
Abstract
Glioblastoma is the most common and aggressive tumor of the central nervous system. Locoregional administration of therapeutic radiopharmaceuticals appears to be a promising modality for recurrent glioblastomas. In this study, fibroblast activation protein alpha (FAPα) targeting molecule fibroblast activation protein inhibitor-04 (FAPI-04) was conjugated to polydopamine (PDA) nanoparticles, and then, α-emitter astatine-211 was labeled onto the nanocomposite to form [211At]At-PDA-FAPI. In vitro, [211At]At-PDA-FAPI was able to significantly reduce the cell viability, induce DSB formation, arrest cell cycle at G2/M phase and promote cell apoptosis. Furthermore, [211At]At-PDA-FAPI exhibited effective tumor inhibition ability in U87MG xenografts. Mice received 0.56 MBq [211At]At-PDA-FAPI showed a reduced tumor volume of approximately 65% on the 9th day after injection, and the median survival in this group (48 days) was obviously improved compared with that in the saline group (18 days). Meanwhile, increased apoptosis was also observed in tumor sites after [211At]At-PDA-FAPI treatment. In addition, H&E analysis of major organs confirmed the biological safety of [211At]At-PDA-FAPI. This study provides an effective and promising strategy for locoregional treatment of glioblastoma.
Similar content being viewed by others

Introduction
Glioblastoma is the most common and aggressive malignant tumor of the central nervous system in adults, carrying a poor prognosis. Constant efforts on improving optimal treatment regimens have been made in the past decades, but a majority of glioblastomas recurred or progressed in a short term, with a median overall survival of only 9–15 months1,2 Due to its propensity for extensive infiltration into surrounding tissues, complete resection of tumor tissues, especially for critically located glioblastomas, is usually unfeasible. Moreover, the hypoxic and heterogeneous microenvironment within the tumor limit the efficacy of chemotherapy and external beam radiotherapy3 Although numerous therapeutic modalities have been explored, the sustained remission of glioblastoma remains an ongoing and formidable challenge.
Currently, targeted radionuclide therapy (TRT) through locoregional administration of radiopharmaceuticals (e.g. [90Y]Y/[177Lu]Lu-DOTAGA-SP, [213Bi]Bi/[225Ac]Ac-DOTA-SP or [211At]At-81C6) appears to offer a promising adjuvant strategy for the recurrent or residual glioblastoma4,5,6 Well-tolerated, stable disease or improved functional status were observed in most cases. In this context, characteristics of the selected target, vector and radionuclide are the most important considerations to maximize the therapeutic effect. Optimal targets for TRT should be highly and specifically expressed in the tumor sites, but rarely presented in normal tissues. Fibroblast activation protein (FAP), highly expressed by multiple cell types within glioblastoma but nearly absent in normal tissues, is considered as a promising theranostic target for glioblastoma7,8 In particular, several recently developed FAP-targeted radiotracers have exhibited excellent diagnostic performance in glioblastoma, further indicating the feasibility of FAP as an ideal target for TRT in glioblastoma9,10.
Given that critically located and chemo/external-radiotherapy resistant tumors are main indications for TRT in glioblastoma, α-emitters with high linear energy transfer (LET) and short tissue range may be more advantageous5 More specifically, the short-tissue-range (40–100 μm) of α-emitters results in a weak “cross-fire effect”, minimizing damage to surrounding tissues. The high LET of α-emitters can induce irreparable DNA double strand breaks (DSB), making them particularly effective in treating hypoxic and resistant tumors11,12,13,14 Among the available alpha radionuclides, astatine-211 (211At) appears to be the optimal option due to its decay characteristics: a moderate half-life (T1/2 = 7.21 h) and absence of a long decay chain, ensuring that the majority of radioactive decays occurs within the tumor site before migrating to other organs15,16,17 In our previous study, [211At]At-FAPI-04, a derivative of the FAP-targeted inhibitor, exhibited effective targeting of glioblastomas and significant cell killing ability in vivo and in vitro18 However, [211At]At-FAPI-04 cleared rapidly from the bloodstream and tumor site, limiting the therapeutic efficacy, thus it is urgent to improve its retention ability within tumor tissues. Recent research has indicated that attaching small molecule drugs onto the surface of nanocarriers can effectively prolong tumor retention and enhance drug utilization efficiency19,20,21 Among various nanocarriers, polydopamine (PDA) has been regarded as an ideal choice for drug delivery applications due to its extraordinary properties. With abundant aromatic rings and catechol/quinone groups on the surface, PDA nanoparticles are easily modified with a range of molecules by physical absorption (π-π stacking, hydrogen bond) or chemical bonding (Michael addition or Schiff-base reactions)22,23 Moreover, PDA nanoparticles exhibited excellent stability, favorable biocompatibility and safety profiles in several previous studies. As reported, PDA-based nanoparticles not only remained stable in water for several months without agglomeration, but also displayed excellent stability in the liver and spleen of mice for at least six weeks24,25 In addition, these nanoparticles showed no obvious cytotoxicity in various type of cells in vitro as well as no notable histological toxicity in the major organs of mice in vivo25,26,27 Hence, in this work, PDA nanoparticles were adopted as a nanocarrier for locoregional astatine-211 targeted-alpha therapy (TAT) of glioblastomas. FAPI-modified nanoparticles (PDA-FAPI) were synthesized and labeled with astatine-211 (Fig. 1), and then the cytotoxicity and therapeutic efficacy of [211At]At-PDA-FAPI were evaluated in U87MG cells and glioma xenografts, respectively. The purpose of this study was to explore a novel approach for 211At-based TRT of glioblastoma.

