
Abstract
This study investigates the impact of iron (III) chloride hexahydrate (FeCl3·6H2O) incorporation on the structural, thermal, and dielectric properties of poly(vinylidene fluoride-co-hexafluoropropylene) [P(VDF-HFP)] nanocomposites, which were prepared using a solution casting method with varying filler concentrations (1–4 wt%). Scanning electron microscopy revealed a systematic increase in porosity—from 0.72% in pure P(VDF-HFP) to 27.5% at 4 wt% FeCl3·6H2O—along with increased pore size and surface heterogeneity. Atomic force microscopy confirmed enhanced surface roughness correlating with increased filler content. Fourier-transform infrared spectroscopy demonstrated a significant α-to-β phase transformation, indicating the formation of the polar β-phase with increasing FeCl3·6H2O content. X-ray diffraction analysis corroborated these findings, revealing a notable increase in crystallinity and β-phase content, with 4 wt% FeCl3·6H2O achieving the highest β-phase fraction (88.99%). Thermogravimetric analysis confirmed thermal stability up to approximately 500 °C, with a gradual shift in degradation onset attributed to FeCl3·6H2O interactions. Dielectric measurements at 10 Hz showed a remarkable enhancement in dielectric constant—from 5.62 in pure P(VDF-HFP) to 19.16 at 4 wt% FeCl3·6H2O—while maintaining a low dielectric loss (< 0.30). These improvements are attributed to the synergistic effects of FeCl3·6H2O on porosity, phase transformation, crystallinity, thermal stability, and dielectric properties. The superior performance of these nanocomposites makes them promising candidates for flexible electronics, energy storage systems, and advanced sensors.
Similar content being viewed by others

Introduction
The escalating urgency of energy crises and environmental concerns has driven researchers to prioritize safer, cleaner, and more sustainable energy solutions. Among these, the conversion of mechanical energy into electrical energy has gained significant interest, with piezoelectric materials emerging as essential components. These materials are extensively used in applications such as sensors, actuators, nanoelectronics, and energy harvesting systems1,2. Within this domain, poly(vinylidene fluoride) (PVDF) and its copolymers have attracted considerable attention due to their affordability, flexibility, and superior piezoelectric properties3,4. Specifically, poly(vinylidene fluoride-co-hexafluoropropylene) [P(VDF-HFP)] is highly regarded for its high dielectric constant, chemical resistance, and mechanical flexibility. Its unique structural combination—amorphous regions enhancing flexibility and crystalline domains ensuring mechanical stability—makes it ideal for use in flexible electronics, energy storage, and sensor applications5. Compared to PVDF, which has a dielectric constant (\(\:{\epsilon\:}_{r}^{}\)) typically ranging from 6 to 12 depending on processing conditions and measurement frequencies6, P(VDF-HFP) demonstrates a higher dielectric constant (\(\:{\epsilon\:}_{r}^{}\) ≈ 11) and enhanced ferroelectric properties primarily attributed to its electroactive β-phase conformation, which facilitates superior polarization and a strong dipole moment7,8. The polymorphic nature of P(VDF-HFP) includes five crystalline phases—α, β, γ, δ, and ε—with the β-phase being particularly sought after due to its ferroelectric, piezoelectric, and pyroelectric properties. The β-phase conformation of this phase contributes to its high dipole moment, making it critical for advanced applications such as energy harvesting, sensors, and ferroelectric devices9.
Various strategies have been explored to enhance β-phase content, including heat treatments, electrical poling, and polymer blending. Among these, incorporating fillers has proven to be an effective approach. Fillers like barium titanate (BaTiO3)10, carbon nanotubes11, ZnO12, and halloysite nanotubes13 have shown the ability to induce β-phase formation through strong polymer-filler interactions, especially ion–dipole interactions. However, challenges such as filler agglomeration and uneven dispersion can hinder their full potential. The intrinsic breakdown strength of pure P(VDF-HFP) typically ranges from 400 to 600 kV/mm, influenced by factors such as molecular weight, crystallinity, processing conditions, and the presence of fillers. Recent advancements by Zeng et al.14 demonstrated that incorporating ultra-thin aluminum oxide (Al₂O₃) nanosheets into P(VDF-HFP) significantly enhances its breakdown strength. Specifically, a sandwich-structured composite film with Al2O3 nanosheets achieved a breakdown strength of approximately 740 kV/mm, representing a 24.8% increase compared to pure P(VDF-HFP).
Hydrated inorganic salts such as Mg(NO3)2·6H2O, Ca(NO3)2·4H2O and Zn(NO3)2·6H2O15, Al(NO3)3∙9H2O16, Co(NO₃)₂·6 H₂O17, MgCl₂·6 H₂O, and AlCl₃·6 H₂O18 have proven to be effective additives for enhancing β-phase nucleation in piezoelectric PVDF and P(VDF-HFP) polymers. Their ability to promote β-phase formation is attributed to mechanisms involving hydrogen bonding and electrostatic interactions with the polymer matrix. The innovative approach of dissolving these salts during the polymer film fabrication process establishes strong interactions, which play a pivotal role in facilitating β-phase nucleation. This strategy significantly enhances the piezoelectric properties of composite materials. Fortunato et al.19 demonstrated the impact of different hexahydrate metal salts on β-phase content and piezoelectric response (d33 = 13.49 pm/V) in PVDF nanocomposites, with Mg(NO3)2·6H2O exhibiting the strongest enhancement due to the synergistic effect of β-phase nucleation and electrostatic interactions with PVDF’s CF2 groups. In previous research20, we investigated the effects of calcium chloride salts (CaCl2, CaCl2·2H2O, and CaCl2·6H2O) on P(VDF-HFP)’s morphology, crystalline transformation, mechanical properties, and dielectric responses. Notably, a composite with 3 wt% CaCl2·6H2O showed the highest β-phase fraction (98.33%) and dielectric constant (21.49 at 10 Hz), along with enhanced flexibility and reduced stiffness compared to pure P(VDF-HFP). These findings emphasize the significant influence of water content in hydrated salts on β-phase formation and the role of dipolar interactions between P(VDF-HFP) and the solvent. Recent advancements include Li et al.‘s work3 on PVDF composite fiber membranes with fillers such as AgNO3, FeCl3·6H2O, and modified nanographene, where an optimal FeCl3·6H2O concentration (0.8 wt%) achieved remarkable piezoelectric properties, including a peak output voltage of 4.8 V. Gong et al.2 developed PVDF/SiC/FeCl3·6H2O nanofiber membranes via electrospinning, achieving significant improvements in β-phase content. The optimized nanogenerators demonstrated ∼12.3 and ∼10.8 times increases in output current and voltage, respectively, compared to pure PVDF. Additionally, they exhibited enhanced moisture resistance and stability, making them suitable for applications such as gait monitoring and self-powered sensors. Despite these advancements, the performance of P(VDF-HFP) nanocomposites incorporating only hydrated FeCl3·6H2O salt remains underexplored, emphasizing the need for further research to optimize these materials for advanced applications. Additionally, limited studies have explored the role of dimethylformamide (DMF) as a solvent in polymer solution preparation. The interaction between the CH₂ groups of P(VDF-HFP) and the carbonyl oxygen (C = O) of DMF may synergize with FeCl3·6H2O to further enhance β-phase formation21. Moreover, the development of iron oxide or chloride particle-reinforced polymer nanocomposites has attracted significant attention due to their superior thermal, mechanical, and dielectric properties, which make them highly promising for advanced applications such as flexible electronics, energy storage systems, and sensors. Among the various metal oxides, magnetite (Fe3O4) and nickel oxide (NiO) nanoparticles have shown remarkable potential in improving the performance of polymer composites. Ramesan et al.22 reported that NiO-reinforced poly(vinyl cinnamate) (PVCin) nanocomposites exhibited enhanced structural, magnetic, and thermal stability, as well as improved DC and AC electrical conductivity, demonstrating their potential for electronic applications. Similarly, chitin/cashew gum-based nanocomposites reinforced with Fe3O4 nanoparticles exhibited enhanced thermal stability and mechanical properties, including tensile strength and hardness, along with significantly improved dielectric properties23. Further research by Ramesan et al.24 demonstrated that poly(ethylene-co-vinyl acetate) (EVA) composites with Fe3O4 nanoparticles exhibited increased tensile strength, modulus, and hardness, despite reduced elongation at break and impact strength at higher filler loadings. Moreover, poly(anthranilic acid) (PANA) nanocomposites with magnetite nanoparticles (Fe3O4) displayed reduced amorphous regions and uniformly dispersed spherical particles, enhancing the thermal stability and dielectric properties25. Jayakrishnan et al.26 further reported that nanocomposites of poly(anthranilic acid-co-indole) (PANA-Co-PIN) exhibited remarkable thermal stability, increased dielectric constant, and improved gas sensing capabilities due to the inclusion of Fe3O4 nanoparticles, making them suitable for nanoelectronic devices.
