
Abstract
Mitochondria play a crucial role in cellular respiration and immune responses. Mitochondrial DNA (mtDNA) haplogroups and variants have been associated with various diseases, including COVID-19. This study analyzed complete mtDNA sequences from 467 Brazilian patients with COVID-19 to investigate associations between mtDNA ancestry and mortality risk. Using classical statistical methods and a machine learning model, we identified key contributors to outcomes, with age as the primary risk factor, followed by male sex. Several mtDNA variants—663G, 1736G, 2706G, 3010A, 4248C, 4824G, 8027A, 8794T, and 10873C—were significantly associated with increased mortality risk. Most are characteristic of haplogroup A2, prevalent in populations with Native American ancestry. Notably, the 8027A allele, a non-synonymous substitution (Alanine > Threonine at position 148 of Cytochrome C Oxidase II), was predicted to be potentially damaging and emerged as the most significant marker. Rather than being disease-causing, these variants may amplify risk through interactions with other genetic, environmental, and clinical factors. Our findings emphasize that mtDNA variants and haplogroups are not phenotypically neutral and could serve as biomarkers of COVID-19 severity. Genetic studies prioritizing Indigenous populations and their descendants, who may be particularly susceptible to certain viruses, are urgently needed, especially given the predominant focus on European populations.
Similar content being viewed by others


Introduction
Coronavirus disease 2019 (COVID-19) is primarily a respiratory illness caused by the SARS-CoV-2 virus. Most cases are mild or moderate, involving symptoms such as fever, cough, fatigue, and loss of taste or smell, with recovery often requiring no specialized treatment. However, severe cases, especially among older adults or individuals with comorbidities (e.g., diabetes, chronic respiratory or cardiovascular disease, or cancer), can lead to life-threatening symptoms, including respiratory failure, confusion, and chest pain1. Although older adults are more vulnerable, the occurrence of severe complications or death from COVID-19 in individuals of all ages1 points to the influence of other contributing risk factors.
SARS-CoV-2's success as a human intracellular pathogen results from a combination of viral properties and host-related factors. For instance, a lack of common genetic variants in key angiotensin-converting enzyme 2 (ACE2) receptor binding sites enables efficient interaction with human cells, promoting transmission2. High viral affinity for host cells, asymptomatic spread, high transmissibility, and relatively low lethality contribute to the evolution of successful viral strains3. As of December 2024, COVID-19 has infected over 777 million people globally, with over 7 million deaths. Brazil ranks second in cumulative deaths after the United States4. Although vaccination campaigns have reduced case numbers, new strains, mainly Omicron-derived lineages, continue to emerge5. Thus, studying genetic variability and environmental factors affecting disease outcomes remains crucial.
Research into genetic susceptibility to COVID-19 has highlighted how host–pathogen interactions evolve and how specific genetic variants can influence disease outcomes. Early studies focused on genes involved in viral entry. More recently, genome-wide association studies and targeted analyses across diverse populations have facilitated the identification of multiple loci influencing both susceptibility to COVID-19 and disease severity5, including polymorphisms in TLR7, TYK2, OAS1, and VIP associated with differential severity6, and survival-associated variants such as rs117011822 and rs72085247. A large-scale meta-analysis further identified 49 loci linked to critical illness and revealed therapeutic targets in immune signaling, immunometabolism, and viral replication pathways8. These findings underscore the importance of integrating both population-level and personalized genomic approaches to comprehensively understand and manage the clinical heterogeneity of COVID-19.
While most genetic studies have focused on nuclear variants, growing evidence suggests that other cellular components, particularly mitochondria, also play a crucial role in modulating COVID-19 outcomes. Mitochondria are among the cellular components most affected by SARS-CoV-2 infection9. Minor alterations in the function of these organelles may contribute to explaining the variability of complex diseases among different populations10. Beyond their well-established role in energy production, mitochondria are key regulators of both innate11,12 and adaptive immune responses12, which are critical in the context of viral infections.
Mitochondrial antiviral signaling proteins (MAVS) are vital for initiating the immune response by promoting the secretion of type I interferons and pro-inflammatory cytokines that help clear viral infections13. Numerous SARS-CoV proteins interact with host mitochondria to disrupt innate immune responses by modulating MAVS activity14. Similarly, SARS-CoV-2 may interfere with this signaling pathway, leading to impaired immune responses. This virus induces mitochondrial dysfunction characterized by changes in mitochondrial morphology, inhibition of oxidative phosphorylation (OXPHOS), increased production of mitochondrial reactive oxygen species (mROS), and activation of mitochondria-mediated apoptosis, exacerbating systemic inflammation and contributing to severe symptoms such as cytokine storm and multi-organ failure in COVID-19 patients15,16,17. Together, these processes, which are closely linked to antiviral immunity and inflammation, underscore the critical role of mitochondria in the pathogenesis of COVID-1917,18.
In addition to their functional role in immunity, mitochondria also exhibit genetic variability that may influence disease outcomes. Mitochondrial DNA (mtDNA) variants, in particular, may act as biomarkers for predicting differential responses to infection19. Human migration out of Africa led to the accumulation of distinct mtDNA mutations, forming haplogroups linked to specific geographic regions20,21. Haplogroups serve as genetic markers of human migration and adaptation, as they may reflect the interplay between environmental pressures and adaptive processes, despite the presence of some neutral changes. Consequently, certain haplogroups are predominantly found in specific regions, reflecting the influence of local environmental conditions on mitochondrial diversity21. As a result of these evolutionary dynamics, certain mutations in the coding region may be uniquely associated with specific haplogroups22, potentially enhancing adaptation to stressors like oxidative stress and inflammation or even conferring an advantage in terms of energy efficiency under specific conditions. Consequently, individuals within these haplogroups may exhibit greater resilience to such challenges, while others may be more susceptible to virus-related diseases, such as COVID-19.
Studies have shown that mtDNA haplogroups and regional variations influence survival under environmental and physiological stress. For example, haplogroup H, which is common in Europe (~ 40%,21), has been linked to a survival advantage in sepsis, highlighting the role of “mitochondrial fitness”23. In addition, specific haplogroups and variants show adaptive benefits under extreme conditions21, such as the 3394C variant in the gene MT-ND1, associated with MT-ND1 complex I defects and common at high altitudes in Tibet24. Moreover, in coronary artery disease (CAD) studies, associations with mtDNA haplogroups varied across regions: no significant links were found in Europe and China25,26, while associations were observed in Japan27.