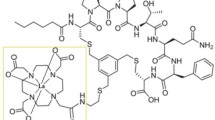
The synthetic diagram of [211At]At-PDA-FAPI.
Materials and methods
General
All commercially-available reagents and chemicals were sourced from Bidepharm (Shanghai, China), J&K Scientific (Beijing, China), Merida Technology (Beijing, China), Gibco (Grand Island, USA) or Beyotime Biotechnology (Shanghai, China) and were used without further purification unless specified otherwise. Astatine-211 was produced on a CS-30 cyclotron via 209Bi(α, 2n)211At reaction at Sichuan University as previously described28 PDA nanoparticles were synthesized based on the method of Liu et al., and Boc-removed FAPI-04 was prepared as previously described29,30 The ultraviolet-visible (UV-Vis) absorption spectra were recorded using a SHIMADZU UV-2450 spectrophotometer (Kyoto, Japan). The hydrodynamic diameter and zeta potential were characterized by Dynamic Light Scattering (DLS, Zetasizer Nano ZS90, Malvern, UK). The micromorphology of PDA-FAPI nanoparticles was visualized using scanning electron microscopy (SEM, ZEISS Gemini 300, Oberkochen, Germany) and transmission electron microscopy (TEM, JEOL JEM 2100 F, Tokyo, Japan). Fluorescence images were captured using a confocal laser microscope (Leica TCS SP5II, Wetzlar, Germany). Flow cytometry analyses were performed using a BD FACScalibur instrument (CA, USA). The radioactivity measurements were conducted using a NaI (Tl) scintillation detector (China National Nuclear Corporation, FH-603, Beijing, China).
Cells and animals
U87MG and MCF-7 cells obtained from ATCC were cultured in DMEM medium supplemented with 10% fetal bovine serum and maintained in a humidified atmosphere containing 5% CO2 at 37 ℃. Four-week-old BALB/c-nude mice were acquired from Chengdu Dossy Experimental Animals Co., Ltd. and inoculated with 5 × 106 U87MG cells in the trunk to establish tumor models. All animal studies were conducted in accordance with the institutional ethics committee regulations and guidelines on animal welfare and approved by the Animal Welfare and Ethics Committee of Sichuan University.
Preparation of [211At]At-PDA-FAPI and Cy5-PDA-FAPI
Boc-removed FAPI-04 (20 mg in 4 mL ethanol) and PDA (15 mg in 20 mL deionized water) were mixed, and then the pH of the mixture was adjusted to 9.0. After stirring for 24 h at room temperature, the mixture was centrifugated (10000 rpm, 10 min), and the dark solid was washed 3 times with water to obtain PDA-FAPI. Subsequently, 1 mg PDA-FAPI nanoparticles resuspended in 200 µL water was added to a mixture solution containing 5 µL acetic acid, 20 µg N-Iodosuccinimide and 56 MBq astatine-211. After reacting for 30 min, a sodium ascorbate solution was added to quench the reaction. Then the mixture was centrifugated (10000 rpm, 10 min) and washed 3 times with deionized water to yield [211At]At-PDA-FAPI nanoparticles. The radiochemical yield was calculated as radioactivity in precipitant / (radioactivity in precipitant and supernatant) × 100%. The radiochemical purity of [211At]At-PDA-FAPI was determined by iTLC using Whatman No. 1 paper as stationary phase and an acetone/water mixture (3:1, v/v) as the developing solvent. The retention factor (Rf) values for [211At]At-PDA-FAPI and astatine-211 were 0 and 0.9, respectively. And the radiochemical purity was calculated as: Radiochemical purity (%) = radioactivity at Rf = 0 / gross radioactivity on the whole chromatography tape. For the evaluation of in vitro stability, [211At]At-PDA-FAPI was incubated with saline at 37 ℃, and then aliquots of each sample were analyzed by iTLC at 3, 6, 12, and 24 h. In addition, Cy5 modified PDA-FAPI nanoparticles were prepared to visually evaluate the cellular binding behavior. Briefly, diSulfo-Cy5 amine (5 µL, 10 mg/mL in deionized water) was mixed with PDA-FAPI nanoparticles (1 mg in 200 µL water), followed by addition of 2 µL DIPEA. After stirring for 2 h at room temperature, the mixture was centrifugated (10000 rpm, 10 min), and the dark solid was washed 3 times with water to obtain Cy5-PDA-FAPI.
Cell uptake assay
U87MG and MCF-7 cells were seeded in cell-culture dishes at a density of 5 × 104 cells/dish and incubated overnight for adherence. Then the cells were incubated with Cy5-PDA-FAPI (10 µg) for 3 and 24 h at 37 ℃. In addition, blocking assay was also performed, with U87MG cells pretreated with 10 µg FAPI ligand 1 h before the addition of Cy5-PDA-FAPI nanoparticles. Next, the cells were washed twice with PBS and subsequently incubated with DiO solution for 20 min to stain the cell membrane. After another round of washing, the cells were fixed with 4% paraformaldehyde and stained with DAPI solution before observed using a confocal laser microscope. Besides, binding capacity of [211At]At-PDA-FAPI to U87MG and MCF-7 cells was also evaluated. In brief, U87MG and MCF-7 cells were seeded in 6-well plates at a density of 7 × 105 cells/well and incubated overnight for adherence. Then the cells were incubated with 5.0 kBq [211At]At-PDA-FAPI for 3 and 24 h at 37 ℃ respectively. For blocking assay, U87MG cells were pretreated with 10 µg FAPI ligand 1 h before [211At]At-PDA-FAPI was added, then the cells were continued to be incubated at 37 ℃ for 3 h. At determined time points, the radioactive medium was collected. Then the cells were washed twice with PBS and subsequently lysed with 1 mL NaOH lysis buffer. The radioactivity for each sample was measured by a γ-counter, and the cell uptake proportion was calculated as percentage of the applied dose.