This study presents a novel approach to significantly enhancing the electroactive β-phase content, crystallinity, and dielectric properties of P(VDF-HFP) nanocomposites by incorporating FeCl3·6H2O as a functional filler. Unlike previous studies that primarily focus on traditional fillers or other hydrated salts, our work introduces FeCl3·6H2O as an innovative filler that not only induces a remarkable transformation from the nonpolar α-phase to the polar β-phase but also significantly improves the dielectric performance and thermal stability of the composite. This novel strategy achieves a notable increase in the β-phase fraction, from 67.18% in pure P(VDF-HFP) to 88.99% at 4 wt% FeCl3·6H2O, accompanied by an increase in crystallinity from 46.64 to 51.56%. Additionally, the dielectric constant at 10 Hz rises from 5.62 to 19.16, maintaining a low dielectric loss (< 0.30), which is unprecedented among similar composite systems. Thermal analysis demonstrates robust stability up to approximately 500 °C, underscoring the composite’s potential for high-temperature applications. The innovation of this work lies in the unique combination of hydrogen bonding, dipolar interactions, and controlled solvent evaporation, which collectively optimize the phase transformation and dielectric properties. These significant advancements position the FeCl3·6H2O/P(VDF-HFP) nanocomposites as promising candidates for next-generation flexible electronics, energy storage devices, and advanced sensor technologies, bridging the gap between fundamental research and practical applications.
Materials and methods
Materials
The materials used for preparing pure P(VDF-HFP) and FeCl3·6H2O/P(VDF-HFP) nanocomposite films included P(VDF-HFP) powder (Solef® 11010/1001) as the polymer matrix, recognized for its outstanding dielectric properties. Supplied by Solvay, this material has a density of 1.78 g/cm3 and a melting temperature range of 158–162 °C. The P(VDF-HFP) used in this study features a weight-average molecular weight (MW) of approximately 171.0 kg/mol, a number-average molecular weight (Mn) of approximately 114.0 kg/mol, and a polydispersity index (PDI) of ~ 1.5. Composed of a 90:10 molar ratio of vinylidene fluoride (VDF) to hexafluoropropylene (HFP), this composition ensures an optimal balance between crystalline and amorphous regions. N, N-Dimethylformamide (DMF), a highly polar solvent with a density of 0.949 g/cm3, was used in analytical reagent (AR) grade with a purity of 99.8% and was procured from RCI Labscan, Thailand. Iron (III) chloride hexahydrate (FeCl3·6H2O), with a purity of 97%, was obtained from PanReac™ (Spain) and was employed as a filler to enhance the nanocomposite’s properties.
Preparation of composites
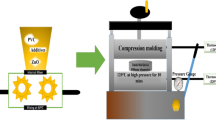
FeCl₃·6 H₂O/P(VDF-HFP) nanocomposite films were prepared using the solution casting method, as illustrated in Fig. 1. Initially, 20 g of P(VDF-HFP) powder was dissolved in 100 ml of DMF solvent, resulting in a concentration of 20 wt% (w/v). The solution was stirred at room temperature using a magnetic stirrer until a homogeneous mixture was achieved. FeCl₃·6 H₂O was then incrementally added to the solution to achieve concentrations of 1 wt% (1 g), 2 wt% (2 g), 3 wt% (3 g), and 4 wt% (4 g), with continuous stirring for 6 h at room temperature. Higher FeCl₃ concentrations exceeding 4 wt% were not pursued due to gelation of the solution, which rendered it unsuitable for film fabrication. The resulting mixture was sonicated (Baku 3050) at 35 W for 600 min and subsequently spread onto a clean glass substrate using an adjustable film applicator (Sheen Instruments). The solvent was evaporated under a controlled airflow at 80 °C for 6 h. After drying, the composite P(VDF-HFP) films were carefully peeled off the glass substrate. The film thickness was measured using a thickness gauge, and the samples were cut into various sizes and shapes as required for different characterization techniques.

Schematic representation of the preparation process for FeCl₃·6 H₂O/P(VDF-HFP) nanocomposite films using the solution casting method
Characterization techniques
The surface morphology of all P(VDF-HFP) samples was analyzed using a scanning electron microscope (SEM; Quanta 400, FEI, Czech Republic). Before imaging, the specimens were securely mounted on carbon tape for stability during observation. The microstructural analysis was conducted at a magnification of 5000× under high vacuum conditions, with an accelerating voltage set at 20 kV. Quantitative assessment of porosity and pore size was performed on the SEM images using ImageJ software (National Institutes of Health, USA), providing detailed evaluations of the pore structures.
The three-dimensional topography of each specimen was characterized using atomic force microscopy (AFM; Nanosurf Easyscan 2, Switzerland) in non-contact mode. Measurements were conducted at room temperature with a tip radius of approximately 10 nm. Each surface was scanned over a 25 × 25 μm area, capturing 256 data points per line. The resulting AFM images were analyzed to calculate the root mean square (RMS) surface roughness (Sq), providing quantitative insights into the surface texture.
To investigate the presence of functional groups and the relative phase composition, particularly the estimation of β-phase fraction, attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopy was performed using a VERTEX 70 spectrometer (Bruker, Germany). Measurements were carried out in the wavenumber range of 400–4000 cm− 1 with a resolution of 4 cm− 1, including both background and sample scans. The β-phase content (Fβ) was calculated using an equation derived from the Lambert-Beer law to analyze the structural composition of the samples based on FTIR spectra. The processed FTIR data allowed for the estimation of the β-phase fraction using the Eq1:
where Aβ represents the absorbance at the wavenumber of 840 cm− 1, corresponding to the β-phase, and Aα represents the absorbance at 764 cm− 1, corresponding to the α-phase. The absorption coefficients are Kβ = 7.7 × 104 cm2 mol− 1 at 840 cm− 1 and Kα = 6.1 × 104 cm2 mol− 1 at 764 cm− 1.
The crystalline structures of the samples were analyzed using X-ray diffraction (XRD) with a Philips X’Pert MPD diffractometer (Netherlands). Measurements were conducted using Cu-Kα radiation (λ = 0.154 nm) at 40 kV and 30 mA, with a scanning rate of 3° per minute. The degree of crystallinity (Xc) was determined from the XRD patterns using the equation27:
In this equation, ΣAcr represents the total integrated area of the crystalline diffraction peaks, and ΣAamr denotes the total integrated area of the amorphous halo. This method is used to quantify the crystalline and amorphous content within the polymer samples. The overall β-phase content in each P(VDF-HFP) specimen was evaluated by calculating the absolute β-phase fraction (\(\:\%\)), defined as the product of Fβ and Xc as expressed in the equation28:
Thermogravimetric analysis (TGA) is a fundamental method for analyzing the thermal properties of materials as they are subjected to increasing temperatures. The thermal stability of P(VDF-HFP) samples was evaluated using a thermogravimetric analyzer (TGA7; Perkin Elmer, USA). This process involved monitoring the temperature differential between the sample and a thermally inert reference material under a nitrogen atmosphere with a flow rate of 20 ml/min. Each sample underwent heating from 50 °C to 700 °C at a uniform rate of 10 °C/min.
Dielectric characterization was conducted to evaluate the electrical properties of polymers using an LCR meter (model IM 3533; HIOKI, Japan). The capacitance (C) of each sample was measured over a frequency range of 1 Hz to 100 kHz at room temperature. From these measurements, the dielectric constant (εr′), dielectric loss tangent (tan δ), and ionic conductivity (σ) were calculated. The dielectric constant, representing the real part of relative permittivity, was determined using the Eq. (4)21:
where t is the film thickness, A is the electrode contact area, and ε0 is the vacuum permittivity (8.854 × 10− 12 F/m). The dielectric loss tangent was derived as:
with εr″ representing the imaginary component of relative permittivity, indicating energy dissipation within the material. The ionic conductivity was calculated using:
where Rb is the bulk resistance obtained from the LCR measurements.
Results and discussion
Structural analysis
2D SEM images
The SEM images of pure P(VDF-HFP) and FeCl3·6H2O/P(VDF-HFP) nanocomposite films, prepared via the solution casting method and shown in Fig. 2a, reveal significant anges in surface morphology as the FeCl3·6H2O concentration increases. The SEM image of pure P(VDF-HFP) displays a dense, smooth, and homogeneous surface, free from visible pores or roughness. This uniformity highlights the intrinsic properties of P(VDF-HFP) and its superior film-forming capability when processed through the solution casting method. The dense morphology results from the effective packing of polymer chains, which remain undisturbed in the absence of external fillers, contributing to the material’s structural stability and mechanical integrity. The introduction of 1% FeCl3·6H2O slightly disrupts this uniformity, as small pores and surface roughness become apparent. These changes suggest that the filler interacts with the polymer chains through hydrogen bonding or ionic interactions, modifying the local packing arrangement. At this low concentration, the filler is well-dispersed due to the compatibility of FeCl3·6H2O with both the polymer and the DMF solvent, ensuring a relatively uniform film. As the FeCl3·6H2O concentration increases to 2%, the porosity of the composite film becomes more pronounced, with larger and more abundant voids visible. This behavior reflects greater disruption of the polymer matrix, as competition between FeCl3·6H2O and P(VDF-HFP) for interaction with DMF results in uneven filler distribution during solvent evaporation. The slow evaporation rate of DMF allows prolonged interaction time, promoting localized phase separation. Regions with higher FeCl₃ concentrations experience microphase separation, forming voids and increasing surface roughness.