Expanding on this, a growing body of research has examined the relationship between mtDNA variability and COVID-19 severity across diverse populations. Certain mtDNA haplogroups and coding-region variants may influence disease severity and could serve as genetic markers for identifying individuals at higher risk of severe outcomes. For example, in Han Chinese populations, Wu et al.28 identified mtDNA variants associated with both increased and reduced severity. Variants such as 5178A (haplogroup D), 6392C, and 10310A (haplogroup F) were linked to lower severity, while 4833G (haplogroup G), 4715G (haplogroup M8), 3394C (haplogroup M9), and 5417A (haplogroup N9a) were associated with increased risk of severity. In Spain, Vázquez-Coto et al.19 found that the mtDNA variant 7028C (haplogroup H) protected against severe COVID-19 in patients ≤ 65 years. In India, Kumari et al.29 showed that haplogroup M3d1a was associated with severe outcomes in deceased patients, while haplogroup W3a1b was linked to severe cases in recovered individuals. They identified 15 mtDNA mutations associated with severity, including six that impacted protein stability and mitochondrial function29.
More recently, Bľandová et al.30 found that haplogroups J1 and clusters of H + U5b and T2b + U5b were associated with reduced severity in the Slovak population, while haplogroups T1, H11, and K increased risk. After Bonferroni correction, only the cluster H + U5b remained statistically significant30. Additionally, another study involving European COVID-19 patients revealed a potentially protective role for the macro-haplogroup HV (comprising H, V, and HV), highlighting that genetic diversity across human populations is not neutral but reflects the action of natural selection over time in response to successive epidemic and pandemic waves caused by viruses31.
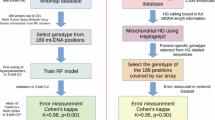
The Brazilian population is highly diverse, with ancestry primarily of European, African, and Native American origin, offering a unique opportunity to investigate associations between haplogroups of these various genetic backgrounds and COVID-19 outcomes. By integrating detailed clinical and mtDNA genetic data, we aimed to assess whether mitochondrial ancestry, specific mtDNA variants, and clinical factors collectively influence COVID-19 severity. In addition to traditional statistical methods, we analyzed this dataset using machine learning approaches to ensure robustness and provide a more comprehensive understanding of disease outcomes in this cohort.
Material and methods
Ethics and samples
The study was conducted following ethical guidelines and was approved by the Research Ethics Committee of the Hospital de Clínicas de Porto Alegre (CAAE: 36974620.3.0000.5327). Participants were contacted to provide informed consent for participation in this study. In cases where participants could not be reached, the requirement for informed consent was waived by the Institutional Review Board (IRB). The study was conducted in accordance with local legislation and institutional requirements. The cohort consisted of individuals aged 18 or older who tested positive for COVID-19 by qPCR testing at the Hospital de Clínicas de Porto Alegre. DNA samples were sourced from the Biobank of the same hospital (https://biobanco-covid-19.hcpa.edu.br/amostras).
A convenience sampling approach was employed, recruiting participants who had their genome sequenced in the DNA do Brasil project (https://www.gov.br/saude/pt-br/composicao/sectics/decit/genomas-brasil). The final cohort consisted of 467 individuals, categorized into 371 surviving patients (survivors) and 96 deceased patients (deceased). All participants were Brazilian residents of Rio Grande do Sul, the southernmost state in Brazil. Among these patients, 277 were identified as carrying one of twelve SARS-CoV-2 strains, predominantly B.1.1.28 and B.1.1.33 during the first wave in early 2020, with P.1 overlapping during the second wave in late 2020 and early 202132. Genomic DNA extraction was performed using the FlexiGene DNA Kit (QIAGEN), followed by quantification through Qubit™ fluorometric assays (Thermo Fisher Scientific) and NanoDrop™ One Microvolume UV–Vis Spectrophotometer (Thermo Fisher Scientific).
Sequencing and variant calling
The mitochondrial variants were called from whole genome sequencing data (WGS; Supplementary Information) using Mutect2 (Mitochondrial mode) from the Genome Analysis Toolkit (GATK) v4.1.133. Variant filtering was performed using the FilterMutectCalls module to eliminate blacklisted sites, contamination, clustered events, duplicate reads, fragment length issues, germline variants, haplotype bias, low allele fraction, mapping quality, multiallelic sites, high N ratio, artifacts in normal, nuclear-mitochondrial DNA variants (NuMTs), orientation bias, slippage, strand bias, weak evidence, and base quality. Variants passing all filters were included in the analysis. Additionally, haplogroup-defining mutations were considered after further verification. Variant Call Format (VCF) files were then used in the subsequent steps.
Haplogroup affiliation
We used the online tool Haplogrep334, based on PhyloTree 17—Forensic Update 1.235, with the default Kulczynski algorithm for the distance function, to classify our mitogenome sequences into haplogroups directly from VCFs. This tool was also used to further verify our sequences for positioning errors and artifacts. Additionally, we used the annotated revised Cambridge Reference Sequence (rCRS)36, as provided on the PhyloTree website37, to assign each variant to its corresponding mtDNA region. We also consulted the EMPOP database (https://empop.online/)38 to examine variant distribution across different haplogroups.
Filtering artifacts, phantom mutations, and editing mtDNA sequences
Since insertions and deletions (InDels) are sometimes detected at incorrect positions or may misalign expected mutations, a thorough manual review was conducted by cross-referencing Haplogrep334 results with the corresponding VCF files. Additionally, misaligned mutations were corrected, and the online tool Check for Phantom Mutations integrated into Haplogrep239 was employed to detect and exclude these artifact mutations40,41. Only variants with a Soares score below 342 that were observed in at least two distinct samples were included in the analysis. Overall, we applied a more conservative approach to minimize spurious mutations and ensure the reliability of the results.