Cell toxicity assay
The CCK-8 kit was applied to assess the effect of [211At]At-PDA-FAPI on viability of U87MG cells. Briefly, U87MG cells were cultured in 96-well plates at a density of 8 × 103 cells/well, and then treated with different doses of [211At]At-PDA-FAPI (23, 46, 92 and 185 kBq), [211At]NaAt (23, 46, 92 and 185 kBq) and PDA-FAPI (0.63, 1.25, 2.5 and 5.0 µg) for 24 h. After replaced with fresh medium, the cells were continued to be incubated for another 24 h. Then CCK-8 reagent was added to each well and incubated for 2 h. The absorbance value at 450 nm of each well was detected using a microplate reader.
Immunofluorescent staining for γ-H2AX
Immunofluorescent detection of γ-H2AX foci formation was performed to monitor the DSB degree induced by [211At]At-PDA-FAPI. Specifically, U87MG cells were seeded (5 × 104 cells/dish) in confocal dishes and allowed to adhere overnight. Thereafter, [211At]At-PDA-FAPI (185 and 370 kBq) and [211At]NaAt (185 and 370 kBq) were added to respective dishes and incubated for 3 h. Then the cells were washed with PBS and fixed for 15 min in 4% paraformaldehyde. After another round of washing, the cells were incubated in blocking buffer for 20 min, followed by incubation with the primary rabbit monoclonal antibody against γ-H2AX at 4°C overnight. Next, the cells were washed three times with PBS, and incubated with a Cy3-conjugated secondary antibody for 1 h. For localization, cell nuclei were stained with DAPI. The quantitative analysis of γ-H2AX foci formation in U87MG cells was counted by the Image-J software.
Cell cycle arrest assay
U87MG cells were seeded in 6-well plates at a density of 5 × 105 cells/well, then incubated with various doses (6.25, 12.5, 23.0 and 46.0 kBq) of [211At]At-PDA-FAPI for 24 h. Thereafter, the cells were collected, washed with PBS and fixed with 75% ethanol. Afterwards, the cells were washed with PBS again, followed by incubation with 500 µL PBS containing PI and RNase A for 30 min. Subsequently, the cell cycle distribution was detected using a flow cytometer.
Cell apoptosis assay
U87MG cells were seeded (5 × 105 cells/well) in 6-well plates and incubated overnight for adherence. Then the cells were treated with different doses (23, 46, 92 and 185 kBq) of [211At]At-PDA-FAPI or [211At]NaAt for 24 h. Thereafter, the cells were collected, washed and suspended in 500 µL PBS, followed by stained with Annexin V-FITC and PI for 15 min. Next, the cell suspension was analyzed by flow cytometry.
Biodistribution studies
[211At]At-PDA-FAPI (0.3 MBq, 7.5 µg) was administrated to U87MG xenografts (n = 4) via intratumoral injection when the tumor volumes reached ~ 500 mm3. Then the mice were euthanatized by isoflurane at 3 and 15 h post-injection, and the main organs including heart, liver, spleen, lung, kidney, etc. were harvested and weighed (thyroid was harvested along with the surrounding tissue). Subsequently, the radioactivity in each sample was quantified using a γ-counter. The distribution of [211At]At-PDA-FAPI in each interested organ was presented as a percentage of the injected dose per gram of tissue (% ID/g) with all radioactivity measurements corrected for decay. In addition, Cy5-PDA-FAPI (7.5 µg) was also intratumorally injected to U87MG xenografts to observe the whole-body distribution of this nano-carrier by fluorescence imaging.
Therapeutic effect in vivo
After 10 days of cell inoculation (tumor volume reached ~ 150 mm3, U87MG xenografts were randomly divided into three groups and intratumorally injected with saline (n = 6), 0.56 MBq (14 µg) [211At]At-PDA-FAPI (n = 6) or [211At]NaAt (n = 4), respectively. Tumor volume calculated by (Length × Width2)/2 and body weight were measured every three days in a blinded manner until reaching the endpoint. Tumor volume larger than 1500 mm3, mortality, ulcerating tumor tissue, or more than 20% weight loss was considered as the endpoint.
Hematoxylin and eosin (H&E) and terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling (TUNEL) staining were also performed on tumor sections to evaluate the in vivo anti-tumor effect of [211At]At-PDA-FAPI. One mouse randomly selected from the saline group and one from the [211At]At-PDA-FAPI group were euthanatized by isoflurane at 24 h post treatment respectively. The tumor tissues were collected, fixed in formalin, embedded by paraffin and cut into sections. Then these sections were stained with H&E and TUNEL respectively. In addition, histopathology examination stained with H&E in the major organs was performed to evaluate the potential toxicity of [211At]At-PDA-FAPI in vivo at 15 days after injection.