a SEM images, b porosity and c pore size analyses by ImageJ of pure P(VDF-HFP) and FeCl₃·6 H₂O/P(VDF-HFP) nanocomposites
At higher FeCl3·6H2O concentrations (3% and 4%), the SEM images reveal highly irregular surfaces with widespread interconnected pores and pronounced agglomeration. Excessive filler concentrations cause significant phase separation, disrupting the continuity of the polymer matrix. Large clusters of FeCl3·6H2O particles form, exacerbating pore formation and structural instability. The hydrophilic nature of FeCl3·6H2O also attracts residual solvent molecules, delaying evaporation and further contributing to surface irregularities. The formation of porosity in these films is driven by several factors1,29,30. Solvent evaporation dynamics play a crucial role, as the slow-evaporating DMF solvent allows extended interaction times between the polymer and filler, leading to localized phase separation. Filler-polymer interactions, particularly between FeCl3·6H2O and the polar groups of P(VDF-HFP), disrupt the dense packing of the polymer matrix, with the degree of disruption increasing with filler concentration. Phase separation occurs due to the limited compatibility between the filler and the polymer, especially at higher concentrations. Additionally, the hydrophilic nature of FeCl3·6H2O causes it to retain residual solvent, further interfering with uniform film formation. Finally, crystallization disruption caused by the filler reduces the compactness of the polymer matrix, leading to increased porosity.
Figure 3 presents the porosity and pore size analyses of pure P(VDF-HFP) and FeCl3·6H2O/P(VDF-HFP) nanocomposites, conducted using ImageJ on SEM images shown in Fig. 2b, c, respectively. These results reveal a systematic increase in both porosity and pore size with higher FeCl3·6H2O concentrations. Pure P(VDF-HFP) exhibits minimal porosity at 0.72%, which progressively increases to 6.4%, 17.7%, 21.6%, and 27.5% for composites with 1%, 2%, 3%, and 4% FeCl3·6H2O, respectively. This increase in porosity is attributed to FeCl3·6H2O acting as a phase modifier, inducing structural rearrangements and creating voids within the polymer matrix. Similarly, the average pore size rises steadily from 0.295 μm for pure P(VDF-HFP) to 1.250 μm at 4% FeCl3·6H2O, as depicted by the curve in Fig. 2. SEM images corroborate these findings, showing increasingly pronounced and heterogeneous pore distributions at higher filler concentrations, with overlays marking the porous regions. The histogram plots derived from ImageJ analysis, shown in Fig. 2c (inset), confirm a Gaussian distribution of pore sizes, with broader distributions observed at higher filler contents. These findings highlight the pivotal role of FeCl3·6H2O in altering the nanocomposite’s microstructure to achieve enhanced.

Porosity and pore size values analyzed by ImageJ of pure P(VDF-HFP) and FeCl₃·6 H₂O/P(VDF-HFP) nanocomposites
3D AFM images
Figure 4 presents the AFM images of pure P(VDF-HFP) and FeCl3·6H2O/P(VDF-HFP) nanocomposites, providing additional insights into surface roughness and its correlation with the SEM observations. The root mean square (RMS) roughness values (Sq) extracted from the AFM images show a significant increase with rising FeCl3·6H2O content, consistent with trends observed in the SEM results. These variations in roughness correspond to structural changes caused by the interaction of FeCl3·6H2O with the polymer matrix and the solvent dynamics during the solution casting process. The AFM image of pure P(VDF-HFP) reveals a smooth surface with an Sq value of 34.78 nm, indicating a dense and homogeneous polymer matrix. The minimal roughness observed in the solvent-cast film may result from the evaporation of the DMF solvent, as similarly reported by Yasar et al.1. This observation is consistent with the SEM image, which shows no visible pores or irregularities. The uniform surface underscores the intrinsic properties of P(VDF-HFP), particularly its excellent film-forming capability, which enhances its mechanical stability and overall structural integrity. At 1% FeCl3·6H2O in the nanocomposite, the Sq value increases to 49.71 nm, signifying a slight increase in surface roughness. This corresponds to the initial disruption of the dense polymer matrix observed in the SEM images, where small pores and surface irregularities appear. The compatibility between FeCl3·6H2O and the polymer matrix, aided by the DMF solvent, ensures relatively uniform filler dispersion, minimizing agglomeration and maintaining moderate roughness. At 2% FeCl3·6H2O, the Sq rises significantly to 163.45 nm, reflecting greater disruption of the polymer matrix. The AFM and SEM images show larger and more abundant pores resulting from uneven filler distribution and phase separation during the solvent evaporation process. The slow evaporation of DMF allows prolonged interaction between the polymer and filler, promoting localized phase separation and increased roughness.

AFM images of pure P(VDF-HFP) and FeCl₃·6 H₂O/P(VDF-HFP) nanocomposites
At higher FeCl3·6H2O concentrations (3% and 4%), the roughness increases sharply. For instance, the Sq value reaches 331.00 nm at 3% FeCl3·6H2O. The SEM and AFM images reveal highly irregular surfaces with significant pore formation and filler agglomeration. Excessive FeCl3·6H2O concentrations cause microphase separation and clustering of filler particles, disrupting the polymer matrix’s continuity. This increased roughness may enhance dielectric properties due to the formation of interfacial regions, but it comes at the expense of mechanical stability. The term “excessive roughness” specifically refers to surface conditions where the Sq value exceeds 163.45 nm, marking a threshold where mechanical integrity and uniformity of the film are compromised. For instance, at 3% and 4% FeCl3·6H2O, roughness values of 331.00 nm were observed, which we define as “excessive roughness.” This level of roughness correlates with visible agglomeration and irregularities in SEM images, indicating disruption of the polymer matrix. While moderate roughness enhances interfacial interactions and dielectric properties, excessive roughness leads to mechanical instability and uneven filler dispersion. As reported in previous studies31, the increased roughness, particularly at higher FeCl3·6H2O concentrations, introduces more interfacial regions, which can enhance dielectric polarization. However, excessive roughness and agglomeration may lead to dielectric losses, which will be discussed in the dielectric properties section. In conclusion, the AFM results complement the SEM findings by quantifying surface roughness and illustrating its impact on material properties. While moderate FeCl3·6H2O concentrations (1-2%) balance roughness and structural integrity, higher concentrations (3-4%) lead to excessive roughness, agglomeration, and potential compromises in mechanical performance.
From SEM analysis, the uniform dispersion of FeCl3·6H2O in the polymer matrix ensures even stress transfer, reducing stiffness and improving flexibility. AFM imaging further highlights strong polymer-filler interactions and increased surface roughness, which reduce elongation at break and mechanical robustness. While tensile strength, elongation at break, and Young’s modulus were not explicitly measured in this study, insights from our prior research20 demonstrated that the incorporation of 3 wt% CaCl2·6H2O into P(VDF-HFP) reduced Young’s modulus from 926 MPa to 780 MPa and tensile strength from 35.94 MPa to 16.77 MPa, while elongation at break decreased from 555.10 to 366.10%. These findings highlight the critical role of filler dispersion and polymer-filler interactions in improving mechanical properties.
FTIR analysis
The FTIR spectra of pure P(VDF-HFP) and FeCl3·6H2O/P(VDF-HFP) nanocomposites, presented in Fig. 5, reveal significant vibrational modes linked to the polymer’s α-, β-, and γ-phases, alongside distinctive stretching, bending, and rocking modes of functional groups. These findings are consistent with earlier studies32,33,34,35 and demonstrate the structural modifications caused by the addition of FeCl3·6H2O. Additionally, reference FTIR spectra of pure FeCl3·6H2O (as reported by Inam at al36 and Sheng et al.37) show characteristic absorption bands due to its coordinated water molecules and Fe–O bonding. Notably, a broad band around 3400–3300 cm− 1 corresponds to O–H stretching from strongly hydrogen-bonded water in the [Fe(H2O)6]3⁺ complex. Bending vibrations of water appear in the 1650–1620 cm− 1 region. Peaks between 1200 and 1000 cm− 1 (antisymmetric Fe–O stretching), 970–880 cm− 1 (symmetric Fe–O stretching), and 750–650 cm− 1 (water libration modes) are evident, confirming the characteristic structural features of FeCl3·6H2O. These assignments serve as a comparative baseline to understand the interactions between FeCl3·6H2O and the polymer matrix. In the FeCl3·6H2O/P(VDF-HFP) composite spectra, subtle shoulders and intensity shifts within the regions corresponding to Fe–O and coordinated water vibrations suggest overlapping contributions with polymer modes—particularly in the 970–880 cm− 1 and 750–650 cm− 1 ranges. While no new distinct peaks attributed solely to FeCl3·6H2O are observed due to these overlaps, the modified intensity patterns indicate successful incorporation and interaction of FeCl3·6H2O within the polymer matrix. These spectral changes also support the hypothesis that FeCl3·6H2O contributes to dipole alignment and enhances polar phase formation via hydrogen bonding and ionic interactions.