Statistical analysis
We compared variables across different case groups using the Pearson Chi-Square test, which is appropriate for categorical variables, or the Fisher’s exact test, when necessary, particularly in cases with low expected frequencies. These analyses were conducted on the clinical and genetic data of the patients. Clinical data included age (non-elderly: < 65 years or elderly: ≥ 65 years), sex, and the following comorbidities: diabetes, obesity, hypertension, chronic diseases (excluding kidney disease), chronic kidney disease, and immunodeficiencies. Correlation analysis using Phi Coefficient, appropriate for binary data, was also performed. Additionally, multivariate logistic regression analyses were performed with adjustments for confounding variables and false discovery rate (FDR) correction, consistently comparing the survivors and deceased patients. For the genetic data, the analyses were performed at multiple levels, including mtDNA ancestry at the continental and macro-haplogroup scales, as well as specific mtDNA variants within the dataset. All data were binarized as follows: 0 indicated the absence of a given condition, survival, female sex, or non-elderly status; 1 indicated the presence of a given condition, death, male sex, or elderly status. For statistical purposes, we filtered out variants with low relative frequency in our dataset (< 5%). Furthermore, only coding-region variants were analyzed, as these are the most relevant for potential functional consequences. All statistical analyses were performed in the R environment43. Details on specific R packages used are provided in the Supplementary Information.
Machine learning analysis
We also employed an alternative method to explore our dataset, going beyond classical statistical methods by applying a machine learning approach. The dataset comprised 467 instances (79.4% survivors and 20.6% deceased) and 102 features, including 94 coding-region variants with a minimum frequency of 5% in the sample, along with eight clinical variables. Data were partitioned into training and test sets using a 70:30 stratified split, preserving the proportional distribution of outcomes. The training set included 259 survivors (79.45%) and 67 deceased (20.55%), while the test set included 112 survivors (79.43%) and 29 deceased (20.57%). A Random Forest classifier was implemented using the RandomForestClassifier from the scikit-learn library with default parameters44. Model performance was evaluated using accuracy, precision, recall (sensitivity), F1-score, and the Receiver Operating Characteristic (ROC) curve. Because interpretability was a key objective, we reported metrics separately for each class, survivor and deceased, rather than focusing solely on the deceased class. The confusion matrix provided class-specific performance metrics, while the ROC curve evaluated the model’s ability to discriminate between survivors and deceased patients by plotting the true positive rate (recall for the deceased class) against the false positive rate. The Area Under the Curve (AUC) was used to quantify classification performance, where higher values indicate better discrimination, with 1.0 indicating perfect classification and 0.5 representing random guessing.
To interpret feature importance, SHAP (SHapley Additive exPlanations) values were computed using the TreeExplainer module from the SHAP library. This method extends standard SHAP calculations by estimating feature contributions while accounting for interactions within the predictive model. Given that the hierarchical structure of decision trees inherently captures feature dependencies, these relationships are reflected in the SHAP values computed by TreeSHAP. Although SHAP values are additive by definition, TreeSHAP effectively models non-linear effects and feature interdependencies learned by the model. Visualization included a global bar plot and a beeswarm plot, ranking the top 15 features influencing predictions. SHAP values offer a quantitative measure of each feature’s contribution to the model’s predictions, offering an interpretable framework to understand the role of individual variables. All analyses were conducted in Python45, with details on specific packages provided in the Supplementary Information.
Structural and functional predictions
We used MutPred246, APOGEE247, the Sorting Intolerant From Tolerant (SIFT)48, and Polymorphism Phenotype v2 (PolyPhen-2)49 scores to predict the pathogenicity of variants significantly associated with mortality, with the last three analysis performed using the MitImpact 3D online tool50. Detailed explanations of how these tools calculate functional impact predictions and how to interpret their results are provided in the Supplementary Information.
Results
Age, sex, comorbidities, and COVID-19 outcomes
Our initial results align with what is already established in the scientific community: older age, male sex, and the presence of comorbidities are associated with an increased risk of mortality from COVID-19 (Supplementary Tables S1-S2). Additionally, our correlation analysis indicated that none of the clinical variables were strongly correlated with each other, thus avoiding multicollinearity and making them suitable covariates for regression adjustment (Supplementary Tables S3-S4, Supplementary Fig. S1).
Logistic regression analysis using only clinical data showed that, among the predictors of mortality, advanced age (≥ 65 years) (OR = 5.09, CI = 2.84–9.24, p = 5.95 × 10–8), male sex (OR = 2.30, CI = 1.38–3.86, p = 1.48 × 10–3), obesity (OR = 1.85, CI = 1.06–3.26, p = 3.13 × 10–2), hypertension (OR = 1.82, CI = 1.01–3.31, p = 4.61 × 10–2), and diabetes (OR = 1.78, CI = 1.00–3.12, p = 4.68 × 10–2) were the strongest and most significant associations. Other comorbidities, including kidney and other chronic diseases, and immunodeficiencies, were not statistically significant. These findings highlight the role of specific pre-existing conditions in increasing the risk of COVID-19 mortality (Supplementary Table S5).
Continental ancestry and macro-haplogroup association with mortality
We first analyzed the distribution of mtDNA continental ancestry (Fig. 1) and macro-haplogroups (Fig. 2) across the different case groups. Survivor and deceased groups differed significantly in the distribution of West Eurasian (χ2 = 6.92, df = 1, p = 0.0085) and Native American (χ2 = 4.23, df = 1, p = 0.0398) mtDNA ancestry, but not African ancestry (χ2 = 0.84, df = 1, p = 0.3595) (Supplementary Table S6). Frequency comparisons suggest that West Eurasian mtDNA ancestry may be associated with a reduced mortality risk, whereas Native American ancestry may be linked to increased risk. However, a clearer understanding emerged from the multivariate logistic regression. After controlling for covariates (age, sex, and comorbidities), no continental ancestry remained statistically significant (Supplementary Table S7). These findings suggest that not all West Eurasian lineages are protective, nor are all Native American lineages associated with increased mortality risk. Building on this finding, the analysis at the macro-haplogroup level revealed intriguing results: the Native American haplogroups A2 (χ2 = 4.613, df = 1, p = 0.032) and D (p = 0.046) appeared to associated with an increased risk of death, while the European haplogroup H appears to be associated with a decreased risk (χ2 = 4.242, df = 1, p = 0.039) (Supplementary Table S6). However, only the association with haplogroup A2 was confirmed in the multivariate logistic regression analysis. After adjusting for covariates and applying FDR correction, the association remained, with haplogroup A2 still associated with increased mortality risk (OR = 3.445, 95% CI = 1.532–7.746, p = 0.0028) (Fig. 3; Supplementary Table S8).