Results and discussion
Preparation and characterization of [211At]At-PDA-FAPI
PDA nanoparticles were firstly synthesized by self-polymerized under alkaline conditions for the preparation of [211At]At-PDA-FAPI. As reported, there are plenty of chemically-active groups including benzoquinone group on the surface of PDA nanoparticles, which are available for Michael addition and Schiff reaction31 Accordingly, Boc-removed FAPI-04 was used to modify the surface of PDA to generate PDA-FAPI. As shown in the UV spectra (Fig. 2A), an obvious characteristic absorption peak of FAPI at 235 nm was observed in PDA-FAPI relative to PDA, indicating the successful functionalization. According to the weight changes before and after FAPI modification, the loading ratio of FAPI onto PDA nanoparticles was calculated to be 0.38 mmol g− 1. Based on the DLS analysis, the hydrodynamic diameter and Zeta potential of PDA-FAPI were 308 (PDI = 0.29) nm and − 25.7 mV, respectively (Fig. 2B). SEM and TEM (Fig. 2C and D) images visually indicated that PDA-FAPI nanoparticles have uniform spherical shape with a geometric diameter of 100 ~ 200 nm. Afterwards, astatine-211 was directly labeled onto PDA-FAPI with a high radiochemical yield about 70% after centrifugation. However, the stability of [211At]At-PDA-FAPI was limited in saline. As shown in Fig. S1, the radiochemical purity of [211At]At-PDA-FAPI treated with saline was 77.0% ± 2.16% and 58.9 ± 1.31% at 3 and 24 h, respectively. It appeared that [211At]At-PDA-FAPI was much stabler in serum and its radiochemical purity was still over 90% at 24 h. A possible reason for this was that serum may include some reducing substances (such as glutathione and cysteine) capable of protecting C-At bond from oxidative cleavage. It was reported that ascorbic acid (a reducing agent) could improve the stability of 211At-labeled compounds32 Thus, although [211At]At-PDA-FAPI exhibited a barely satisfactory stability in saline, adding a reducing agent could be attempted to enhance its stability in future studies.

(A) UV spectrum of PDA and PDA-FAPI nanoparticles. (B) DLS data of PDA-FAPI. (C) SEM and (D) TEM images of PDA-FAPI. (E) Confocal images of FAP-negative MCF-7 and FAP-positive U87MG cells incubated with Cy5-PDA-FAPI for 3 and 24 h. Cy5-PDA-FAPI indicated as red, nuclei stained with DAPI (blue) and cytomembrane stained with DiO (green).
Cellular uptake assay
The cellular uptake assay was conducted using U87MG (FAPα-positive) and MCF-7 (FAPα-negative) cells, with their different FAPα expression profiles demonstrated in our previous studies18,30 For visualization, Cy5 modified PDA-FAPI nanoparticles were used to evaluate the cellular binding behavior. Meanwhile, the cell membrane was stained green with DiO, the nuclei were stained blue with DAPI. As illustrated in Fig. 2E, red fluorescence signals were observed in U87MG cells after incubation with Cy5-PDA-FAPI for 3 h, suggesting efficient uptake of the nanoparticles. In addition, the red signals in U87MG cells increased after incubation for 24 h, and most of them were inside the cytoplasm, indicating a strong cellular internalization and prolonged intracellular retention of the nanoparticles. Meanwhile, no obvious red fluorescence signal was detected in MCF-7 cells at both timepoints, confirming the specific affinity of these nanoparticles towards U87MG cells. In accordance with fluorescence imaging, [211At]At-PDA-FAPI also showed significant binding ability to U87MG cells (Fig. S2). A binding rate of 38.1 ± 0.94% was detected after 3 h of incubation, which increased to 66.1 ± 0.10% after 24 h of incubation. In contrast, MCF-7 cells exhibited very low uptake of [211At]At-PDA-FAPI at both time points. In addition, in U87MG cells pretreated with excess FAPI ligand, the accumulation of Cy5-PDA-FAPI and [211At]At-PDA-FAPI was significantly inhibited after 3 h of incubation (Fig. S2). Rapid and specific cell binding, as well as prolonged intracellular retention ability are critical characteristics of ideal target vector, which are beneficial for realizing effective TRT.

(A) Cell viability of U87MG after incubated with different doses of [211At]NaAt and [211At]At-PDA-FAPI. (B) Quantitative analysis of γ-H2AX foci formation in U87MG cells treated with different doses of [211At]NaAt and [211At]At-PDA-FAPI. (C) Representative immunofluorescent microscopy images of γ-H2AX foci in U87MG cells untreated or treated with different doses of [211At]At-PDA-FAPI for 3 h. γ-H2AX foci were shown as red, nuclei were stained with DAPI (blue).
Cytotoxicity of [211At]At-PDA-FAPI on U87MG cells
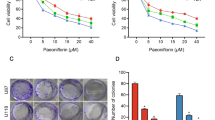
As expected, a dose-dependent decrease of cell viability was observed in both [211At]At-PDA-FAPI and [211At]NaAt treated groups (Fig. 3A). After incubated with 23, 46, 92, and 185 kBq [211At]At-PDA-FAPI, the viability of U87MG cells decreased to 47.5 ± 3.65%, 31.7 ± 1.24%, 17.2 ± 3.64%, and 9.60 ± 0.60%, respectively. In [211At]NaAt treated group, the decrease of viability was less obvious compared to the same dose as [211At]At-PDA-FAPI, with a cell viability of 73.7 ± 5.17%, 61.2 ± 4.32%, 53.8 ± 3.18%, and 52.2 ± 6.11% at 23, 46, 92, and 185 kBq, respectively. In contrast, the viability of cells treated with PDA-FAPI was near to 100% even at the maximum determined concentration (Fig. S3), suggesting the safety of PDA-FAPI to cells. In [211At]NaAt treated group, the decrease of cell viability was mainly due to the nonspecific external radiation of astatine-211. Contrarily, stronger cytotoxicity induced by [211At]At-PDA-FAPI may be attributed to its specific cell targeting ability and favorable intracellular retention capacity.