ATR-FTIR spectra of pure P(VDF-HFP) and FeCl3·6H2O/P(VDF-HFP) nanocomposites with different filler concentrations (1–4 wt%). Peaks corresponding to α-, β-, and γ-phases are marked. Increasing FeCl3·6H2O content enhances the β-phase (840 cm− 1) while reducing α-phase signals, indicating phase transformation. The inset highlights these spectral changes for improved clarity
The α-phase is identified by vibrational bands at 431 cm− 1, 530 cm− 1, 614 cm− 1, 762 cm− 1, and 797 cm− 1, which correspond to CF2 bending, skeletal bending, and CH2 rocking modes. These peaks in the pure P(VDF-HFP) spectrum confirm the retention of the α-phase. Peaks at 511 cm− 1 (CF2 bending) and 834 cm− 1 (CH2 rocking and CF2 asymmetric stretching) indicate the presence of the polar β-phase. A peak at 875 cm− 1, associated with CH2 rocking vibrations from both β-and γ-phases, further confirms the retention of less polar phases in the pure polymer. In the FeCl3·6H2O/P(VDF-HFP) nanocomposites, the disappearance of α-phase peaks and the increased intensity of β-phase-specific peaks indicate that FeCl3·6H2O facilitates dipole alignment within the polymer matrix, driving the phase transition. The γ-phase, identified by a peak at 1231 cm− 1 (CF out-of-plane deformation), remains unaffected, highlighting that FeCl3·6H2O selectively enhances the β-phase while maintaining the stability of the γ-phase. Peaks at 1070, 1170, and 1402 cm− 1, associated with CF3 out-of-plane deformation, CF2 antisymmetric stretching, and CH2 scissoring vibrations, remain consistent across all samples, indicating that the polymer backbone retains its structural integrity32. Minor intensity variations suggest localized interactions between FeCl3·6H2O and the polymer chains. The inclusion of FeCl3·6H2O significantly enhances the β-phase content by promoting dipole alignment and encouraging polar interactions. This enhancement is evident from the increased intensity of β-phase-specific peaks, such as those at 511 and 834 cm− 1, and the suppression of α-phase peaks, such as those at 614 and 797 cm− 1. FeCl3·6H2O achieves this phase transformation through hydrogen bonding and ionic interactions, which destabilize the nonpolar α-phase and stabilize the polar β-phase.
The β-phase fraction, F(β), was calculated for pure P(VDF-HFP) and FeCl3·6H2O/P(VDF-HFP) nanocomposites using Eq. (1), with the results summarized in Table 1. The data reveal a clear trend of increasing β-phase fraction with rising FeCl3·6H2O content, highlighting the effectiveness of FeCl3·6H2O as a phase-modifying agent. In pure P(VDF-HFP), the β-phase fraction was determined to be 67.18%, reflecting the inherent presence of the polar β-phase. However, this value also underscores the dominance of the nonpolar α-phase, as corroborated by FTIR spectra, which display prominent α-phase peaks alongside moderate β-phase peaks. From Fig. 6a, the formation of the β-phase in pure P(VDF-HFP) films is primarily influenced by the polymer’s intrinsic properties and its interaction with the polar solvent DMF during the solution casting process38. DMF, as a polar aprotic solvent, ensures uniform dispersion of the polymer components, disrupts the crystalline packing of the nonpolar α-phase, and promotes molecular rearrangement and dipole alignment through its strong interactions with the CF2 dipoles. Additionally, hydrogen bonding between the CH2 groups of P(VDF-HFP) and the carbonyl oxygen (C = O) of DMF enhances chain flexibility and mobility, further stabilizing the β-phase. These specific interactions between DMF and the polymer matrix play a crucial role in reducing chain entanglement and facilitating molecular rearrangement, which promotes dipole alignment along the polymer backbone21,39,40. Another contributing factor is the self-hydrogen bonding between the P(VDF-HFP) chains, which can also promote the formation of the β-phase. In this interaction, the hydrogen atoms of the CH2 groups are attracted to the fluorine atoms of adjacent –CF2 groups (C–H···F–C). These combined effects result in a substantial β-phase fraction of 67.18% in pure films, providing a baseline for further enhancement. However, this interaction may not play a significant role in the phase transformation due to the relatively weak interactions within this polymer-solvent system, leading to a mixture of α- and β-phases in the film.

Schematic representation of hydrogen bonding interaction for a pure P(VDF-HFP) and b FeCl₃·6 H₂O/P(VDF-HFP) nanocomposites.
The addition of only 1% FeCl3·6H2O significantly increases the β-phase fraction to 86.55%, representing a substantial transformation from the α-phase to the β-phase. This enhancement is attributed to the strong ionic interactions between Fe3+ ions and the electronegative fluorine atoms in the CF2 groups, as well as hydrogen bonding between the coordinated water molecules of FeCl3·6H2O and the polymer chains41. These interactions disrupt the symmetrical crystalline packing of the α-phase, promote dipole alignment, and stabilize the polar β-phase, as evidenced by the enhanced intensity of β-phase peaks, such as the one at 840 cm− 1. At 2% FeCl3·6H2O, the β-phase fraction increases slightly to 88.17%, indicating near-optimal interactions between FeCl3·6H2O and the polymer matrix. This level of interaction facilitates efficient dipole alignment and a near-complete phase transformation from α to β. The FTIR spectra show the suppression of α-phase peaks and further enhancement of β-phase peaks, demonstrating the efficiency of FeCl3·6H2O at this concentration. The small increase in β-phase fraction from 1 to 2% suggests that most of the phase transformation has already occurred by 2% FeCl3·6H2O.
At higher concentrations of FeCl3·6H2O (3% and 4%), the β-phase fraction increases only marginally to 88.68% and 88.99%, respectively. These results indicate that the phase transformation reaches a saturation point at approximately 2% FeCl3·6H2O, beyond which additional filler has minimal impact on further increasing the β-phase fraction. This saturation can be attributed to filler agglomeration, as observed in SEM and AFM images, which limits effective interaction between FeCl3·6H2O and the polymer chains. Agglomeration also introduces structural irregularities, leading to increased surface roughness and uneven phase distribution. The β-phase fraction data confirm the ability of FeCl3·6H2O to induce and stabilize the polar β-phase in P(VDF-HFP) nanocomposites. The significant transformation observed at low filler concentrations (1-2%) highlights the importance of ionic and hydrogen bonding interactions in facilitating dipole alignment and phase transition. However, the near-saturation of the β-phase fraction at higher concentrations emphasizes the importance of optimizing filler loading to achieve a balance between phase enhancement and structural uniformity. These findings align with structural and spectral data, providing a comprehensive understanding of how FeCl3·6H2O interacts with P(VDF-HFP) to tailor its properties for advanced applications such as flexible electronics and energy storage devices.
XRD analysis
The XRD patterns of pure P(VDF-HFP) and FeCl3·6H2O/P(VDF-HFP) nanocomposites, as shown in Fig. 7, provide crucial insights into the crystalline structure and phase composition. Based on standard XRD data for P(VDF-HFP)1,8, diffraction peaks at 2θ values of 18.6°, 19.7°, 20.6°, 26.5°, and 38.8° are indexed as (100), (020), (110), (110), and (021) planes, respectively. These peaks confirm the crystallization of P(VDF-HFP) units in the copolymer and its semi-crystalline nature. Specifically, the peaks at 18.6°, 19.7°, 26.5°, and 38.8° are attributed to the nonpolar α-phase, while the prominent peak at 20.6° corresponds to the polar β-phase42. In the XRD spectrum of pure P(VDF-HFP), strong diffraction peaks at approximately 18.6°, 26.5°, and 38.8° indicate that the α-phase dominates the crystalline structure. However, the presence of a minor peak at 20.6° suggests a partial contribution from the β-phase, confirming a combination of α- and β-phases in the pure P(VDF-HFP) films. This finding aligns with the β-phase fraction determined from the FTIR analysis. Furthermore, the XRD pattern of pristine FeCl3·6H2O exhibits distinct peaks at approximately 13.2°, 18.5°, 24.7°, and 28.1° (2θ), corresponding to the (011), (020), (111), and (121) crystallographic planes, respectively, as reported by Chen et al.43 and Louvain et al.44. These reflections are characteristic of its monoclinic crystal structure and reflect the layered arrangement of chloride ions and coordinated water molecules surrounding Fe3+ centers in the [Fe(H2O)4Cl2]+ complex. In the FeCl3·6H2O/P(VDF-HFP) composite films, these distinct FeCl3·6H2O peaks are not prominently observed, likely due to peak broadening, overlap with the polymer matrix, and the nanoscale dispersion of the salt within the composite. Nonetheless, subtle shifts in peak intensity and position within the composite XRD patterns suggest successful incorporation of FeCl3·6H2O and its interaction with the P(VDF-HFP) lattice, contributing to the observed structural modifications. The incorporation of FeCl3·6H2O into the polymer matrix induces significant changes in the crystalline structure. The intensity of the β-phase peak at 20.6° increases with higher concentrations of FeCl3·6H2O, while the intensities of the α-phase peaks at 18.6°, 26.5°, and 38.8° decrease. Figure 6b illustrates the synergistic mechanisms driving the transformation from the nonpolar α-phase to the polar β-phase in FeCl3·6H2O/P(VDF-HFP) nanocomposites. This transition is facilitated by multiple interactions45,46,47, starting with self-hydrogen bonding within P(VDF-HFP) chains, where hydrogen atoms in CH2 groups form weak hydrogen bonds with fluorine atoms in adjacent – CF2 groups, partially aligning dipoles. Interactions with the solvent, DMF, further enhance this effect; the carbonyl oxygen (C = O) of DMF forms hydrogen bonds with the polymer’s CH2 groups, while dipolar interactions between DMF and CF2 groups improve chain mobility, creating an environment conducive to β-phase nucleation. FeCl3·6H2O plays a crucial role by establishing strong hydrogen bonding between Fe3+ ions and CF2 dipoles, disrupting the compact α-phase structure and aligning dipoles for β-phase crystallization. The coordinated water molecules in FeCl3·6H2O increase polymer chain flexibility, facilitating dipole rearrangement and stabilizing the β-phase. Additionally, the slow solvent evaporation during film formation allows adequate time for these molecular interactions28, while the uniform dispersion of FeCl3·6H2O prevents filler aggregation, ensuring efficient α-to-β phase transition. Together, these interactions optimize the piezoelectric and ferroelectric properties of the nanocomposites, making them suitable for advanced applications in energy harvesting and flexible electronics.