Frequency of mitochondrial DNA continental ancestry. Pie charts depicting the distribution of mtDNA continental ancestry in the overall dataset and within each COVID-19 case group (survivors and deceased).

Frequency of mitochondrial macro-haplogroups by case group. Column chart showing the distribution of mtDNA macro-haplogroups across COVID-19 case groups (survivors and deceased).

Adjusted multivariate logistic regression for macro-haplogroups. Bar chart displaying odds ratios (ORs) and 95% confidence intervals for variables that remained statistically significant in the multivariate regression model, which included mtDNA macro-haplogroups and clinical covariates. The dashed vertical line at OR = 1.0 indicates the null value (no association).
Variant association with mortality
Of the 1572 variants identified in our dataset, 1265 remained after filtering to include only coding regions: 1041 in protein-coding regions, 80 in rRNA-coding regions, and 144 in tRNA-coding regions. Further filtering for common variants (those with a relative frequency > 5%) resulted in 94 coding-region variants. Of these, 70 were in protein-coding regions, 14 in tRNA regions, and 10 in rRNA regions.
In relation to mortality, our multivariate logistic regression analysis identified ten variants significantly associated with an increased risk of death. However, after FDR correction, only the variants 1736G, 4248C, 4824G, 663G, 8794T, and 8027A remained statistically significant (Fig. 4; Supplementary Table S9). The clinical variable age remained the strongest predictor of mortality (OR = 5.134, CI = 2.756–10.276, p = 5.970 × 10–8), while male sex also showed a significant association (OR = 2.316, CI = 1.326–4.269, p = 1.429 × 10–3).

Adjusted multivariate logistic regression for mitochondrial DNA variants. Bar chart displaying odds ratios (ORs) and 95% confidence intervals for variables that remained statistically significant in the multivariate logistic regression model, which included mtDNA variants and clinical covariates. The dashed vertical line at OR = 1.0 represents the null value (no association).
Machine learning on variants
The Random Forest classifier achieved an overall accuracy of 78% on the test set using both genetic and clinical variables. For the survivor class, precision, recall, and F1-scores were 0.82, 0.92, and 0.87, respectively, while for the deceased class, these values were 0.44, 0.24, and 0.31, as derived from the confusion matrix (Supplementary Fig. S2). Although the F1-score was higher for the survivor class, likely due to the larger sample size, we chose not to balance the dataset by randomly downsampling this group. Instead, we prioritized maximizing the model’s learning capacity over artificially balancing the class distribution at the cost of losing valuable information. The model’s discriminative ability was quantified by an AUC of 0.73 (Supplementary Fig. S3).
The machine learning analysis identified age as the most influential feature, with higher SHAP values reflecting a stronger impact on predictions (Supplementary Table S10). Hypertension was the second most important feature, followed by diabetes and sex. Additional clinical variables—including obesity, chronic diseases (excluding kidney disease), chronic kidney disease, and immunodeficiencies—also significantly contributed to the model’s predictions.
Several genetic alleles—including 8027A, 2706G, 11914A, 3010A, 10873C, and 1736G—were critical in distinguishing between survival and mortality outcomes (Fig. 5). Notably, three of these alleles—11914A, 3010A, and 10873C—had not been previously identified by traditional statistical methods. The allele 11914A was the only one associated with survival rather than mortality (Supplementary Fig. S4). This allele is present in several haplogroups but was observed in 100% of individuals belonging to macro-haplogroups C and L0 in our patient sample (Supplementary Fig. S6). The 3010A variant was present in all patients belonging to the Native American haplogroup D, as well as in some individuals of West Eurasian and African ancestry, while 10873C is associated with one of the most basal haplogroups found in sub-Saharan Africa.

Global feature importance based on mean SHAP values. Bar chart showing the average contribution of each feature to the model’s predictions, as measured by SHAP (SHapley Additive exPlanations) values. Age was the most influential variable, followed by specific comorbidities (hypertension and diabetes), sex, and other comorbidities (obesity and chronic conditions, including kidney-related and non-kidney-related diseases). Several mitochondrial DNA (mtDNA) variants—particularly 8027A, 2706G, 11914A, 3010A, 10873C, and 1736G—also contributed substantially, with the first three ranking above immunodeficiencies. The combined contribution of 88 additional mtDNA variants accounted for approximately 0.17, reflecting their collective impact on model predictions.
The 8027A variant emerged as the most influential genetic feature among the top 15 contributors to model predictions (Fig. 5). The 8027A variant, which defines haplogroup A2, is prevalent in Native Americans and their descendants, including admixed populations. However, 8027A was also observed in other haplogroups, particularly L1c (Supplementary Fig. S6). The 8027A allele had a greater overall impact than immunodeficiencies (Fig. 5; Supplementary Table S10). Furthermore, a SHAP value of 0.013 (Supplementary Table S10) suggests that, on average, the 8027A allele exerts a modest influence on model predictions, though it may still be significant when interacting with other genetic or clinical factors.
The global bar plot (Fig. 5) highlighted the cumulative contribution of features, with the combined influence of 88 additional mtDNA variants playing a substantial role in the model’s predictions. Our results indicate that a specific combination of mitochondrial genetic variants significantly influenced survival outcomes in COVID-19 patients. This combination, not necessarily limited to additive effects, may underlie the observed association between haplogroup A2 and increased mortality risk from COVID-19 in Brazilian patients.
Key variants in regression and random forest
All mtDNA variants that were statistically significant in the regression analysis or identified as key predictors in the random forest feature importance analysis are highlighted in Supplementary Table S11, which also presents their general characteristics and frequencies. The distribution of each variant across case groups is detailed in Supplementary Table S12 and Supplementary Figure S5.