DSB formation in U87MG cells induced by [211At]At-PDA-FAPI
It is widely known that the most severe DNA damage induced by α irradiation, DSB, constituted the primary cause of cell death during TAT33,34 Immediately followed by DSB formation, large numbers of H2AX molecules in the chromatin around the break sites will be phosphorylated as an early response triggered by DSB35 Therefore, detection of phosphorylated H2AX (γ-H2AX) formation is extensively used to evaluate the extent of DSB36,37 In this study, we visualized the γ-H2AX foci formation by immunofluorescence assay using a confocal microscopy. Noticeable γ-H2AX foci formation was observed in [211At]At-PDA-FAPI treated cells (Fig. 3B and C), and the foci number in each cell increased with exposed dose. After incubation with 185 and 370 kBq [211At]At-PDA-FAPI for 3 h, 113 ± 20 and 158 ± 41 γ-H2AX foci per cell were counted, respectively. Increased γ-H2AX foci formation was also observed in cells exposed to [211At]NaAt (Fig. 3B and Fig. S4), but the foci number was lower than that in [211At]At-PDA-FAPI group, counted as 73 ± 34 and 146 ± 27 foci per cell for 185 and 370 kBq, respectively. Almost all cells in [211At]At-PDA-FAPI or [211At]NaAt group exhibited significant formation of γ-H2AX foci, while only a few cells produced γ-H2AX foci in untreated group. These results reveal that [211At]At-PDA-FAPI could induce serious DSB, which might trigger the DNA damage response, including cell cycle arrest and cell apoptosis.
Cell cycle arrest in U87MG cells triggered by [211At]At-PDA-FAPI
After DNA damage occurs, cell cycle checkpoint is activated to repair the damaged DNA38 Thus, cell cycle distribution assay was carried out to evaluate the corresponding response of U87MG to DSB. As shown in Fig. 4A, the cycle distribution of U87MG was obviously changed even after incubation with a low dose (6.25 kBq) of [211At]At-PDA-FAPI. The percentage of G2/M phase increased from 16.4 ± 0.29% to 26.3 ± 3.39% while the proportion of S phase decreased from 15.0 ± 1.02% to 4.32 ± 0.48% (Fig. 4B). The same trend was also observed in the higher dose groups, and the percentage of G2/M phase increased with dose. After incubation with 46 kBq [211At]At-PDA-FAPI, the proportion of G2/M phase increased to 36.4 ± 0.35%, indicating that [211At]At-PDA-FAPI can significantly arrest cell cycle at G2/M phase. The decrease in the proportion of S phase suggested the stopped DNA replication induced by [211At]At-PDA-FAPI. Meanwhile, the increase percentage of G2/M phase reflected the examination and repair of erroneous DNA. Overall, [211At]At-PDA-FAPI can effectively affect the DNA replication ability of U87MG cells, induce significant cell cycle arrest, which might trigger the apoptosis process if the damaged DNA cannot be effectively repaired.
Apoptosis of U87MG cells after treated with [211At]At-PDA-FAPI
[211At]At-PDA-FAPI exhibited a favorable capacity to induce cell apoptosis in a dose-dependent manner (Fig. 4C). In addition, the percentage of apoptotic cells in [211At]At-PDA-FAPI group was higher than that in [211At]NaAt group treated with the same dose (Fig. S5). After incubation with 23 and 185 kBq of [211At]At-PDA-FAPI for 24 h, the apoptosis proportion (Q2 + Q3) was 15.3 ± 1.42% and 32.7 ± 2.49%, respectively (Fig. 4D). In contrast, the apoptosis proportion was 10.9 ± 2.94% and 25.8 ± 1.64% in [211At]NaAt treated group, respectively. Compared with [211At]NaAt, [211At]At-PDA-FAPI demonstrated higher proportion of apoptosis, mainly attributed to more irreparable DNA damage that can activate cell death pathways. Additionally, higher necrosis percentage (Q1) was observed in both treated group when compared with saline group. It has been reported that effective radiation may induce serious DNA damage, subsequently resulting in cell cycle arrest, apoptosis and necrosis38 Consistently, we also observed significant DSB, G2/M phase arrest, apoptosis and reduced viability in U87MG cells after incubation with [211At]At-PDA-FAPI, suggesting excellent anti-tumor ability of [211At]At-PDA-FAPI in vitro.

(A) Representative cell cycle distribution of U87MG after incubation with different doses of [211At]At-PDA-FAPI for 24 h. (B) Quantitative analysis of cycle distribution after incubation with different doses of [211At]At-PDA-FAPI. (C) Representative flow cytometric analysis of apoptosis in U87MG cells treated with different doses of [211At]At-PDA-FAPI. (D) Graphic representation of flow cytometry data showing the percentage of U87MG cells undergoing apoptosis after treated with different doses of [211At]At-PDA-FAPI for 24 h.
Biodistribution of [211At]At-PDA-FAPI in U87MG xenografts
There is no doubt that the therapeutic effect of radiopharmaceuticals depends on the radioactivity accumulated in tumor sites. According to the in vitro cell assay described above, [211At]At-PDA-FAPI could induce severe DNA damage within U87MG cells after only 3 h of incubation. Thus, we evaluated the biodistribution of [211At]At-PDA-FAPI in U87MG xenografts 3 h after intratumoral injection to determine the retention of [211At]At-PDA-FAPI in tumor tissues and its metabolic distribution in other organs. As illustrated in Fig. 5A, tumor sites showed the highest retention of [211At]At-PDA-FAPI ( 60.9 ± 26.5% ID/g), which was significantly higher than that of [211At]At-FAPI-04 at 2 h after intratumoral injection (19.24 ± 3.84% ID/g) reported in our previous study18 The improved tumor retention may be attributed to the introduction of PDA nanocarriers. Undoubtedly, the cellular uptake and biological behavior of nanoparticles are necessarily different from those of small molecules due to comparatively large size39 Targeting ligands modified nanoparticles could be internalized into cells through receptor-mediated endocytosis, and usually remained trapped in endosomes, exhibiting a long-term retention in cells39 Higher radioactivity accumulation indicated the improvement in tumor retention, which is beneficial to enhancing the utilization efficiency of astatine-211 and delivering more radiation dose to tumor sites. At 3 h post-injection, favorable tumor-to-organ ratios (insert of Fig. 5A) were displayed in U87MG xenografts, including tumor-to-blood ratio of 23.8 ± 11.4, tumor-to-liver ratio of 19.4 ± 13.7, tumor-to-kidney ratio of 9.58 ± 4.13 and tumor-to-muscle ratio of 42.3 ± 21.2, respectively. Consistent with the biodistribution data of [211At]At-PDA-FAPI, the strongest fluorescent signal was also observed in the tumor sites in U87MG xenografts injected with Cy5-PDA-FAPI by fluorescence imaging at 3 h post-injection (Fig. 5B). However, the lung, spleen and stomach exhibited mildly high radioactivity at 3 h post-injection, and an obvious decrease of activity was observed in all organs of interest except for the thyroid and stomach at 15 h post-injection (Fig. S6), suggesting the deastatination in vivo. Hence, therapeutic effect as well as the biosafety of [211At]At-PDA-FAPI were both evaluated in further study.