XRD patterns of pure P(VDF-HFP) and FeCl3·6H2O /P(VDF-HFP) nanocomposites with varying FeCl3·6H2O concentrations (1–4 wt%). Characteristic diffraction peaks corresponding to the α-phase (2θ ≈ 18.6°, 19.7°) and β-phase (2θ ≈ 20.6°) are indicated. The inset provides a magnified view of the 2θ range from 15° to 25°, highlighting the subtle increase in the β-phase peak intensity at 20.6° with increasing FeCl3·6H2O content.
The degree of crystallinity (Xc) of pure P(VDF-HFP) and FeCl3·6H2O/P(VDF-HFP) nanocomposites was determined using Eq. (2), with the results summarized in Table 1. The data reveal a progressive increase in crystallinity as the FeCl3·6H2O content increases. For pure P(VDF-HFP), the crystallinity was measured at 46.64%, reflecting its semi-crystalline structure, which is characterized by a combination of ordered crystalline regions (associated with α- and β-phases) and disordered amorphous regions. The introduction of FeCl3·6H2O enhanced crystallinity, increasing it to 48.44% at 1 wt% and 50.86% at 2 wt%. This improvement can be attributed to FeCl3·6H2O’s ability to promote molecular alignment and dipole interactions within the polymer matrix, thereby encouraging the alignment of polymer chains into the β-phase crystalline structure. These findings align with XRD and FTIR analyses, which also indicated an increase in β-phase content and a corresponding reduction in nonpolar α-phase peaks. As the FeCl3·6H2O concentration increased further, the crystallinity exhibited a slower growth rate, reaching 51.15% and 51.56% for 3 wt% and 4 wt% FeCl3·6H2O, respectively. This suggests that the crystallinity approaches a saturation point, where the influence of FeCl3·6H2O on further ordering becomes limited. The plateau is likely due to filler agglomeration at higher concentrations, which disrupts the uniform distribution of FeCl3·6H2O within the polymer matrix and reduces its efficiency in inducing crystallinity. Therefore, the trend of increasing crystallinity highlights the effectiveness of FeCl3·6H2O as a phase-modifying agent. The enhanced crystallinity contributes to improved structural properties, including better mechanical stability and dielectric performance. However, the reduced effectiveness at higher concentrations underscores the importance of optimizing filler content to achieve maximum crystallinity without compromising uniformity in the polymer matrix.
The absolute β-phase fraction values (%β) presented in Table 1 provide valuable insights into the distribution of the polar β-phase within the crystalline regions of pure P(VDF-HFP) and FeCl3·6H2O/P(VDF-HFP) nanocomposites. This fraction, calculated using Eq. (3) based on the degree of crystallinity and the β-phase content, represents the proportion of the β-phase within the crystalline domains of the polymer matrix. For pure P(VDF-HFP), the %β value is calculated to be 31.33%, indicating that approximately one-third of the crystalline regions consist of the polar β-phase, while the remainder corresponds to the nonpolar α-phase. This demonstrates the inherent β-phase content in pure P(VDF-HFP), despite the crystalline structure being predominantly dominated by the α-phase. With the addition of FeCl3·6H2O, a significant increase in the %β value is observed, rising to 41.92% at 1 wt% FeCl3·6H2O. This notable enhancement is attributed to the filler’s ability to facilitate the transformation of the nonpolar α-phase to the polar β-phase by promoting dipole alignment and increasing chain mobility through ionic and hydrogen bonding interactions. At 2 wt% FeCl3·6H2O, the %β further increases to 44.84%, reflecting continued improvement in β-phase content within the crystalline regions. This trend aligns with the structural changes observed in XRD and FTIR analyses, which reveal a reduction in α-phase peaks alongside an increase in β-phase-specific peaks. At higher filler concentrations (3 wt% and 4 wt%), the %β reaches 45.36% and 45.88%, respectively, suggesting a plateau in the transformation of the α-phase to the β-phase. This indicates that the maximum achievable β-phase fraction within the crystalline region has been reached, with additional FeCl3·6H2O having minimal impact. The slight increase at these higher concentrations may result from diminishing filler efficiency due to agglomeration and uneven distribution, which limit further interactions with the polymer matrix. These findings underscore the effectiveness of FeCl3·6H2O as a phase-modifying agent in promoting the β-phase within the crystalline regions of P(VDF-HFP). The observed trend highlights the potential for optimizing filler content to achieve a balance between maximum β-phase enhancement and maintaining uniform structural integrity.
Thermal analysis
The determination of thermal stability is essential for applications requiring high processing temperatures, particularly in flexible electronics, actuators, and energy storage devices. The thermal stability, based on TGA and DTA (inset) curves presented in Fig. 8 and data in Table 2, provides a comprehensive overview of the thermal behavior of pure P(VDF-HFP) and FeCl3·6H2O/P(VDF-HFP) nanocomposites. These analyses reveal a single-step degradation mechanism for all samples within the temperature range of 400–500 °C, as confirmed by the DTG peaks that indicate polymer-based thermal degradation. The degradation onset temperature (T5%) was determined at 5% of mass loss. Additionally, the degradation endset temperature (T70%) was assessed at 70% of mass loss, while the maximum degradation temperature (Tmax) was determined at the peak of the first derivative of the DTG curve. Pure P(VDF-HFP) exhibits high inherent thermal stability, as evidenced by its T5% (458.73 °C) and Tmax (496.53 °C), which are the highest among all samples. However, the incorporation of FeCl3·6H2O introduces coordinated water molecules, leading to a reduction in T5% as the filler content increases. For instance, T5% decreases to 419.83 °C at 3 wt% FeCl3·6H2O due to the evaporation or decomposition of water. This early-stage destabilization is primarily attributed to the plasticizing effect of water, which enhances polymer chain mobility and lowers thermal resistance. Despite this, the Fe³⁺ ions in FeCl3·6H2O act as stabilizing agents, mitigating the destabilizing effects of water by promoting dipole alignment, β-phase crystallinity, and filler-polymer interactions48,49. Interestingly, a slight deviation from the general thermal degradation trend is observed at 3 wt% FeCl3·6H2O, where the T₅% value (419.83 °C) is lower than that of the 4 wt% sample (429.89 °C). This unexpected drop can be attributed to localized agglomeration at the 3% loading, which likely leads to uneven dispersion and retention of more coordinated water. This retained water acts as a plasticizer, enhancing polymer chain mobility and promoting early-stage thermal degradation. However, at 4 wt%, the dispersion of FeCl3·6H2O appears to improve, and the formation of thermally stable iron oxide residues becomes more effective, resulting in increased thermal stability. This is supported by the highest T₇₀% (557.27 °C) and residue (24.44%) at 4 wt%, indicating enhanced thermal shielding and char retention. These findings highlight the delicate balance between destabilizing effects of coordinated water and the stabilizing influence of Fe³⁺ ions and iron oxide residues, emphasizing the importance of optimizing filler concentration for maximal thermal performance.

