixture results, Principal Component Analysis and barrier analyses showed clear genetic differentiation between the three peripheral populations and the other main populations. The genetic patterns and structures of the three peripheral populations were most likely a consequence of their isolation from the main populations and their small effective population sizes. Geographic and hydrological barriers between peripheral and central populations effectively interfere with gene flow among populations. Habitat protection should be strengthened for the entire distribution of S. koreensis to maintain its genetic diversity and unique genotypes, including peripheral populations.
Similar content being viewed by others
Introduction
Determining genetic diversity is a critical aspect of population genetics that helps us understand the genetic variation that exists within and between the populations of a particular species. Genetic variation within a population is maintained through various genetic events, such as gene flow, natural selection, mutation, and genetic drift, which can either maintain or disrupt the genetic equilibrium in a population1. In small populations, genetic drift can have an even greater impact, leading to the loss of genetic diversity over time2. Similarly, fragmentation and the founder effect can result in a loss of genetic diversity by reducing gene flow between populations, ultimately decreasing the overall genetic diversity within a species3,4. Clonal or asexual reproduction can also decrease genetic diversity within a population owing to a lack of recombination and limited reproductive potential, ultimately reducing the adaptive potential of the population and increasing its vulnerability to environmental changes5. Therefore, while clonal reproduction has ecological advantages, such as rapid dispersal and avoidance of the costs associated with sexual reproduction, it is not as effective in maintaining genetic diversity as sexual reproduction6,7. Small populations, especially those of threatened species, are more vulnerable to extinction due to genetic drift that decreases heterozygosity and inbreeding that increases homozygosity within populations8,9,10. Therefore, assessing genetic diversity and structure within a population is crucial for understanding the evolutionary processes of endangered plants and developing effective conservation strategies. Molecular analyses, such as microsatellite genotyping or DNA sequencing, are typically used to measure the structure of genetic diversity within populations and provide valuable insights into the genetic architecture of species, which has important implications in a range of fields.
Sophora L. contains approximately 50–70 species of the Fabaceae family and is known for its high species diversity in tropical and temperate regions worldwide11. However, despite their widespread distribution, some of these species are geographically restricted. Sophora koreensis Nakai, Korean necklace pod, is a rare species in East Asia that is endemic to the Korean Peninsula; it is a small deciduous shrub (0.5–1 m in height) and it grows yellow papillinaceous flowers from April to May (Fig. 1). Its legume is elongated, cylindrical, or slightly curved, maturing between July and September, and it contains 1–4 seeds12. Although effective pollinators and the seed dispersal mechanisms of S. koreensis have not been identified, it has been reported that bumblebees, Bombus diversus diversus, frequently visit yellow flowers of this species during the blooming season12. It was initially assigned to the genus Sophora but was later elevated to its own Korean endemic genus (Echinosophora Nakai) because of the unique characteristics of its shoots, which grow from creeping rhizomes and four-winged pods13 (Fig. 1). However, molecular and embryological analyses have suggested that Echinosophora should be treated as S. koreensis Nakai, and the generic name Echinosophora has now been eliminated14. This species is distributed from the central to northern parts of the Korean Peninsula. Most southern populations were located in Gangwon province of South Korea (Fig. 2). Owing to its limited distribution, the International Union for Conservation of Nature Red List classifies it as endangered (EN)15.

Habit of Sophora koreensis in a natural population. (a) yellow flowers, (b) four-winged pods, and (c) seeds.

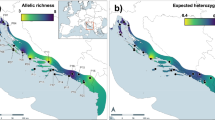
Distribution map and genetic clustering results of S. koreensis. Pie charts represent the ADMIXTURE Q-values for best the clustering K = 4 result. The sizes of the charts reflect the sample size per population. Genetic barriers among 12 S. koreensis populations, predicted using BARRIER (version 2.2). The genetic barriers are shown in bold red lines. Orange lines indicate Voronoi tessellation which is the optimal arrangement of barriers in the environment to maximize protection. The map source was obtained from the Public Data Portal of the Republic of Korea (data.go.kr), which provides open-source shapefiles (.shp). The map was further refined and customized using QGIS (version 3.42, qgis.org).