In vivo anti-tumor effects of [211At]At-PDA-FAPI
In the case of glioma, locoregional administration of radiopharmaceuticals has been proved to be a feasible treatment modality in previous studies5,6 Compared with intravenous injection, localized intracranial delivery enables drugs to act directly at the site of tumor resection, which can help to achieve high therapeutic drug concentration at the residual tumor sites while minimizing toxicity and side effects to normal tissues simultaneously40 Meanwhile, the intratumoral administration of nanoparticles was reported to facilitate improved therapeutic outcomes compared to systemic delivery by promoting their tumor-specific accumulation and retention41 Based on the significant cytotoxicity in vitro and improved tumor retention in vivo, the anti-tumor effect of [211At]At-PDA-FAPI in U87MG xenografts was subsequently evaluated. As depicted in Fig. 5C, mice in saline group exhibited rapidly increased tumor volume with a poor median survival of 18 days (Fig. 5D). In contrast, mice received 0.56 MBq [211At]At-PDA-FAPI treatment showed significantly inhibited tumor growth compared with the saline group. The tumor volume in [211At]At-PDA-FAPI group reduced approximately 65% within 9 days after treatment, which was not observed in our previous study18 These findings can certainly demonstrate the excellent therapeutic efficacy of [211At]At-PDA-FAPI in U87MG xenografts. Despite tumor regrowth being observed at 12 days post treatment, a significantly prolonged median survival of 48 days was still observed in [211At]At-PDA-FAPI treated mice. Although inhibited tumor growth and improved median survival were also observed in [211At]NaAt treated mice, the tumor suppression effect of [211At]NaAt was markedly lower than that of the same dose of [211At]At-PDA-FAPI. Owing to the excellent nuclide properties of astatine-211 and high tumor retention provided by PDA-FAPI, [211At]At-PDA-FAPI exhibited superior anti-tumor efficacy in vivo.

Biodistribution and therapeutic efficacy of [211At]At-PDA-FAPI in U87MG xenografts. (A) Biodistribution and tumor-to-organ ratios (insert) of [211At]At-PDA-FAPI in U87MG xenografts at 3 h post-injection (n = 4). T/B: tumor-to-blood, T/L: tumor-to-liver, T/K: tumor-to-kidney and T/M: tumor-to-muscle. (B) Fluorescence imaging of Cy5-PDA-FAPI in U87MG xenografts at 3 h post-injection. (C) Tumor volume changes in three groups (saline, [211At]NaAt and [211At]At-PDA-FAPI) during the therapeutic period. (D) Kaplan-Meier survival plot of the three groups (saline, [211At]NaAt and [211At]At-PDA-FAPI) during the whole treatment period.
In addition, both H&E and TUNEL staining of tumor sections demonstrated abundant cell necrosis and apoptosis induced by [211At]At-PDA-FAPI at 24 h post-treatment (Fig. 6A and B). Specifically, significantly increased proportion of TUNEL positive cells was detected in [211At]At-PDA-FAPI treated group relative to saline group. Taken together, above results further confirmed the excellent anti-tumor efficacy of [211At]At-PDA-FAPI. Nevertheless, the tumor regrowth suggested multiple doses may be required for curing the tumor.
Regarding TAT, the biological safety was of comparable importance to its therapeutic efficacy. In cellular uptake assay, [211At]At-PDA-FAPI displayed rapid and specific FAPα targeting ability to U87MG cells compared with MCF-7 cells, suggesting its favorable binding characteristics. Intratumoral administration mimicked the approach used in cell experiments, enabling the drug to directly contact and be taken up by tumor cells. Benefiting from the short range of astatine-211, the radiation damage caused by the “cross-fire effect” to surrounding tissue was considered negligible[5]. However, deastatination was a common challenge with 211At-labeled compounds, which was also observed in our study. It was reported that astatine-211 could induce colloidal depletion and necrosis of follicular epithelial cells at high dose, while safe at dose below 1 MBq42 In accordance with the study of Tadashi et al., no noticeable abnormalities or damage were observed in the thyroid of mice treated with 0.56 MBq [211At]At-PDA-FAPI (Fig. 6C). According to the biodistribution data, histological analysis of the heart, liver, spleen, lung, kidney, stomach and intestine in [211At]At-PDA-FAPI group was also performed by H&E staining at 15 days post injection. As illustrated in Fig. 6C, no significant differences in histological appearance were observed between the [211At]At-PDA-FAPI and saline groups, with no evident morphological alterations or tissue damage detected in either group. Meanwhile, there was unnoticeable changes in body weight between the groups over the experimental period (Fig. S7), revealing the ideal biosafety of [211At]At-PDA-FAPI for glioblastoma treatment via local administration.