TGA and DTG (inset) curves of pure P(VDF-HFP) and FeCl₃·6 H₂O/P(VDF-HFP) nanocomposites
The role of Fe3+ ions was central to the stabilization mechanism in these nanocomposites. Fe3+ promotes β-phase crystallinity by interacting with the polymer chains through ionic bonding, specifically with electronegative fluorine atoms in the CF2 groups. This interaction disrupts the α-phase, facilitating dipole alignment and enhancing β-phase content, which contributes to greater thermal resistance50. Furthermore, Fe3+ restricts polymer chain mobility by forming ionic and dipole-aligned interactions with the polymer matrix, which increases the energy required for bond dissociation and delays thermal decomposition. As a nucleating agent, Fe3+ reinforces crystalline regions within the polymer, improving its structural and thermal stability. Additionally, during thermal degradation, Fe3+ decomposes to form thermally stable iron oxides (e.g., Fe2O3 or Fe3O4), which remain as residual products and contribute to higher residual weight observed in TGA analyses51. The DTA curves further confirm the stabilizing role of Fe3+ ions, as evidenced by the gradual weight loss profile in the composites. Tmax values for composites, such as 493.06 °C at 1 wt% FeCl3·6H2O and 489.92 °C at 2 wt%, reflect the contribution of Fe3+ to thermal stability through ionic bonding and improved crystalline phase development. Additionally, the T70% values, which increase with FeCl3·6H2O concentration and reach 557.27 °C at 4 wt%, highlight superior resistance during later degradation stages.
Residual weight analysis further underscores the stabilizing role of FeCl3·6H2O. Pure P(VDF-HFP) has a residue of 15.10%, attributed to carbonaceous char, whereas composites exhibit significantly higher residues, such as 24.44% at 4 wt% FeCl3·6H2O. This increase is due to the formation of thermally stable iron oxides during FeCl₃ decomposition. These oxides enhance thermal stability and structural integrity at elevated temperatures. Thus, the thermal behavior trends highlight the dual role of FeCl3·6H2O in modifying the polymer matrix. The coordinated water molecules reduce stability at lower temperatures, while Fe3+ ions counteract this effect by reinforcing the polymer matrix through dipole alignment, enhanced β-phase crystallinity, reduced chain mobility, and the formation of stable iron oxides as reported by Martins et al.51. These findings confirm FeCl3·6H2O’s effectiveness as a stabilizing agent and underscore the importance of optimizing its concentration to achieve a balance between thermal resistance and degradation for applications in flexible electronics, energy storage, and other high-temperature environments.
Dielectric properties
The dielectric properties of pure P(VDF-HFP) and FeCl3·6H2O/P(VDF-HFP) nanocomposites, as presented in Fig. 9; Table 3, reveal the significant impact of FeCl3·6H2O filler content on dielectric constant, dielectric loss, and conductivity. The dielectric constant demonstrates a decreasing trend with increasing frequency across all samples, reflecting typical dielectric dispersion behavior caused by the lag of polarization mechanisms at higher frequencies. At 10 Hz, the dielectric constant for pure P(VDF-HFP) is 5.62, which increases significantly with the addition of FeCl3·6H2O, reaching 19.16 for the 4 wt% filler concentration. This enhancement can be attributed to the influence of Fe3+ ions, which promote ionic and electronic polarization while facilitating dipole alignment within the polymer matrix. The increased filler concentration introduces more polymer-filler interfaces, which trap charges and amplify interfacial polarization, while the improved β-phase crystallinity induced by Fe3+ ions contribute to greater dipole density and alignment in good agreement with the reports made available in the literature2,52. At higher frequencies, the dielectric constant decreases as polarization mechanisms become less effective due to reduced response times53.

a Dielectric constant, b dielectric loss and c conductivity of pure P(VDF-HFP) and FeCl₃·6 H₂O/P(VDF-HFP) nanocomposites
The dielectric loss follows a similar trend, decreasing with increasing frequency, which indicates reduced energy dissipation at higher frequencies. At 10 Hz, pure P(VDF-HFP) exhibits a dielectric loss of 0.16, increasing to 0.30 for the 4 wt% filler concentration. This increase is attributed to higher ionic conductivity and charge carrier mobility introduced by Fe3+ ions. At lower filler concentrations, the uniform dispersion of FeCl3·6H2O minimizes defects and keeps dielectric loss relatively low. However, at higher filler concentrations, localized agglomeration can create regions with increased charge dissipation, leading to slightly higher dielectric loss. Despite this, the loss values remain within acceptable limits, indicating efficient energy utilization in the composites. The balance between filler-induced improvements and the slight increase in energy dissipation highlights the need for optimized filler content to maintain high performance while minimizing losses. Compared to previously reported systems, the dielectric performance of the FeCl3·6H2O/P(VDF-HFP) nanocomposites in this study shows a notable improvement. At 10 Hz, the dielectric constant increases from 5.62 for pure P(VDF-HFP) to 19.16 with 4 wt% FeCl3·6H2O, while maintaining a low dielectric loss of 0.30. This balance between high permittivity and low loss is rarely achieved in other systems. For instance, P(VDF-HFP) composites with 20 vol% BaTiO3 or Fe2O3@BaTiO3 have reported dielectric constants of up to 31.7 at 1 kHz but require much higher filler loadings and exhibit increased dielectric loss54. Similarly, flower-like TiO₂-based nanocomposites showed over 200% enhancement in dielectric constant but with a compromise in loss performance at low frequencies55. Moreover, PVDF-based systems filled with magnetite nanoparticles (Fe3O4) also demonstrated improved dielectric properties, but their dielectric constant remained lower than that achieved in the present study56. Additionally, in a study by Sobola et al.15, PVDF fibers doped with Ca(NO3)2, Mg(NO3)2, and Zn(NO3)2 achieved β-phase fractions of 92.29%, 95.75%, and 85.30%, respectively, but their dielectric constants at 10 Hz were relatively low—approximately 3.9, 3.6, and 3.5. Furthermore, their electrospinning-based fabrication method limits scalability. In contrast, our solution-cast FeCl3·6H2O/P(VDF-HFP) films achieved a β-phase fraction of 88.99% and a dielectric constant of 19.16 at 10 Hz with excellent film uniformity and scalability. Likewise, Fortunato et al.19 reported that among several hexahydrate salts, FeCl3·6H2O led to the lowest β-phase fraction (Fβ ≈ 65.7%) in PVDF nanofibers. In our P(VDF-HFP) matrix, however, FeCl3·6H2O facilitated strong hydrogen bonding and polar interactions, enabling effective α-to-β phase transformation and superior dielectric enhancement. These findings clearly demonstrate the superior efficiency of FeCl3·6H2O as a dielectric modifier and present our composite as a scalable, high-performance material suitable for energy storage, flexible electronics, and advanced sensor technologies.
Conductivity exhibits a clear frequency-dependent increase across all samples, consistent with the behavior typical of polymer composites. At 10 Hz, pure P(VDF-HFP) shows the lowest conductivity of 9.26 × 10− 8 S/cm, which rises progressively with higher filler concentrations, reaching 3.07 × 10− 7 S/cm for the 4 wt% FeCl3·6H2O composite. This improvement is driven by several synergistic mechanisms: Fe3+ ions act as charge carriers, facilitating ionic conductivity by enabling efficient charge transport within the matrix57. The increased β-phase crystallinity further enhances charge mobility by aligning dipoles, while interfacial polarization at polymer-filler interfaces generates space charge polarization, contributing to higher conductivity. As filler content increases, these effects are magnified through the creation of additional interfaces and conductive pathways. However, at higher frequencies, conductivity stabilizes, reflecting the transition to the intrinsic properties of the polymer matrix. Together, these results demonstrated that FeCl3·6H2O/P(VDF-HFP) nanocomposites achieve enhanced dielectric properties and conductivity through the combined influence of polarization mechanisms, making them promising candidates for applications in energy storage, flexible electronics, and sensors.
Conclusion
This study demonstrates the effective incorporation of FeCl3·6H2O as a novel functional filler for enhancing the electroactive β-phase content, crystallinity, and dielectric performance of P(VDF-HFP) nanocomposites. The composite with 4 wt% FeCl3·6H2O achieved a substantial increase in β-phase fraction (from 67.18 to 88.99%) and dielectric constant (from 5.62 to 19.16 at 10 Hz), while maintaining a low dielectric loss (< 0.30). These enhancements are attributed to strong hydrogen bonding, dipolar interactions between Fe3+ ions and CF2 groups, and the structural influence of coordinated water molecules. Thermal analysis confirmed the composite’s stability with degradation onset temperatures above 400 °C, supporting their applicability in high-temperature environments. The innovation of this work lies in achieving significant dielectric and structural improvements using a low filler concentration, thereby preserving the polymer’s flexibility and processability. These results are further supported by quantitative analysis: the degree of crystallinity increased from 46.64 to 51.56%, and the significant rise in dielectric constant is attributed to improved dipole alignment, β-phase formation, and filler-polymer interfacial interactions. The ability to achieve such improvements at low filler content emphasizes the material’s efficiency and preserves flexibility and processability—critical for device integration. Compared to other fillers such as BaTiO3, TiO2, and Fe3O4, FeCl3·6H2O offers a superior balance between performance and compatibility with the polymer matrix. The practical implications of this work extend to a wide array of advanced applications, including flexible electronics, energy storage systems, and sensor technologies. The significant improvement in β-phase content and dielectric properties makes these composites highly suitable for energy harvesting devices, such as piezoelectric nanogenerators (PENGs), enabling efficient conversion of mechanical energy into electrical energy. Their thermal stability further supports their deployment in industrial sensors, while their inherent flexibility highlights their potential for wearable technologies and flexible capacitors in energy storage systems. These findings establish a strong bridge between academic advancements and practical applications, paving the way for future innovation in next-generation flexible and sustainable technologies. Future research will focus on the integration of these composites into device-level applications, the exploration of other hydrated metal salts or hybrid fillers, and the development of scalable fabrication techniques to enable commercial viability in energy storage and flexible electronic systems.