Previous studies on S. koreensis have mainly focused on pollen morphology16,17, ecology, and native habitats18,19,20,21, chemical components22,23,24,25,26, mass propagation27, genetic diversity12,28,29,30. Several population genetic studies using traditional molecular markers (i.e., allozymes, inter-simple sequence repeat [ISSR], and simple sequence repeat) have suggested that S. koreensis exhibits low-to-moderate levels of genetic variation12,28,29,30. To preserve endangered plant species such as S. koreensis, it is vital to understand the geographic patterns of genetic variation, population structure, and differentiation of the species. However, the genetic variation and population structure of S. koreensis across its distribution range in South Korea have not yet been thoroughly addressed using fine-scale genomic data. Therefore, we evaluated the genetic diversity, structure, and differentiation patterns of S. koreensis in South Korea using genotyping-by-sequencing (GBS). Genome-wide single nucleotide polymorphisms (SNPs) offer several advantages, including broader genomic coverage, higher reproducibility, and suitability for large-scale analyses. Moreover, SNP data enable fine-scale resolution of population structure and gene flow. Thus, SNP-based analyses provide a more robust and comprehensive understanding of the genetic diversity and evolutionary dynamics of S. koreensis. In this study, we reveal previously unreported patterns of genetic isolation in peripheral populations and identify several contributing factors underlying their formation. We also propose constructive suggestions for enhanced protection and follow-up management of this endemic and endangered species.
Results
Habitat characteristics of S. koreensis
The habitat characteristics of the 12 populations of S. koreensis were examined. S. koreensis mainly grows at low elevations between 169 m and 561 m above sea level (Table 1). Among these populations, the SB population was concentrated at the lowest elevations (i.e., 169–230 m), whereas the SA, SH, and SN populations extended to over 500 m. Population sizes varied from 8,000 m2 (SC) to 528,000 m2 (SN) (Table 1). S. koreensis thrives predominantly in vegetation types of pine, oak, and mixed pine-oak forests. Unlike most endangered plant species, most S. koreensis populations were found in artificially disturbed forest areas such as forest selection-cutting sites. Particularly, SA, SD, and SF populations were found in forest-fired areas, suggesting S. koreensis mainly grows in open forests where sunlight is allowed to reach the forest floor.
Levels of genetic and clonal diversity
In total, 1,014,297 contigs were generated from the trimmed SN_09 individual and assembled to a length range of at least 200 bp to a maximum of 22,783 bp (SAMN43487288). The sequenced GBS library of 94 S. koreensis produced 406,407,034 reads. The maximum genome size of S. koreensis was estimated to be approximately 571.8 Mb based on K-mer analysis of Illumina sequencing reads. While no prior research has been conducted on the genome size of this species, other species of Sophora have an average genome size of 399.9 Mb31. The estimated genome size was larger than the assembled genome size. In this study, the genome was assembled with a total sequencing coverage of 239.5×. Additionally, the short-reads used for assembly cover 95.7% of the assembled genome. Trimmed reads ranged from 581,892 to 12,587,806 and the average percentage mapped to the scaffold contig was 92.72% as Supplementary Table S1 online. No raw reads were generated from samples SD_09 or SF_03 during demultiplexing, and these samples were eliminated for subsequent analysis. Through a series of filtering processes, we filtered 1,463 SNPs across 92 individuals from 12 natural populations. Genetic variation and clonal diversity indices were calculated (Table 1). The mean observed heterozygosity (Ho) was 0.015, which was significantly lower than the average expected heterozygosity (He) of 0.150 (P < 0.001). The Ho ranged from 0.010 (SE) to 0.017 (SF and SG), while the He ranged from 0.101 (SF) to 0.217 (SL). All studied populations of S. koreensis had significantly low Ho, indicating that not all populations were in Hardy-Weinberg equilibrium. Nucleotide diversity (Pi) ranged from 0.107 (SF) to 0.226 (SL), with an average of 0.163. Heterozygote deficiency and low genetic diversity were detected in S. koreensis; these were supported by a high inbreeding coefficient (FIS).
We used a genetic distance matrix to estimate the clonal assignment and diversity in S. koreensis. Distance peak analysis revealed a small peak that was attributed to somatic mutations or genotyping errors within genets, whereas the long-distance peak represented variation between genets (see Supplementary Fig. S1 online). A distance window of 723 was selected as the threshold between the two peaks, in which the assigned gene number remained constant with increasing genetic distance. Using this approach, we identified 1 (SC, SE, and SF) to 10 (SL) genotypes (number of genotypes [Num]) in 12 populations. Clone diversity analysis revealed that half of the clones were assigned to populations SC, SE, and SF, which had only one genotype each and thus had very low diversity values (e.g., genotypic diversity [Div], Nei’s uncorrected genetic diversity [Diu], Shannon index corrected for sample size [Shc], and Shannon-Wiener index [Shu]) (Table 1). These three populations (i.e., SC, SE, and SF) are located in the southeastern peripheral region of S. koreensis distribution in South Korea and have significantly lower genetic diversity indices (e.g., Diu = 0, Shc = 0, and Shu = 0) than other populations. Contrastingly, the SL and SN populations showed high clonal diversity indices.