Representative images of H&E (A) and TUNEL (B) staining of tumor sections at 24 h post-treatment in saline and [211At]At-PDA-FAPI group (For immunofluorescence analysis of TUNEL, nuclei were stained with DAPI, TUNEL were shown as red). (C) H&E staining of the major organs at 15 days post-treatment in saline and [211At]At-PDA-FAPI group.
Conclusion
In summary, PDA-FAPI nanoparticles with uniform size and specific FAPα targeting ability were synthesized and labeled with astatine-211. In vitro cell assay confirmed the excellent ability of [211At]At-PDA-FAPI in inducing DSB, arresting cell cycle, promoting apoptosis and reducing cell viability. Therapeutic study in U87MG xenografts further demonstrated the excellent anti-tumor effect and biosafety of [211At]At-PDA-FAPI. These favorable properties indicated the potential application of this 211At labeled compound as an adjuvant strategy in the treatment of glioblastoma. Our future work will focus on optimizing the dosing frequency and exploring combination with other therapies to eradicate glioblastoma.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Obrador, E. et al. Glioblastoma therapy: past, present and future. Int. J. Mol. Sci. 25 (5), 2529 (2024).
Omuro, A. & DeAngelis, L. M. Glioblastoma and other malignant gliomas: a clinical review. Jama 310 (17), 1842–1850 (2013).
Genoud, V., Kinnersley, B., Brown, N. F., Ottaviani, D. & Mulholland, P. Therapeutic targeting of glioblastoma and the interactions with its microenvironment. Cancers (Basel). 15 (24), 5790 (2023).
Bolcaen, J. et al. A perspective on the radiopharmaceutical requirements for imaging and therapy of glioblastoma. Theranostics 11 (16), 7911–7947 (2021).
Kunikowska, J., Morgenstern, A., Pełka, K., Bruchertseifer, F. & Królicki, L. Targeted alpha therapy for glioblastoma. Front. Med. (Lausanne). 9, 1085245 (2022).
Cordier, D., Krolicki, L., Morgenstern, A. & Merlo, A. Targeted radiolabeled compounds in glioma therapy. Semin Nucl. Med. 46 (3), 243–249 (2016).
Ebert, L. M. et al. Endothelial, pericyte and tumor cell expression in glioblastoma identifies fibroblast activation protein (FAP) as an excellent target for immunotherapy. Clin. Transl Immunol. 9(10), e1191 (2020).
Shi, Y. et al. Microenvironment modulation and clinical potentiality of FAP in glioblastoma: lessons learned from other solid tumors. Cells 10 (5), 1142 (2021).
Chen, H. et al. Comparison of [68Ga]Ga-DOTA-FAPI-04 and [18F] FDG PET/CT for the diagnosis of primary and metastatic lesions in patients with various types of cancer. Eur. J. Nucl. Med. Mol. Imaging. 47 (8), 1820–1832 (2020).
Röhrich, M. et al. FAP-specific PET signaling shows a moderately positive correlation with relative CBV and no correlation with ADC in 13 IDH wildtype glioblastomas. Eur. J. Radiol. 127, 109021 (2020).
Kim, Y. S. & Brechbiel, M. W. An overview of targeted alpha therapy. Tumour Biol. 33 (3), 573–590 (2012).
Gudkov, S. V., Shilyagina, N. Y., Vodeneev, V. A. & Zvyagin, A. V. Targeted radionuclide therapy of human tumors. Int. J. Mol. Sci. 17 (1), 33 (2015).
Makvandi, M. et al. Alpha-Emitters and targeted alpha therapy in oncology: from basic science to clinical investigations. Target. Oncol. 13 (2), 189–203 (2018).
Parker, C. et al. Targeted alpha therapy, an emerging class of Cancer agents: A review. JAMA Oncol. 4 (12), 1765–1772 (2018).
Albertsson, P. et al. Astatine-211 based radionuclide therapy: current clinical trial landscape. Front. Med. (Lausanne). 9, 1076210 (2022).
Guérard, F., Gestin, J. F. & Brechbiel, M. W. Production of [211At]-astatinated radiopharmaceuticals and applications in targeted α-particle therapy. Cancer Biother Radiopharm. 28 (1), 1–20 (2013).
Li, F. Z., Yang, Y. Y., Liao, J. L. & Liu, N. Recent progress of astatine-211 in endoradiotherapy: great advances from fundamental properties to targeted radiopharmaceuticals. Chin. Chem. Lett. 33 (7), 3325–3338 (2022).
Ma, H. et al. In vitro and in vivo evaluation of 211At-labeled fibroblast activation protein inhibitor for glioma treatment. Bioorg. Med. Chem. 55, 116600 (2022).
Shipunova, V. O. et al. Dual regioselective targeting the same receptor in Nanoparticle-Mediated combination Immuno/Chemotherapy for enhanced Image-Guided Cancer treatment. ACS Nano. 14 (10), 12781–12795 (2020).
Wiraja, C. et al. Framework nucleic acids as programmable carrier for transdermal drug delivery. Nat. Commun. 10 (1), 1147 (2019).
Braunová, A. et al. Tumor-targeted micelle-forming block copolymers for overcoming of multidrug resistance. J. Control Release. 245, 41–51 (2017).
Huang, Z. G. et al. RGD-modified pegylated Paclitaxel nanocrystals with enhanced stability and tumor-targeting capability. Int. J. Pharm. 10 (556), 217–225 (2019).
Lin, X. et al. Folic acid-modified Prussian Blue/polydopamine nanoparticles as an MRI agent for use in targeted chemo/photothermal therapy. Biomater. Sci. 7 (7), 2996–3006 (2019).