Data availability
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
During the preparation of this work the authors used ChatGPT to check grammar. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
References
Yasar, M., Hassett, P., Murphy, N. & Ivankovic, A. β phase optimization of solvent cast PVDF as a function of the processing method and additive content, (in eng), ACS Omega, 9(24), 26020–26029 (2024). https://doi.org/10.1021/acsomega.4c01221
Gong, Z. et al. Flexible PVDF/SiC/FeCl3 nanofiber membrane generators with synergistically enhanced piezoelectricity. Nano Energy. 122, 109290. https://doi.org/10.1016/j.nanoen.2024.109290 (2024).
Li, C. et al. Enhancement research on piezoelectric performance of electrospun PVDF fiber membranes with inorganic reinforced materials. Coatings, 11(12), 1495 (2021).
Dani, S., Channabasavanna, P. N., Kulkarni, S., Manohara, S. R. & Udayakumar, K. The electromagnetic interference shielding effectiveness and dielectric response of PVDF-nTiO2Nanocomposites thin films. Trends Sci. 19 (21), 1101. https://doi.org/10.48048/tis.2022.3046 (2022).
Liu, Y. et al. Phase separation of a PVDF–HFP film on an ice substrate to achieve self-polarisation alignment. Nano Energy. 106, 108082. https://doi.org/10.1016/j.nanoen.2022.108082 (2023).
Bouharras, F. E. et al. Dielectric characterization of core-shell structured poly(vinylidene fluoride)-grafted-BaTiO(3) nanocomposites, (in eng), Polymers (Basel), 15(3), (2023). https://doi.org/10.3390/polym15030595
Kim, J. Y. Phase behavior of binary and ternary fluoropolymer (PVDF-HFP) solutions for single-ion conductors. RSC Adv. 12 (33), 21160–21171. https://doi.org/10.1039/D2RA04158H (2022).
Divya, S. & Hemalatha, J. Study on the enhancement of ferroelectric Β phase in P(VDF-HFP) films under heating and poling conditions. Eur. Polymer J. 88, 136–147. https://doi.org/10.1016/j.eurpolymj.2017.01.016 (2017).
Adaval, A. et al. 2D nanomaterials incorporated Poly(vinylidene fluoride) nanocomposites: morphology, crystalline structure, and dielectric, ferroelectric, and piezoelectric properties. J. Phys. Chem. C. 127 (13), 6483–6502. https://doi.org/10.1021/acs.jpcc.2c08689 (2023).
Bužarovska, A. et al. PVDF/BaTiO3 composite foams with high content of Β phase by thermally induced phase separation (TIPS). J. Polym. Res. 29 (7), 272. https://doi.org/10.1007/s10965-022-03133-z (2022).
Cerrada, M. L., Arranz-Andrés, J., Caballero-González, A., Blázquez-Blázquez, E. & Pérez, E. The β form in PVDF nanocomposites with carbon nanotubes: structural features and properties, Polymers 15(6). https://doi.org/10.3390/polym15061491
Peleš Tadić, A. et al. Nanomechanical properties of PVDF–ZnO polymer nanocomposite. Mater. Sci. Eng. B. 287, 116126. https://doi.org/10.1016/j.mseb.2022.116126 (2023).
Wang, B. & Huang, H. X. Incorporation of Halloysite nanotubes into PVDF matrix: nucleation of electroactive phase accompany with significant reinforcement and dimensional stability improvement. Compos. Part A: Appl. Sci. Manuf. 66, 16–24. https://doi.org/10.1016/j.compositesa.2014.07.001 (2014).
Zeng, Y., Pan, H., Shen, Z., Shen, Y. & Liu, Z. Improved breakdown strength and restrained leakage current of sandwich structure ferroelectric polymers utilizing Ultra-Thin Al2O3 nanosheets. Nanomaterials 13(21), 2836 (2023).
Sobola, D. et al. PVDF fibers modification by nitrate salts doping, Polymers 13(15). https://doi.org/10.3390/polym13152439
Sarkar, R. & Kundu, T. K. Hydrogen bond interactions of hydrated aluminum nitrate with PVDF, PVDF-TrFE, and PVDF-HFP: A density functional theory-based illustration. Int. J. Quant. Chem. 120, e26328. https://doi.org/10.1002/qua.26278 (2020).
Hwangbo, S., Kang, J. M., Min, W. M., Lee, J. H. & Hwang, K. S. Preparation of β-Phase Poly(vinylidene fluoride) films on aluminum substrate with the addition of hydrated metal salts, (in eng). J. Nanosci. Nanotechnol. 19 (3), 1824–1828. https://doi.org/10.1166/jnn.2019.16167 (Mar 1 2019).
Yuennan, J., Sukwisute, P. & Muensit, N. Effect of hydrated salts on the microstructure and phase transformation of poly(vinylidenefluoride-hexafluoropropylene) composites. Mater. Res. Express. 5 (5), 055702. https://doi.org/10.1088/2053-1591/aabf4d (2018).
Fortunato, M. et al. Piezoelectric effect and electroactive phase nucleation in self-standing films of unpoled PVDF nanocomposite films, Nanomaterials 8(9). https://doi.org/10.3390/nano8090743
Yuennan, J., Tohluebaji, N., Putson, C., Muensit, N. & Channuie, P. Enhanced electroactive β-phase and dielectric properties in P(VDF-HFP) composite flexible films through doping with three calcium chloride salts: CaCl, CaCl·2HO, and CaCl·6HO. Polym. Adv. Technol. 35 (6), e6437. https://doi.org/10.1002/pat.6437 (2024).
Ekbote, G. S., Khalifa, M., Mahendran, A. & Anandhan, S. Cationic surfactant assisted enhancement of dielectric and piezoelectric properties of PVDF nanofibers for energy harvesting application. Soft Matter. 17 (8), 2215–2222. https://doi.org/10.1039/D0SM01943G (2021).
Ramesan, M. T. & Jayakrishnan, P. Role of nickel oxide nanoparticles on magnetic, thermal and temperature dependent electrical conductivity of novel Poly(vinyl cinnamate) based nanocomposites: applicability of different conductivity models. J. Inorg. Organomet. Polym Mater. 27 (1), 143–153. https://doi.org/10.1007/s10904-016-0456-x (2017).
Ramesan, M. T. et al. Influence of magnetite nanoparticles on electrical, magnetic and thermal properties of Chitin/cashew gum biopolymer nanocomposites. Polymer Compos., 39(S1), E540–E549. https://doi.org/10.1002/pc.24688 (2018).
Ramesan, M. T. Fabrication, characterization, and properties of poly(ethylene-co-vinyl acetate)/magnetite nanocomposites. J. Appl. Polym. Sci. 131 (7). https://doi.org/10.1002/app.40116 (2014).
Jayakrishnan, P. & Ramesan, M. T. Synthesis, structural, magnetoelectric and thermal properties of poly (anthranilic acid)/magnetite nanocomposites, Polym. Bull. 74(8), 3179–3198. https://doi.org/10.1007/s00289-016-1883-0 (2017).
Jayakrishnan, P., K K, J., Ramesan, M. T. & M. K, and Synthesis, characterization, magnetoelectric properties and gas sensing application of Poly(anthranilic acid- co- indole)/ magnetite nanocomposites. J. Thermoplast. Compos. Mater. 36, 2523–2542. https://doi.org/10.1177/08927057221098969 (2023).
Tohluebaji, N., Yuennan, J., Putson, C. & Muensit, N. Improved electroactive Β phase nucleation and dielectric properties of P(VDF-HFP) composite with Al(NO3)3·9H2O fillers. Integr. Ferroelectr. 224 (1), 181–191. https://doi.org/10.1080/10584587.2022.2035608 (2022).
Low, Y. K. A., Tan, L., Tan, L. P., Boey, F. & Ng, K. Increasing solvent polarity and addition of salts promote b-Phase poly(vinylidene fluoride) formation. J. Appl. Polym. Sci. 128 (05), 2902–2910. https://doi.org/10.1002/app.38451 (2013).
Fu, H. et al. Phase separation in intrinsically stretchable electronics: mechanisms, functions and applications. Mater. Sci. Eng. R: Rep. 161, 100863. https://doi.org/10.1016/j.mser.2024.100863 (2024).
Magalhães, R. et al. The role of solvent evaporation in the microstructure of electroactive β-Poly(Vinylidene Fluoride) membranes obtained by isothermal crystallization. Soft Mater. 9, 1–14. https://doi.org/10.1080/1539445X.2010.525442 (2011).
Ha, J. Y. et al. Thickness dependent ferroelectric properties of BSTO thin films deposited by RF Magnetron sputtering. J. Electroceram. 17, 141–144. https://doi.org/10.1007/s10832-006-9734-2 (2006).