Genetic structure and differentiation
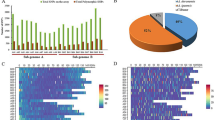
Genetic clustering analyses of the populations were performed using ADMIXTURE software (version 1.3.0). We applied K-values ranging from 2 to 12, and the optimal K-value was selected according to the Cross Validation (CV) error. In this study, K = 4 was the most optimal (see Supplementary Fig. S2 online); however, we visualized the results of K = 2–6 to show the genetic structure patterns of S. koreensis. When the CV was 0.74857, the optimal K was 4, indicating that 92 S. koreensis individuals from the 12 populations could be classified into four groups with the highest probability. The first group comprised the SA, SB, SD, SH, SK, SL, SM, and SN populations of all individuals and two individuals of SG, the second group comprised the SC population with seven SG individuals; and two peripheral populations (SE and SF) were assigned unique genotypes (Figs. 2 and 3). Interestingly, despite the large distance between SG and SC, the second group showed a mixed distribution with the first group in the SG population (Figs. 2 and 3).

Genetic clustering patterns of S.koreensis using 1,463 SNPs with ADMIXTURE at a specific range of K (2–6). Each individual is represented by vertical bars divided into K-colored proportions of the results. Optimal K = 4 values of Cross-validation errors was the best in explaining the data.
Principal Component Analysis (PCA) was conducted to analyze the genetic relationships and population structure within the 12 populations. The first three eigenvalues represented approximately 18.79% of the total genetic variation with values of 6.81%, 6.45%, and 5.53%, respectively (see Supplementary Fig. S3 online). Four groups were identified in the comparison between PC1 and PC2. The peripheral populations (SC, SE, and SF) were grouped independetly, and the remaining populations were grouped together as a single cluster (Fig. 4a). A comparison between PC2 and PC3 showed a similar pattern; however, some individuals in the SD population were separated from the main cluster (Fig. 4b). Population structure analysis and PCA strongly supported the differentiation of the three peripheral populations (SC, SE, and SF), which were independently grouped from the main populations (SA, SB, SD, SG, SH, SK, SL, SM, and SN).

Results of principal component analysis (PCA) of genetic distances for all 92 genotyped individuals. (a) PC1 versus PC2; (b) PC2 versus PC3.
Pairwise genetic distance (FST) among the 12 populations of S. koreensis was measured (Fig. 5). The results showed that genetic differentiation among the populations ranged from 0.0615 to 0.3423, with an average of 0.2. Relatively high FST values were observed in pairwise comparisons between populations from peripheral regions (SC, SE, and SF) and other populations, whereas lower FST values were observed between the largest and central populations (SL and SN) and other populations. Furthermore, the mean FST value among peripheral populations (SC, SE, and SF) was 0.3363, indicating that peripheral populations were genetically differentiated from each other. Historical gene flow barriers based on the pair of FST matrices also suggest the isolation of SC, SE, and SF populations. (Fig. 2, bold red line). Additionally, the results from analysis of molecular variance (AMOVA) (Table 2) revealed that 27.58% of the total genetic variance was due to population differentiation and that the majority of differentiation was within populations (i.e., 67.39%). Pairwise Mantel tests revealed a significant correlation between FST and geographical distance (Euclidean distance: P = 0.616, R2 = 0.1625; Fig. 6).

Pairwise population differentiation (FST) among 12 populations of S. koreensis based on the 1,463 single nucleotide polymorphisms (SNPs).

Pairwise Mantel test of isolation by distance for 1,463 single nucleotide polymorphisms (SNPs). Each point represents one population pairwise FST/(1 − FST) plotted against straight line geographic distance between paired populations. FST, Pairwise genetic distance.
Discussion
The neutral theory provides a valuable theoretical framework for understanding genetic diversity under the assumption of selective neutrality32. However, it does not account for adaptive processes or selection pressures acting on specific loci. In wild plant populations, particularly under conditions of environmental heterogeneity or anthropogenic disturbance, some loci may deviate from neutrality due to selection33. Although this study does not directly investigate selection signatures, it is important to interpret patterns of genetic diversity with awareness of possible violations of neutrality assumptions.