The variants identified in this study are located in mitochondrial genes that are essential for oxidative phosphorylation, mitochondrial protein synthesis, and other critical cellular processes. Variants in NADH dehydrogenase subunits 1, 2, and 4 (MT-ND1, MT-ND2, MT-ND4) affect Complex I, which catalyzes the transfer of electrons from NADH to ubiquinone. The variant in cytochrome c oxidase subunit 2 (MT-CO2) affects Complex IV, which is responsible for the final reduction of oxygen to water. The variant in ATP synthase membrane subunit 6 (MT-ATP6) affects Complex V, which synthesizes ATP through proton translocation. Variants in the mitochondrially encoded 12S and 16S rRNA genes (MT-RNR1, MT-RNR2) affect mitochondrial rRNAs that are essential for the translation of respiratory chain components.
Structural and functional predictions
Among the significant variants after correction and the essential variants in the machine learning model, the protein-coding variants 4248C (I314I), 10873C (P38P), and 11914A (T385T) are synonymous mutations, whereas 4824G (T119A), 8027A (A148T), and 8794T (H90Y) are non-synonymous. Although scores from APOGEE2, SIFT, and Grantham suggest that the three non-synonymous variants may exert neutral or mild effects, other structural and functional predictions indicate potential impact: 8027A was classified as potentially damaging (PolyPhen-2 score = 1.0), and 8794T as possibly pathogenic (MutPred2 score = 0.608) (Supplementary Table S13). These conflicting predictions likely arise from differences in prediction methods of the programs and sensitivity to specific structural contexts. For instance, PolyPhen-2 evaluates amino acid substitutions by integrating sequence conservation, structural context, and functional domains, making it more sensitive to structural changes within proteins. PolyPhen-2 often classifies variants as potentially damaging when protein structural integrity is affected, even in cases where the impact on sequence conservation is minimal, which may lead to discrepancies between prediction methods49.
Ancestry and variant relationships
Most of the statistically significant variants associated with mortality are derived alleles, differing from the ancestral state represented by the Reconstructed Sapiens Reference Sequence (RSRS)51, with the exceptions of 2706G, 10873C, and 11914A (Supplementary Table S11). The RSRS belongs to macro-haplogroup L and was introduced by Behar et al.51 as an alternative to the rCRS36, with the aim of representing the ancestral sequence of modern humans. It is rooted in haplogroup L3, the ancestor of all non-African haplogroups (M, N, and their branches), as well as several African lineages. Thus, the RSRS reflects the genetic point of origin of modern humans prior to their migration out of Africa.
Variants 663G, 1736G, 4248C, 4824G, and 8794T are associated with the Asian macro-haplogroup A, while the variant 8027A defines haplogroup A2—prevalent in Native Americans—and also appears in haplogroup L1c, found in Africans and their admixed descendants. According to mtDNA Tree Build 17, variants 1736G and 8794T are exclusive to macro-haplogroup A, whereas the others are not exclusive and recur in multiple haplogroups (Supplementary Tables S14-S15; Supplementary Fig. S6). Variants identified as important predictors by the random forest method are less haplogroup-defining and frequently recur across numerous distinct lineages (Supplementary Tables S14-S15). Notably, the variant 2706G, although present in multiple macro-haplogroups, was found in all individuals belonging to haplogroup A2. Finally, it is important to highlight that some of these variants found in haplogroup A2 result in amino acid substitutions in genes of recognized functional importance, as previously mentioned, MT-ND1 (4248C), MT-ND2 (4824G), MT-CO2 (8027A), and MT-ATP6 (8294T) (Supplementary Tables S11 and S14).
Discussion
Our analysis of mitochondrial macro-haplogroups in relation to COVID-19 outcomes revealed a significant association between macro-haplogroup A and increased COVID-19-related mortality. Additionally, variant-based regression analysis revealed that five of ten relevant variants (A663G, A1736G, T4248C, A4824G, and C8794T) are defining markers of macro-haplogroup A (Supplementary Table S14). All are present in sublineage A2, along with G8027A. Although none of these variants are exclusive to a single haplogroup (Supplementary Tables S14–15), each occurs at 100% frequency among patients with the Native American haplogroup A2, except 1736G (~ 98%) (Supplementary Table S15, Supplementary Fig. S6). Another key variant, 2706G, although not diagnostic of A or A2, was present in all A2 carriers in our sample. The presence of G8027A in all COVID-19 patients carrying the African haplogroup L1c is noteworthy, as approximately 29% of these patients died. This finding warrants further investigation, particularly because these individuals also carry the 2706G and 10873C variants. Haplogroup L1c is most prevalent in Central Africa, particularly in Gabon, Cameroon, and the Republic of the Congo38.
Haplogroup A2 is commonly found among Indigenous Brazilian groups and in the admixed population (Supplementary Table S16). In Rio Grande do Sul, it is particularly observed in present-day Guarani and Kaingang individuals, who represent the native peoples of the region. Therefore, the presence of A2 in the admixed population of this State likely reflects Indigenous maternal ancestry.
Our findings are particularly relevant given the well-documented elevated mortality associated with SARS-CoV-2 infection among Native Americans, although the complexity of its underlying causes is still poorly understood. The genetic basis of susceptibility or protection remains largely unknown, and multiple confounding factors—such as health inequities, low socioeconomic status, limited access to healthcare, and cultural influences—further complicate its assessment and implications for infectious disease outcomes.
Most studies indicate that Native Americans face a higher risk of COVID-19-related mortality compared to other groups, particularly individuals classified as “White”, who generally exhibit a greater proportion of European ancestry, both in Brazil52,53,54,55,56,57 and the United States58,59,60,61. A similar trend has been observed for African ancestry, which may also represent a risk factor for severe clinical outcomes54,55,57. These disparities underscore the structural and socioeconomic vulnerabilities disproportionately affecting non-“White” populations, exacerbating health inequalities among them and historically marginalized groups, such as Native communities. Such inequities may also obscure other determinants, including genetic factors that modulate disease severity. For instance, haplogroup H has been reported to reduce the risk of early-onset critical COVID-19 and, in combination with U5b, to be associated with a less severe disease course30. Cabrera-Alarcón31 developed a random forest model for mtDNA haplogroup classification during the COVID-19 pandemic and found that the HV branch (H, V, and HV) functioned as a protective factor against SARS-CoV-2 severity in European patients, independent of comorbidities, age, or sex. Our study revealed a similar trend, particularly for macro-haplogroup H, which is characteristic of Europeans and their descendants. However, its association lost statistical significance after adjustment for confounding variables. Sacuena et al.62 conducted the first genetic study on 263 Amazonian Indigenous individuals to investigate factors influencing SARS-CoV-2 susceptibility. Their findings suggest that these populations exhibit lower frequencies of risk alleles in three nuclear genes involved in SARS-CoV-2 cellular entry, potentially contributing to asymptomatic or milder clinical courses observed among the Araweté, Kararaô, Kayapó, Munduruku, Parakanã, and Xikrin peoples. However, due to the relatively small sample size, the authors emphasized the need for larger-scale genetic studies to better assess susceptibility or protection and disease outcomes in infectious diseases among Indigenous populations62. In contrast, our study identified the mtDNA variant 8027A, present in both the African L1c and Native American A2 haplogroups, as the most relevant genetic factor in the machine learning analysis. While macro-haplogroup A was significantly associated with mortality, none of the haplogroups commonly found in sub-Saharan populations exhibited a similar association.