Mishra, K., Hashmi, S. A. & Rai, D. K. Protic ionic liquid-based gel polymer electrolyte: structural and ion transport studies and its application in proton battery. J. Solid State Electrochem. 18 (8), 2255–2266. https://doi.org/10.1007/s10008-014-2475-2 (2014). /08/01 2014.
Bormashenko, Y., Pogreb, R., Stanevsky, O. & Bormashenko, E. Vibrational spectrum of PVDF and its interpretation. Polym. Test. 23 (7), 791–796. https://doi.org/10.1016/j.polymertesting.2004.04.001 (2004). /10/01/ 2004, doi.
Lanceros-Méndez, S., Mano, J. F., Costa, A. M. & Schmidt, V. H. FTIR and DSC studies of mechanically deformed β-PVDF films. J. Macromolecular Sci. Part. B. 40, 3–4. https://doi.org/10.1081/MB-100106174 (2001).
Sim, L. N., Majid, S. R. & Arof, A. K. FTIR studies of PEMA/PVdF-HFP blend polymer electrolyte system incorporated with LiCF3SO3 salt. Vib. Spectrosc. 58, 57–66. https://doi.org/10.1016/j.vibspec.2011.11.005 (2012).
Inam, M. A., Khan, R., Park, D. R., Lee, Y. W. & Yeom, I. T. Removal of Sb(III) and Sb(V) by ferric chloride coagulation: implications of Fe solubility, Water 10(4). https://doi.org/10.3390/w10040418
Sheng, X., Wang, B., Mao, C., Sha, X. & Zhou, Y. Influence of FeCl3-modified chloroaluminate ionic liquids on long-chain alkenes alkylation. Appl. Organomet. Chem. 35 (1), e6055. https://doi.org/10.1002/aoc.6055 (2021).
Ruan, L. et al. Properties and applications of the β phase poly(vinylidene fluoride), (in eng), Polymers (Basel) 10(3). https://doi.org/10.3390/polym10030228 (2018).
Marshall, J. E. et al. On the solubility and stability of polyvinylidene fluoride, Polymers 13(9). https://doi.org/10.3390/polym13091354
Lederle, F., Härter, C. & Beuermann, S. Inducing Β phase crystallinity of PVDF homopolymer, blends and block copolymers by anti-solvent crystallization. J. Fluorine Chem. 234, 109522. https://doi.org/10.1016/j.jfluchem.2020.109522 (2020).
Song, R. et al. Modification of polymorphisms in polyvinylidene fluoride thin films via water and hydrated salt. J. Colloid Interface Sci. 401, 50–57. https://doi.org/10.1016/j.jcis.2013.03.021 (2013)
Molla, S. et al. Electroactive CTAB/PVDF composite film based photo-rechargeable hybrid power cell for clean energy generation and storage. Sci. Rep. 12 (1), 22350. https://doi.org/10.1038/s41598-022-26865-w (2022).
Chen, Y., Yu, H. Y. & Li, Y. Highly efficient and superfast cellulose dissolution by green chloride salts and its dissolution mechanism. ACS Sustain. Chem. Eng. 8 (50), 18446–18454. https://doi.org/10.1021/acssuschemeng.0c05788 (2020).
Louvain, N. et al. One-shot versus stepwise gas–solid synthesis of iron trifluoride: investigation of pure molecular F2 fluorination of chloride precursors, CrystEngComm 15(18), 3664–3671. https://doi.org/10.1039/C3CE27033E (2013).
Chen, S., Yao, K., Tay, F. & Liow, C. Ferroelectric Poly(Vinylidene Fluoride) thin films on Si substrate with the Beta phase promoted by hydrated magnesium nitrate. J. Appl. Phys. 102 (20), 104108–104108. https://doi.org/10.1063/1.2812702 (2007).
Jana, S., Garain, S., Sen, S. & Mandal, D. The influence of hydrogen bonding on dielectric constant and piezoelectric energy harvesting performance of hydrated metal salt mediated PVDF films. Phys. Chem. Chem. Phys. 17, 08. https://doi.org/10.1039/C5CP01820J (2015).
Mammeri, F. Chapter 3 - Nanostructured flexible PVDF and fluoropolymer-based hybrid films, in Frontiers of Nanoscience, (Eds. Benelmekki, M. & Erbe, A.) 14 67–101 (Elsevier, 2019).
Gopika, M. S., Bindhu, B., Sandhya, K. Y. & Reena, V. L. Impact of surface-modified molybdenum disulphide on crystallization, thermal and mechanical properties of polyvinylidene fluoride. Polym. Bull. 77 (2), 757–773. https://doi.org/10.1007/s00289-019-02765-4 (2020).
Liu, P. et al. Facile preparation of α-Fe2O3 nanobulk via bubble electrospinning and thermal treatment. Therm. Sci. 20 (01), 967–972. https://doi.org/10.2298/TSCI1603967L (2016).
AlAhzm, A., Alejli, M., Ponnamma, D., Elgawady, Y. & Al-Maadeed, M. Piezoelectric properties of zinc oxide/iron oxide filled polyvinylidene fluoride nanocomposite fibers. J. Mater. Sci.: Mater. Electron. https://doi.org/10.1007/s10854-021-06020-3 (2021).
Martins, P., Costa, C., Benelmekki, M. & Lanceros-Méndez, S. Preparation of magnetoelectric composites by nucleation of the electroactive β-Phase of Poly(vinylidene fluoride) by NiZnFe2O4 nanoparticles. Sens. Lett. 11, 0101. https://doi.org/10.1166/sl.2013.2808 (2013).
Reddy, M. & Reddy, L. R. M. Structural, dielectric, optical and magnetic properties of Ti3+, Cr3+, and Fe3+: PVDF polymer films. J. Polym. https://doi.org/10.1155/2013/295291 (2013).
Ji, W. et al. The enhancement in dielectric properties for PVDF based composites due to the incorporation of 2D TiO2 nanobelt containing small amount of MWCNTs. Compos. Part A: Appl. Sci. Manufac. 149, 106493. https://doi.org/10.1016/j.compositesa.2021.106493 (2021).
Jiang, Y., Zhang, Z., Zhou, Z., Yang, H. & Zhang, Q. Enhanced dielectric performance of P(VDF-HFP) composites with satellite–core-structured Fe2O3@BaTiO3 nanofillers, Polymers 11(10). https://doi.org/10.3390/polym11101541
Yu, E., Zhang, Q., Xu, N. & Yang, H. F-TiO2/P(VDF-HFP) hybrid films with enhanced dielectric permittivity and low dielectric loss. RSC Adv. 7 (7), 3949–3957. https://doi.org/10.1039/C6RA26772F (2017).
Tsonos, C. et al. Polyvinylidene fluoride/magnetite nanocomposites: dielectric and thermal response. J. Phys. Chem. Solids. 129, 01. https://doi.org/10.1016/j.jpcs.2019.01.025 (2019).
Tawansi, A., Oraby, A., Abdelkader, H. & Abdelaziz, M. FeCl3–CoCl2 mixed fillers effects on the structural, electrical and magnetic properties of PVDF films. J. Magn. Magn. Mater. 262, 203–211. https://doi.org/10.1016/S0304-8853(02)00590-5 (2003).
Acknowledgements
This work (grant no. RGNS 65–169) was financially supported by the Office of the Permanent Secretary, Ministry of Higher Education, Science, Research and Innovation (OPS MHESI), Thailand Science Research and Innovation (TSRI). Additionally, the authors would like to thank Faculty of Science and Technology, Nakhon Si Thammarat Rajabhat University, Faculty of Science and Technology, Princess of Naradhiwas University, Division of Physical Science (Physics), Faculty of Science, Prince of Songkla University and Center of Excellence in Nanotechnology for Energy (CENE), Prince of Songkhla University for instrumental supports.
Author information
Authors and Affiliations
Contributions
Jureeporn Yuennan: Supervision, Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing; Nantakan Muensit: Supervision, Validation; Nikruesong Tohluebaji: Formal analysis, Writing – original draft; Wichain Chailad: Investigation; Liu Yang: Writing – original draft; Nathapong Sukhawipat: Writing – original draft; Ghulam Abbas Ashraf: Writing – original draft; Phongpichit Channuie: Writing – original draft, Writing – review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Informed consent
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yuennan, J., Muensit, N., Tohluebaji, N. et al. Tailoring dielectric properties and crystallinity in poly(vinylidene fluoride-co-hexafluoropropylene) nanocomposites via iron (III) chloride hexahydrate incorporation. Sci Rep 15, 17810 (2025). https://doi.org/10.1038/s41598-025-02895-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-02895-y
Keywords
This article is cited by
-
Investigations on novel bio-compatible PVDF-Sc composites for Low-frequency energy harvesting using electrospinning
Journal of Materials Science: Materials in Electronics (2025)
-
Improved piezoelectric and conductive properties of bent-core liquid crystal-polymer composites for sustainable energy harvesting
Advanced Composites and Hybrid Materials (2025)
-
Silane-modified waste amethyst as a functional filler in PVDF-HFP composites for flexible dielectric and energy applications
Journal of Materials Science (2025)