Various analyses have been performed to estimate levels of genetic differences and species diversity. The development of next-generation sequencing technology and the advent of multiplex sequencing have enabled individual SNP screening at the genomic level and are widely used for comparisons between groups. Although genetic evaluations of wild plants using this method have accumulated in recent years, the need for conservation measures in many species has not been addressed because of a lack of samples, reference genomes, and available markers. In this study, we analyzed the genetic and clonal diversity of S. koreensis, which is a species endemic to Korea. However, as S. koreensis is distributed in both South and North Korea on the Korean Peninsula, it is important to note a limitation in the scope of this study. This study focused primarily on distribution within the South Korean region, which is a potential limitation for providing a comprehensive understanding of the full distribution of S. koreensis across the Korean Peninsula. Genetic diversity analysis revealed significant heterozygote deficiency (P < 0.001) and low levels of genetic diversity (He = 0.150, Ho = 0.015, Pi = 0.163) in S. koreensis, whereas clonal diversity was moderate to high (Div = 0.581, Shu = 0.402). Previous studies on the same species using ISSRs (Cheon30; He = 0.087, Shannon’s index [SI] = 0.132) and allozymes (Chung28; A = 1.14, He = 0.026) showed lower levels of genetic diversity, whereas another study using ISSRs (Chung12; He = 0.204) showed moderate levels of genetic diversity. However, Chung12 surveyed only two central populations of Yanggu-gun to characterize the fine-scale genetic structure of this species; therefore, it is possible that a higher genetic variation was observed. The population-level genetic diversity of the congeneric species S. moorcroftiana (Bentham) Bentham ex Baker (He = 0.3095, SI = 0.310) and S. alopecuroides L. (Ne = 1.81, He = 0.39) is higher than that of S. koreensis34,35. Generally, the reproductive strategy of a species, such as selfing and clonal reproduction, is an important factor affecting its genetic diversity36. While some asexually reproducing species, such as Taraxacum officinale, have been reported to maintain high genetic diversity through mechanisms such as mutation accumulation and hybridization36, asexually reproductive species often exhibit lower genetic diversity than sexually reproductive species. S. koreensis exhibits both sexual and asexual reproductive strategies12. This species is well known for its asexual propagation via the rhizome (an approximately 180 cm long rhizome was identified) and is self-compatible under greenhouse conditions12. The low genetic diversity of S. koreensis can be attributed to the biological traits of this species, such as a higher rate of vegetative reproduction than sexual reproduction, a feature commonly observed in this genus18,37, as supported by the high FIS (0.373).
Notably, three peripheral populations (SC, SE, and SF) located at the southeastern margin of S. koreensis distribution in South Korea showed lower levels of genetic diversity than the other populations (Table 1). Furthermore, clonal diversity analysis results indicated that these three populations were composed of only one effective genotype. However, a previous study showed different genetic patterns. Cheon30 indicated that other peripheral populations (Dochang-ri, Jinae-ri, and Sungdong-ri) and one central population (Gullyang-ri) had lower values than the others. Jinae-ri, Sungdong-ri, and Gullyang-ri in Cheon30 had the same populations as SB, SC, and SN, respectively, in this study. However, Dochang-ri, approximately 40 km northwest of SG, was excluded from this study because of population extinction. New populations (SE and SG) were found and included in this study. These differences can be attributed to the different sampling ranges and genetic markers (i.e., 6 ISSRs and 1,463 SNPs).
The evidence of the population genetic structure and differentiation presented in this study provides a novel understanding of S. koreensis. Population admixture and PCA showed that the 12 populations could be classified into four groups based on the large-scale sequencing data. Although seven individuals of the SG populations were grouped differently in the admixture and PCA results, clear genetic differentiation was observed between the three peripheral populations (SC, SE, and SF) and the other main populations (Figs. 2 and 4). Genetic barrier analysis, based on pairwise FST values, indicated that these three populations were isolated from the main populations (Fig. 2). As shown in the AMOVA, a statistically significant among-population genetic differentiation was observed, probably because of the three populations. The observed genetic pattern is consistent with the ‘central-marginal’ hypothesis38, which suggests that populations at the edge of the species’ distribution have lower genetic diversity and higher genetic differentiation than core populations. The genetic patterns and structures of the three peripheral populations are most likely a consequence of isolation from the main populations and their small effective population sizes37. Geographic (e.g., mountains) and hydrological (e.g., rivers) barriers between peripheral and central populations effectively interfere with gene flow among populations. Although foraging distances of bumble bees differ among species, they generally exhibit a preference for foraging within 1–2 km from their nest39,40,41. The restricted movement of pollinators between populations suggests a limited capacity for pollen-mediated gene flow, which could lead to increased inbreeding and reduced genetic diversity within peripheral populations.
As it is an endemic and endangered species of the Korean Peninsula, it is important to protect its natural habitats and maintain genetic diversity. However, low levels of genetic diversity and high FIS of S. koreensis were observed in this study. Furthermore, very low fruiting (0.0038) and seedling rates in natural populations of S. koreensis have been reported18, which can be attributed to the lack of effective pollinators or/and clonal reproduction42. Lack of pollinators can lead to increased selfing or clonal reproduction and restricted gene flow in mixed mating systems43, which can lead to sexual extinction9. Therefore, it is necessary to identify effective pollinators and the seed dispersal mechanisms of S. koreensis to establish careful conservation measures.
Several strategies can be implemented to promote the genetic diversity of S. koreensis. First, habitat protection should be strengthened for the entire distribution of S. koreensis to maintain its genetic diversity and unique genotypes, including peripheral populations. Second, habitat restoration of peripheral populations may enhance gene flow among populations by creating favorable conditions for the establishment and connectivity of S. koreensis individuals, thereby supporting natural dispersal mechanisms and promoting genetic exchange. Furthermore, regular genetic monitoring and community involvement are also critical to ensure long-term conservation success.