The machine learning analysis identified mitochondrial coding variants associated with COVID-19 outcomes, revealing patterns that might be overlooked by traditional statistical methods. Notably, it confirmed the relevance of the 8027A allele (Fig. 5), a non-synonymous mutation causing an alanine-to-threonine substitution at position 148 of MT-CO2. This variant emerged as the most statistically significant marker, with PolyPhen-2 predicting it to be potentially damaging. Our findings, supported by both regression analysis and feature importance assessment (Supplementary Tables S9–S10), reinforce its potential role in MT-CO2 function, despite ongoing debate over the pathogenicity of certain previously reported mtDNA variants63.
Another key variant identified in our analysis is the non-synonymous mutation C8794T in the MT-ATP6 gene, which encodes subunit 6 of ATP synthase, a critical enzyme involved in mitochondrial ATP production. The 8794T allele, a diagnostic marker of haplogroup A2, results in a histidine-to-tyrosine substitution at position 90 of MT-ATP6 and is predicted to be potentially pathogenic by in silico analyses. MT-ATP6 facilitates proton translocation through a rotational mechanism during ATP synthesis, and mutations in this gene have been associated with severe multisystem disorders (see Supplementary Information).
In addition to non-synonymous mutations, other relevant variants identified in this study merit consideration due to their potential biological significance. For instance, the 663G variant in MT-RNR1 (12S rRNA) was also reported by Wu et al.28 in a comparison between moderate COVID-19 cases and controls, remaining significant after adjustment for age, sex, smoking, and comorbidities, as well as Bonferroni correction. Additionally, Vázquez-Coto et al.19 identified the 7028C variant (MT-CO1), which was linked to a reduced risk of early-onset critical COVID-19. Interestingly, in our study, prior to FDR correction, the 7028T variant at the same position was associated with increased mortality risk after controlling for covariates.
Macro-haplogroup A and the variants identified as significant in our study, except for 663G, represent novel findings in the context of respiratory diseases. However, several of these variants have previously been associated with other medical conditions. Macro-haplogroup A has been linked to atherothrombotic cerebral infarction64,65, type 2 diabetes mellitus66, and coronary atherosclerosis27 in studies conducted in Japanese populations. Variants 2706G and 3010A have been shown to induce significant structural alterations in mitochondrial 16S rRNA, supporting their proposed role in the pathophysiology of Leber’s hereditary optic neuropathy (LHON)67.
Variant 3010A has also been extensively investigated in cyclic vomiting syndrome (CVS)68,69, although some studies have challenged this association70,71,72. Interestingly, the 3010G variant has been linked to high-altitude adaptation in Tibetan populations73, suggesting that in respiratory diseases such as COVID-19, 3010A allele may confer a disadvantage. Similarly, preliminary evidence suggests that 2706G is associated with slower cognitive decline in Parkinson’s disease74 and may contribute to the pathogenesis of type 2 diabetes mellitus75. Variants 663G and 8794T, both diagnostic of and A2 lineages, have also been reported as genetic risk factors for severe coronary atherosclerosis in elderly Japanese individuals27. Nishigaki, Fuku, and Tanaka65 proposed that the H90T amino acid substitution, resulting from the C8794T mutation, may have functional implications in ATPase subunit 6, as suggested by its Grantham value (83.54). Given the critical role of this protein in proton translocation, such alterations could have broader implications for mitochondrial physiology.
Furthermore, Zhu et al.76 identified a potential association between the 4248C variant, a diagnostic marker of macro-haplogroup A/A2, and maternally inherited essential hypertension (MIEH) in a Chinese population. In a preliminary analysis, Li et al.77 suggested that the 10873C variant may contribute to the pathophysiology of type 2 diabetes. Additionally, the 11914A variant has been positively associated with bullous pemphigoid, an autoimmune blistering skin disease, in German patients78. Further details on the genes associated with these variants are provided in the Supplementary Information.
Our results do not suggest that these variants directly cause mortality; rather, they may contribute to an increased risk of death. Potential interactions with nuclear genome variants, whether additive or epistatic, may further modulate disease susceptibility. Studies on nuclear genes and COVID-19 severity in Brazilian patients have begun to explore these associations79. The mtDNA variants identified in our study, many of which have been corroborated by previous clinical research, suggest a role in modulating protein function and contributing to disease susceptibility. Their association with increased mortality risk—particularly when co-occurring within the same geographic haplogroups, such as A2—suggests the presence of underlying biological mechanisms rather than coincidence, reinforcing the role of mtDNA in infectious disease susceptibility31. The distribution of mtDNA lineages across human populations may have been shaped by historical selective pressures, potentially driven by infectious disease challenges.
Our study has certain limitations that should be acknowledged. A larger cohort would enhance statistical power, enabling more refined stratification of mtDNA subgroups and the inclusion of additional variables associated with COVID-19 outcomes. Integrating nuclear DNA with mtDNA and clinical data could also yield a more comprehensive understanding of disease susceptibility. Another limitation concerns the exclusion of rare variants, which may significantly influence both common80 and rare diseases81. Due to the limited sample size, the low frequency of these variants hindered both statistical power and the predictive performance of our machine learning model. Furthermore, the lack of SARS-CoV-2 strain data for most patients represents another limitation, as viral lineage differences are known to impact disease severity and clinical outcomes32.