Wu, H. et al. PDA-Based drug delivery nanosystems: A potential approach for glioma treatment. Int. J. Nanomed. 17, 3751–3775 (2022).
Liu, X. et al. Mussel-inspired polydopamine: a biocompatible and ultrastable coating for nanoparticles in vivo. ACS Nano. 7 (10), 9384–9395 (2013).
Ho, C. C. & Ding, S. J. The pH-controlled nanoparticles size of polydopamine for anti-cancer drug delivery. J. Mater. Sci. Mater. Med. 24 (10), 2381–2390 (2013).
Zhang, Y. et al. Engineering Metal-Organic frameworks for photoacoustic Imaging-Guided Chemo-/Photothermal combinational tumor therapy. ACS Appl. Mater. Interfaces. 10 (48), 41035–41045 (2018).
Qin, Y. L. et al. Understanding the extraction behavior and mechanism of Th(IV) and U(VI) by the irradiated tri-iso-amyl phosphate with At-211 as an α-source. Sep. Purif. Technol. 336, 126242 (2024).
Liu, Y. et al. Dopamine-melanin colloidal nanospheres: an efficient near-infrared photothermal therapeutic agent for in vivo cancer therapy. Adv. Mater. 25 (9), 1353–1359 (2013).
Ma, H. et al. Synthesis and preliminary evaluation of 131I-Labeled FAPI tracers for Cancer theranostics. Mol. Pharm. 18 (11), 4179–4187 (2021).
Cheng, W. et al. Versatile polydopamine platforms: synthesis and promising applications for surface modification and advanced nanomedicine. ACS Nano. 13 (8), 8537–8565 (2019).
Watabe, T. et al. Enhancement of At-211 uptake via the sodium iodide symporter by the addition of ascorbic acid in targeted alpha-Therapy of thyroid Cancer. J. Nucl. Med. 60 (9), 1301–1307 (2019).
Morgan, K. A. et al. Tumor targeted alpha particle therapy with an actinium-225 labelled antibody for carbonic anhydrase IX. Chem. Sci. 15 (9), 3372–3381 (2024).
Claesson, A. K., Stenerlöw, B., Jacobsson, L. & Elmroth, K. Relative biological effectiveness of the alpha-particle emitter 211At for double-strand break induction in human fibroblasts. Radiat. Res. 167 (3), 312–318 (2007).
Natale, F. et al. Identification of the elementary structural units of the DNA damage response. Nat. Commun. 8, 15760 (2017).
Chu, P. M. et al. Enhancement of radiosensitivity in human glioblastoma cells by the DNA N-mustard alkylating agent BO-1051 through augmented and sustained DNA damage response. Radiat. Oncol. 6, 7 (2011).
Jia, T. et al. Anti-Cancer and Radio-Sensitizing properties of new bimetallic (N-Heterocyclic Carbene)-Amine-Pt(II) complexes. J. Med. Chem. 66 (10), 6836–6848 (2023).
Zhang, D. et al. The interplay between DNA repair and autophagy in cancer therapy. Cancer Biol. Ther. 16 (7), 1005–1013 (2015).
Mosquera, J., García, I. & Liz-Marzán, L. M. Cellular uptake of nanoparticles versus small molecules: A matter of size. Acc. Chem. Res. 51 (9), 2305–2313 (2018).
Alomari, S. et al. Drug repurposing for glioblastoma and current advances in drug Delivery-A comprehensive review of the literature. Biomolecules 11 (12), 1870 (2021).
Yun, W. S. et al. Recent studies and progress in the intratumoral administration of Nano-Sized drug delivery systems. Nanomaterials (Basel). 13 (15), 2225 (2023).
Liu, Y. W. et al. Preclinical evaluation of Radiation-Induced toxicity in targeted alpha therapy using At-211 NaAt in mice: A revisit. Transl Oncol. 13 (4), 100757 (2020).
Acknowledgements
The authors would like to thank Mr. Xiaodong Liao for his assistance in the production of astatine-211.
Funding
This study was financially supported by the National Natural Science Foundation of China (12475349), the Youth Talent Fund of Sichuan Academy of Medical Sciences and Sichuan Provincial People’s Hospital (2022QN32) and the Fundamental Research Funds for the Central Universities.
Author information
Authors and Affiliations
Contributions
H. M. and T. Y. performed the experiments and prepared the main manuscript, G. Q. and Y. Q. were ressponsible for separating radionuclide, J. L., Y. Y. and W. Z. supervised the experiments and verified the data analysis, N. L. and F. L. designed the research and edited the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Ethical approval
All animal studies were conducted in accordance with ARRIVE guidelines and approved by the Animal Welfare and Ethics Committee of Sichuan University. This article does not contain any studies with human participants performed by any of the authors.
Supporting information
Cell viability of U87MG after incubated with different doses of PDA-FAPI, Representative immunofluorescent microscopy images of γ-H2AX foci in U87MG cells untreated or treated with different doses of astatine-211 for 3 h, Representative flow cytometric analysis of apoptosis in U87MG cells treated with different doses of astatine-211 and body weight changes in three groups (saline, astatine-211 and [211At]At-PDA-FAPI) during the therapeutic period.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, H., Ye, T., Qu, G. et al. Locoregional radionuclide therapy of glioblastoma with [211At]At-PDA-FAPI. Sci Rep 15, 18248 (2025). https://doi.org/10.1038/s41598-025-03356-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-03356-2
Keywords
This article is cited by
-
Advances in tumor-targeted ²¹¹At radiopharmaceuticals: preclinical investigations, challenges, and future perspectives
Clinical and Translational Imaging (2025)