Methods
Vegetation survey and sampling of S. koreensis
Field surveys were conducted between May and August of 2018 and 2024 to determine the distribution and population size of S. koreensis in South Korea. Based on previous studies21,44, 12 populations were examined. The habitat size was accurately estimated and population distribution of this endemic species characterized using Global Positioning System tracking (Oregon 550, Garmin). An accurate distribution map of each population was created using MapSource (Garmin), which extracted the coordinates of the surveyed trials and distribution points. The coordinates were then used in QGIS Desktop 2.0.1 to generate the distribution map. For the population genomic analysis, young leaves of 94 individuals from 12 populations were collected in 2021 (Fig. 2; Table 1). To avoid collecting individuals with vegetative reproduction, we maintained a minimum distance of 10 m between individuals. Formal identification of specimens and samples were conducted by J-B An and H-Y Gil. Voucher specimens (KHB1643375-1643441) were deposited in the Herbarium of Korea National Arboretum. Permission was obtained to collect endemic species of S. koreensis under the rules and policies of the Korea Forest Service.
GBS library construction and sequencing
Genomic DNA was isolated from silica-dried plant material using the QIAGEN Plant Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. The DNA quality and quantity of each sample were assessed using a TapeStation 4200 (Agilent Technologies, Waldbronn, Germany) and a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). GBS libraries were used to identify SNPs from 94 individuals of S. koreensis and were prepared by SEEDUS Inc. (Daejeon, South Korea) with minor modifications from a previously described protocol (Elshire et al., 2011). Barcodes, common adapters, and DNA samples (100 ng in a volume of 10 µL) were aliquoted into a 96-well PCR plate. Double digesting DNA was performed using PstI and MspI (New England Biolabs, Ipswich, MA, USA) restriction enzymes and incubated at 75 °C for 2 h. Adaptor ligation was conducted using T4 DNA ligase (200 U; MGMED, Korea) and incubated at 22 °C for 1 h, followed by heating to 65 °C for 30 min. The digested DNA samples, each with a different barcode adapter, were combined and purified using a commercial kit (QIAquick PCR Purification Kit; Qiagen, USA) according to the manufacturer’s instructions. Amplification was performed to enrich the adapter-ligated DNA fragments, with PCR conditions as follows: 72 °C for 5 min, 98 °C for 30 s, followed by 18 cycles of 98 °C for 30 s, 65 °C for 30 s, 72 °C for 30 s, with a final Taq extension step at 72 °C for 5 min. Libraries were sequenced using an Illumina HiSeq X-10 platform with 2 × 150 paired-end sequences at Macrogen Inc., Korea. Additionally, we sequenced whole genome of one individual (SN_09) to apply a reference guide assembly platform in Stacks ver. 2.6245. The extracted gDNA was run on an Illumina HiSeq X-10 platform by 2 × 150 paired-end sequencing (Macrogen Inc., Korea).
Reference genome assembly, demultiplexing, and genotyping
We checked the quality of the sequencing raw data of SN-09 and trimmed it using the SolexaQA v.1.13 software package46. The resulting 50Gb of high-quality reads from the sequencing data were used to assemble scaffold contigs using the SOAPdenovo2 software47 with the k-mer size (K) set to 31 and a minimum contig length cutoff of 200. The genome size of S. koreensis was estimated based on the occurrences of K-mers with a peak depth using filtered Illumina reads. The estimation was performed with JELLYFISH 2.3.1 using a 31 K-mer, and the maximum genome size was calculated by dividing the total number of K-mers by the peak depth48. Generated contigs were used as reference sequences in Stacks software. Raw data of the GBS sequence were demultiplexed using “Process_radtags” module in Stacks software with the options for clean data (c) and discarding reads with low quality (q). The demultiplexed data were trimmed using Trimmomatic ver.0.3649 with the following options: LEADING, 3 TRAILING, 3 SLIDINGWINDOW, 4:15 MINLEN, and 36. The resulting trimmed and cleaned reads of each sample were aligned against the reference contigs using the “bwa-mem” algorithm with the default options after constructing an index file using BWA software (version 0.7.17)50. The generated.sam files were then converted to.bam files using the SAMtools software (version 1.10.; 35) with default options. To identify the SNPs from metapopulations, we used the “Gstacks” module in Stacks ver.2.6245 with default options.
SNP calling and filtering
The “Populations” module in Stacks software utilizes SNP calling and calculates the genetic diversity within populations. For SNP calling, we set 10 as ‘-p’ option, indicating the minimum number of populations required to process a locus, and 0.8 as ‘-r’ option, indicating the minimum percentage of individuals in a population. Additionally, we set the minimum minor allele frequency to 0.05 (–min_maf 0.05) and the maximum observed heterozygosity to 0.95 (–max_obs_het 0.95) and restricted the study to the first SNP per locus (–write-single-snp).