In conclusion, this study highlights the significant impact of mtDNA haplogroups—particularly haplogroup A2, which is associated with Native American ancestry—on COVID-19 outcomes in an admixed Brazilian population. Some of the identified alleles have been previously associated with disease susceptibility, while others are newly reported in this context. The complex genetic mosaic of admixed populations, combining distinct mtDNA and nuclear ancestry components, poses challenges for genetic studies and underscores the need for population-specific analyses, revealing the limitations of generalized conclusions. By integrating machine learning with classical statistical approaches, we achieved greater robustness in identifying clinically relevant variants. Our findings align with recent studies in European populations, reinforcing that geographic and population-specific mtDNA haplogroups influence disease susceptibility. This is potentially due to population-specific genetic backgrounds shaped by evolutionary pressures, maintaining a delicate balance between mitochondrial bioenergetic function and responses to viral infections throughout human evolution. Expanding genetic research beyond populations of European descent is essential to advance precision medicine and promote health equity, in alignment with the United Nations Sustainable Development Goals 3 (Good Health and Well-being) and 10 (Reduced Inequalities).
Data availability
Our database consists of individual-level genomic data from study participants, and sharing this information would involve disclosing sensitive data that cannot be fully anonymized. Additionally, we do not have ethical approval to publicly share these genomic sequences. As such, this constitutes an exception to data availability. However, the data can be made available upon reasonable request and with authorization from the relevant third party. The key mitochondrial genetic variants that support the main conclusions of the study are listed in Supplementary Table S17. For inquiries regarding the dataset, readers may contact the corresponding author, F.S.L.V.
References
World Health Organization. Coronavirus disease (COVID-19). World Health Organization. https://www.who.int/health-topics/coronavirus#tab=tab_1 (2024).
Fam, B. S., Vargas-Pinilla, P., Amorim, C. E. G., Sortica, V. A. & Bortolini, M. C. ACE2 diversity in placental mammals reveals the evolutionary strategy of SARS-CoV-2. Genet. Mol. Biol. 43, e20200104. https://doi.org/10.1590/1678-4685-gmb-2020-0104 (2020).
Yépez, Y. et al. Evolutionary history of the SARS-CoV-2 Gamma variant of concern (P. 1): A perfect storm. Genet. Mol. Biol. 45, e20210309. https://doi.org/10.1590/1678-4685-gmb-2021-0309 (2022).
World Health Organization. WHO COVID-19 dashboard. World Health Organization. https://www.who.int/health-topics/coronavirus#tab=tab_1 (2024).
Biancolella, M. et al. COVID-19 annual update: A narrative review. Hum. Genom. 17, 68. https://doi.org/10.1186/s40246-023-00515-2 (2023).
Delgado-Wicke, P. et al. Genetic variants regulating the immune response improve the prediction of COVID-19 severity provided by clinical variables. Sci. Rep. 14, 20728. https://doi.org/10.1038/s41598-024-71476-2 (2024).
Minnai, F. et al. A genome-wide association study for survival from a multi-centre European study identified variants associated with COVID-19 risk of death. Sci. Rep. 14, 3000. https://doi.org/10.1038/s41598-024-53310-x (2024).
Pairo-Castineira, E. et al. GWAS and meta-analysis identifies 49 genetic variants underlying critical COVID-19. Nature 617, 764–768. https://doi.org/10.1038/s41586-023-06034-3 (2023).
Rurek, M. Mitochondria in COVID-19: From cellular and molecular perspective. Front. Physiol. 15, 1406635. https://doi.org/10.3389/fphys.2024.1406635 (2024).
Urzúa-Traslaviña, C. G., Moreno-Treviño, M. G., Martínez-Treviño, D. A., Barrera-Saldaña, H. A. & León-Cachón, R. B. R. Relationship of mitochondrial DNA haplogroups with complex diseases. J. Genet. Genome Res. 1, 1–5. https://doi.org/10.23937/2378-3648/1410011 (2014).
Koshiba, T. Mitochondrial-mediated antiviral immunity. Biochim. Biophys. Acta Mol. Cell Res. 1833, 225–232. https://doi.org/10.1016/j.bbamcr.2012.03.005 (2013).
Weinberg, S. E., Sena, L. A. & Chandel, N. S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 42, 406–417. https://doi.org/10.1016/j.immuni.2015.02.002 (2015).
Belgnaoui, S. M., Paz, S. & Hiscott, J. Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Curr. Opin. Immunol. 23, 564–572. https://doi.org/10.1016/j.coi.2011.08.001 (2011).
Shi, C. S. et al. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 193, 3080–3089. https://doi.org/10.4049/jimmunol.1303196 (2014).
Ragab, D., Salah Eldin, H., Taeimah, M., Khattab, R. & Salem, R. The COVID-19 cytokine storm; what we know so far. Front. Immunol. 11, 551898. https://doi.org/10.3389/fimmu.2020.01446 (2020).
Yang, Y. et al. SARS-CoV-2 membrane protein causes the mitochondrial apoptosis and pulmonary edema via targeting BOK. Cell Death Differ. 29, 1395–1408. https://doi.org/10.1038/s41418-022-00928-x (2022).
Guarnieri, J. W. et al. Mitochondrial antioxidants abate SARS-CoV-2 pathology in mice. Proc. Natl. Acad. Sci. U. S. A. 121, e2321972121. https://doi.org/10.1073/pnas.2321972121 (2024).
Prasun, P. COVID-19: A mitochondrial perspective. DNA Cell Biol. 40, 713–719. https://doi.org/10.1089/dna.2020.6453 (2021).
Vázquez-Coto, D. et al. Common mitochondrial haplogroups as modifiers of the onset-age for critical COVID-19. Mitochondrion 67, 1–5. https://doi.org/10.1016/j.mito.2022.09.001 (2022).
Wallace, D. C., Brown, M. D. & Lott, M. T. Mitochondrial DNA variation in human evolution and disease. Gene 238, 211–230. https://doi.org/10.1016/s0378-1119(99)00295-4 (1999).
Stewart, J. B. & Chinnery, P. F. Extreme heterogeneity of human mitochondrial DNA from organelles to populations. Nat. Rev. Genet. 22, 106–118. https://doi.org/10.1038/s41576-020-00284-x (2021).