Genetic diversity and clonal analysis
Filtered SNPs were calculated for the genetic diversity within populations, and Pi, Ho, He, FST, and FIS were calculated using the “populations” module. Utilizing Monmonier’s maximum difference algorithm, which consiters both the geographical coordinates and genetic distance of each population as inputs, the historical barriers suggested by the estimation of genetic barriers analysis based on FST matrices were visualized using Barrier v.2.251.
Clone assignment and diversity were estimated using the GENODIVE 3.0 program52. An infinite allele model was used to calculate the frequency distribution of the FST. To determine the genet assignment threshold from the two observed peaks of genetic distance, we selected a distance window between the two peaks, where the assigned genet number was kept constant with increments in genetic distance (threshold: 723). To test clonal structure, we applied the corrected Nei’s diversity index and set the number of permutations to 999. The remaining options were set to default. We also calculated several indices, including the count of distinct multilocus genotypes, the effective number of multilocus genotypes utilizing rarefaction, Simpson’s diversity, evenness, and Shannon index adjusted for sample size. To determine differences in clonal diversity among the various stage classes, we conducted bootstrap tests (with 1000 replicates).
Population structure or differentiation, geographic and genetic correlations analysis
Bayesian model-based clustering was employed to infer genetic clustering using ADMIXTURE Linux (ver. 1.3.0). The number of putative population sets from the K values ranged from 1 to 12. The best K value corresponds to the lowest cross-validation error. A graphical display of the population structure was generated using DISTRUCT ver. 1.153. PCA and AMOVA were performed in R v3.454 with the following packages: ape55, adegenet56, Poppr57 and vcfR58.
Isolation by distance was evaluated using the Mantel test59 based on pairwise population differentiation (calculated as FST/(1FST)) and log-transformed geographic distances among population pairs using GenAlEx version 6.4160. The statistical significance of the correlation coefficient (r) was tested using 1000 random permutations.
Data availability
All sequencing data analyzed in this study are publicly available from the National center of Biotechnology Information (https://www.ncbi.nlm.nih.gov/) under the Biosample ID: SAMN43487288 (Reference genome) and SAMN43501657-43501748 (SRA data).
References
Futuyma, D. J. & Kirkpatrick, M. Mutation and Variation. Evolution (Fourth ed.)79–102 (Sinauer Associates, Inc, 2017).
Fontdevila, A. Springer,. Founder effects in colonizing populations: the case of Drosophila buzzatii. in Evolutionary biology of transient unstable populations 74–95 (1989).
Tremblay, R. L. & Ackerman, J. D. Gene flow and effective population size in Lepanthes (Orchidaceae): A case for genetic drift. Biol. J. Linn. Soc. 72, 47–62 (2001).
Frei, E. S., Scheepens, J. F. & Stöcklin, J. High genetic differentiation in populations of the rare alpine plant species Campanula thyrsoides on a small mountain. Alp. Bot. 122, 23–34 (2012).
Meloni, M. et al. Effects of clonality on the genetic variability of rare, insular species: the case of Ruta microcarpa from the Canary Islands. Ecol. Evol. 3, 1569–1579 (2013).
Smith, J. M. & Maynard-Smith, J. The Evolution of Sex vol. 4 (Cambridge University Press Cambridge, 1978).
Klimeš, L., Klimešová, J., Hendriks, R. & van Groenendael, J. Clonal plant architecture: a comparative analysis of form and function. Ecol. Evol. Clonal Plants 1–29 (1997).
Frankham, R. Stress and adaptation in conservation genetics. J. Evol. Biol. 18, 750–755 (2005).
Honnay, O. & Bossuyt, B. Prolonged clonal growth: escape route or route to extinction? Oikos 108, 427–432 (2005).
Lee, S. R., Kim, B. Y. & Kim, Y. D. Genetic diagnosis of a rare myrmecochorous species, Plagiorhegma dubium (Berberidaceae): historical genetic bottlenecks and strong spatial structures among populations. Ecol. Evol. 8, 8791–8802 (2018).
Mattapha, S., Suddee, S. & Rueangruea, S. Sophora huamotensis, a new species of Sophora (Fabaceae-Papilionoideae-Sophoreae) from Thailand. Thai For. Bull. (Botany). 46, 4–9 (2018).
Chung, J. M. et al. Fine-scale genetic structure among genetic individuals of the clone-forming monotypic genus Echinosophora koreensis (Fabaceae). Ann. Bot. 98, 165–173 (2006).
Nakai, T. Notulae ad Plantas japoniae et Coreae XIX. Shokubutsugaku Zasshi. 33, en1–en11 (1919).
Lee, W. K., Tokuoka, T. & Heo, K. Molecular evidence for the inclusion of the Korean endemic genus ‘Echinosophora’ in Sophora (Fabaceae), and embryological features of the genus. J. Plant. Res. 117, 209–219 (2004).
Chang, C. S., Kim, H., Son, S. & Kim, Y. S. The Red List of Selected Vascular Plants in Korea (Korea National Arboretum and Korean Plant Specialist Group, 2016).
Ikuse, M. Pollen grains of Leguminosae obtained in Japan, Especailly of their unusual forms. Journ Jpn Bot. 29 (1-10), Preprintat (1954).
Song, U. Pollen Morphology of the Woody Fabaceae in Korea. Korean Journal of Plant Taxonomy vol. 37 87–108 Preprint at (2007). https://doi.org/10.11110/kjpt.2007.37.2.087
Kim, S. et al. Population characteristics of Echinosophora koreensis (Nakai) Nakai, a endemic plants in Korea. Korean J. Environ. Ecol. 34, 18–26 (2020). (in Korean)
Shim, K. K., Ha, Y. M., Son, C. J., Han, D. S. & Lee, S. A. A study on development of materials for native landsape tree-native area survey and characteristic of Korean endemic plant, Echinosophora koreensis Nakai. J. Kor Inst. Traditional Landsc. Archit. 24, 32–42 (2006).
Kim, S. J. et al. Flora of Mt. Samyeong (Yanggu-gun, Gangwon-do) in DMZ area of Korea. Korean J. Plant. Resour. 30, 191–213 (2017).
Cheon, K. S., Jang, S. K., Lee, W. T. & Yoo, K. O. The natural habitat and distribution of Echinosophora koreensis (Nakai) Nakai in Korea. Korean J. Plant. Taxonomy. 39, 254–263 (2009). (in Korean)
Byun, J. H. et al. Triterpenoid saponins from the roots of Sophora koreensis. Chem. Pharm. Bull. (Tokyo). 52, 870–873 (2004).
Sohn, H. Y., Son, K. H., Kwon, C. S., Kwon, G. S. & Kang, S. S. Antimicrobial and cytotoxic activity of 18 prenylated flavonoids isolated from medicinal plants: Morus alba L., Morus mongolica Schneider, Broussnetia papyrifera (L.) Vent, Sophora flavescens Ait and Echinosophora koreensis Nakai. Phytomedicine 11, 666–672 (2004).
Iinuma, M., Ohyama, M., Tanaka, T., Mizuno, M. & Soon-Keun, H. Five flavonoid compounds from Echinosophora koreensis. Phytochemistry 33, 1241–1245 (1993).
Choi, E. J. et al. Four flavonoids from Echinosophora koreensis and their effects on alcohol metabolizing enzymes. Arch. Pharm. Res. 32, 851–855 (2009).
Cho, S. H. et al. Anti-inflammatory activity of Echinosophora koreensis Nakai root extract in lipopolysaccharides-stimulated RAW 264.7 cells and carrageenan-induced mouse paw edema model. J. Ethnopharmacol. 302, 115940 (2023).
Jeong, U., Lee, H., Park, S. & Cheong, E. J. Optimizing in vitro propagation of Sophora koreensis Nakai using statistical analysis. J. Korean Soc. For. Sci. 110, 53–63 (2021).
Chung, M. Y. & Chung, M. G. Low levels of allozyme variation in the Southern populations of the Korean endemic shrub Sophora koreensis (Fabaceae): implications for conservation. Collect Bot 36, (2017).
Lee, J. Y., Lee, D. H. & Choi, B. H. Isolation and characterization of 13 microsatellite loci from a Korean endemic species, Sophora koreensis (Fabaceae). Int. J. Mol. Sci. 13, 10765–10770 (2012).
Cheon, K. S. Distribution, Natural Habitat Characteristics and Genetic Diversity of Echinosophora Koreensis (Nakai) Nakai (Kangwon National University, 2010).
Espejo, J. et al. Exploratory karyological and genome size studies in Chilean Sophora species. N Z. J. Bot. 54, 311–322 (2016).
Kimura, M. The Neutral Theory of Molecular Evolution (Cambridge University Press, 1983). https://doi.org/10.1017/CBO9780511623486
Luikart, G., England, P. R., Tallmon, D., Jordan, S. & Taberlet, P. The power and promise of population genomics: from genotyping to genome typing. Nat. Rev. Genet. 4, 981–994 (2003).
Liu, Y. et al. Geographic population genetic structure and diversity of Sophora moorcroftiana based on genotyping-by-sequencing (GBS). PeerJ 8, (2020).
Wang, Y. et al. The genetic diversity and population structure of Sophora alopecuroides (Faboideae) as determined by microsatellite markers developed from transcriptome. PLoS One. 14, 1–17 (2019).
Majeský, L., Vašut, R. J., Kitner, M. & Trávníček, B. The pattern of genetic variability in apomictic clones of Taraxacum officinale indicates the alternation of asexual and sexual histories of apomicts. PLoS One. 7, e41868 (2012).
Robertson, A. W., Kelly, D. & Ladley, J. J. Futile selfing in the trees Fuchsia excorticata (Onagraceae) and Sophora microphylla (Fabaceae): inbreeding depression over 11 years. Int. J. Plant. Sci. 172, 191–198 (2011).
Eckert, C. G., Samis, K. E. & Lougheed, S. C. Genetic variation across species’ geographical ranges: the central-marginal hypothesis and beyond. Mol. Ecol. 17, 1170–1188 (2008).
Osborne, J. L. et al. A landscape-scale study of bumble bee foraging range and constancy, using harmonic radar. J. Appl. Ecol. 36, 519–533 (1999).
Redhead, J. W. et al. Effects of habitat composition and landscape structure on worker foraging distances of five bumblebee species. Ecol. Appl. 26, 726–739 (2016).
Knight, M. E. et al. An interspecific comparison of foraging range and nest density of four bumblebee (Bombus) species. Mol. Ecol. 14, 1811–1820 (2005).
Ward, M. & Johnson, S. D. Pollen limitation and demographic structure in small fragmented populations of brunsvigia radulosa (Amaryllidaceae). Oikos 108, 253–262 (2005).
Surina, B. et al. Lack of pollinators selects for increased selfing, restricted gene flow and resource allocation in the rare mediterranean Sage Salvia brachyodon. Sci. Rep. 14, 1–17 (2024).
An, J. B. et al. Distribution and potential suitable habitats of an endemic plant, Sophora Koreensis in Korea. Korean J. Environ. Ecol. 35, 154–163 (2021). (in Korean)
Rochette, N. C., Rivera-Colón, A. G. & Catchen, J. M. Stacks 2: analytical methods for paired-end sequencing improve RADseq-based population genomics. Mol. Ecol. 28, 4737–4754 (2019).
Cox, M. P., Peterson, D. A. & Biggs, P. J. SolexaQA: At-a-glance quality assessment of illumina second. BMC Bioinform. 11, 485 (2010).
Luo, R. et al. Erratum to ‘SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler’ [GigaScience, 1, 18]. Gigascience 4, 1 (2015). (2012).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Manni, F., Guérard, E. & Heyer, E. Geographic patterns of (Genetic, morphologic, Linguistic) variation: how barriers can be detected by using monmonier’s algorithm. Hum. Biol. 76, 173–190 (2004).
Meirmans, P. G. Genodive version 3.0: Easy-to-use software for the analysis of genetic data of diploids and polyploids. Mol. Ecol. Resour. 20, 1126–1131 (2020).
Rosenberg, N. A. & DISTRUCT A program for the graphical display of population structure. Mol. Ecol. Notes. 4, 137–138 (2004).
R Core Team. R: A language and environment for statistical computing. Preprint at. (2020).
Paradis, E. & Schliep, K. Ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2019).
Jombart, T. & Adegenet A R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405 (2008).
Kamvar, Z. N., Tabima, J. F., Gr̈unwald, N. J. & Poppr An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 1–14 (2014). (2014).
Knaus, B. J. & Grünwald, N. J. Vcfr: a package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 17, 44–53 (2017).
Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 27, 209–220 (1967).
Peakall, R. & Smouse, P. E. GENALEX 6: genetic analysis in excel. Population genetic software for teaching and research. Mol. Ecol. Notes. 6, 288–295 (2006).
Acknowledgements
The authors thank Jong-Won Lee, Young Min Choi, Gi-Ppeum Bak, Seungju Jo, Jin-Heon Song and Tae-Young Hwang for their efforts in collecting the samples used in this study. We would also like to express our gratitude to Eun-Ho Lee for her invaluable contribution to the laboratory work. Their expertise and dedication greatly helped to make this study successful. This study was supported by the Korea National Arboretum [KNA1-1-13, 14−1, KNA1-2-45, 23−3].
Author information
Authors and Affiliations
Contributions
H.-Y.G. conceived the experiments; J.-B.A., J.-W.Y., and J.C. conducted the field survey; J.-B.A. and H.-Y.G. conducted formal identification of leaf samples and specimens; Y.-H.H. conducted the experiments and analyzed the results; and Y.-H.H. and H.-Y.G. wrote the manuscript. All authors reviewed the manuscript and agreed to the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ha, YH., An, JB., Chung, J. et al. Genotyping-by-sequencing reveals low genetic diversity and peripheral isolation in Southern populations of Sophora koreensis, a Korean endemic shrub. Sci Rep 15, 17996 (2025). https://doi.org/10.1038/s41598-025-02703-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-02703-7

{kind=link}
{kind=link}
{kind=link}