Ruiz-Pesini, E., Mishmar, D., Brandon, M., Procaccio, V. & Wallace, D. C. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science 303, 223–226. https://doi.org/10.1126/science.1088434 (2004).
Baudouin, S. V. et al. Mitochondrial DNA and survival after sepsis: A prospective study. Lancet 366, 2118–2121. https://doi.org/10.1016/s0140-6736(05)67890-7 (2005).
Ji, F. et al. Mitochondrial DNA variant associated with Leber hereditary optic neuropathy and high-altitude Tibetans. Proc. Natl. Acad. Sci. U. S. A. 109, 7391–7396. https://doi.org/10.1073/pnas.1202484109 (2012).
Benn, M., Schwartz, M., Nordestgaard, B. G. & Tybjærg-Hansen, A. Mitochondrial haplogroups: Ischemic cardiovascular disease, other diseases, mortality, and longevity in the general population. Circulation 117, 2492–2501. https://doi.org/10.1161/CIRCULATIONAHA.107.756809 (2008).
Xiao, F. et al. Association between mitochondrial DNA haplogroup variation and coronary artery disease. Nutr. Metab. Cardiovasc. Dis. 30, 960–966. https://doi.org/10.1016/j.numecd.2020.03.006 (2020).
Sawabe, M. et al. Mitochondrial haplogroups A and M7a confer a genetic risk for coronary atherosclerosis in the Japanese elderly: An autopsy study of 1,536 patients. J. Atheroscler. Thromb. 18, 166–175. https://doi.org/10.5551/jat.6742 (2011).
Wu, Y. et al. Common mtDNA variations at C5178A and A249D/T6392C/G10310A decrease the risk of severe COVID-19 in a Han Chinese population from Central China. Mil. Med. Res. 8, 1–10. https://doi.org/10.1186/s40779-021-00351-2 (2021).
Kumari, D. et al. Mitochondrial pathogenic mutations and metabolic alterations associated with COVID-19 disease severity. J. Med. Virol. 95, e28553. https://doi.org/10.1002/jmv.28553 (2023).
Bľandová, G. et al. Mitochondrial DNA variability and Covid-19 in the Slovak population. Mitochondrion 75, 101827. https://doi.org/10.1016/j.mito.2023.101827 (2024).
Cabrera-Alarcon, J. L. et al. Shaping current European mitochondrial haplogroup frequency in response to infection: The case of SARS-CoV-2 severity. Commun. Biol. 8, 33. https://doi.org/10.1038/s42003-024-07314-y (2025).
Fam, B. S. D. O. et al. SARS-CoV-2 strains and clinical profiles of COVID-19 patients in a Southern Brazil hospital. Front. Immunol. 15, 1444620. https://doi.org/10.3389/fimmu.2024.1444620 (2024).
McKenna, A. et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. https://doi.org/10.1101/gr.107524.110 (2010).
Schönherr, S., Weissensteiner, H., Kronenberg, F. & Forer, L. Haplogrep 3—an interactive haplogroup classification and analysis platform. Nucleic Acids Res. 51, W263–W268. https://doi.org/10.1093/nar/gkad284 (2023).
Dür, A., Huber, N. & Parson, W. Fine-tuning phylogenetic alignment and haplogrouping of mtDNA sequences. Int. J. Mol. Sci. 22, 5747. https://doi.org/10.3390/ijms22115747 (2021).
Andrews, R. M. et al. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 23, 147–147. https://doi.org/10.1038/13779 (1999).
Van Oven, M. & Kayser, M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 30, E386–E394. https://doi.org/10.1002/humu.20921 (2009).
Huber, N., Parson, W. & Dür, A. Next generation database search algorithm for forensic mitogenome analyses. Forensic Sci. Int. Genet. 37, 204–214. https://doi.org/10.1016/j.fsigen.2018.09.001 (2018).
Weissensteiner, H. et al. HaploGrep 2: Mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res. 44, W58–W63. https://doi.org/10.1093/nar/gkw233 (2016).
Bandelt, H. J., Quintana-Murci, L., Salas, A. & Macaulay, V. The fingerprint of phantom mutations in mitochondrial DNA data. Am. J. Hum. Genet. 71, 1150–1160. https://doi.org/10.1086/344397 (2002).
Brandstätter, A. et al. Phantom mutation hotspots in human mitochondrial DNA. Electrophoresis 26, 3414–3429. https://doi.org/10.1002/elps.200500307 (2005).
Soares, P. et al. Correcting for purifying selection: An improved human mitochondrial molecular clock. Am. J. Hum. Genet. 84, 740–759. https://doi.org/10.1016/j.ajhg.2009.05.001 (2009).
R Core Team. R: A language and environment for statistical computing. Foundation for Statistical Computing. https://www.r-project.org/ (2024).
Pedregosa, F. et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830. https://doi.org/10.5555/1953048.2078195 (2011).
Van Rossum, G. & Drake Jr, F. L. Python Tutorial, Vol. 620 (Centrum voor Wiskunde en Informatica, 1995).
Pereira, L., Soares, P., Radivojac, P., Li, B. & Samuels, D. C. Comparing phylogeny and the predicted pathogenicity of protein variations reveals equal purifying selection across the global human mtDNA diversity. Am. J. Hum. Genet. 88, 433–439. https://doi.org/10.1016/j.ajhg.2011.03.006 (2011).
Bianco, S. D. et al. APOGEE 2: Multi-layer machine-learning model for the interpretable prediction of mitochondrial missense variants. Nat. Commun. 14, 5058. https://doi.org/10.1038/s41467-023-40797-7 (2023).
Ng, P. C. & Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874. https://doi.org/10.1101/gr.176601 (2001).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 76, 7–20. https://doi.org/10.1002/0471142905.hg0720s76 (2013).
Castellana, S. et al. MitImpact 3: Modeling the residue interaction network of the Respiratory Chain subunits. Nucleic Acids Res. 49, D1282–D1288. https://doi.org/10.1093/nar/gkaa1032 (2021).
Behar, D. M. et al. A “Copernican” reassessment of the human mitochondrial DNA tree from its root. Am. J. Hum. Genet. 90, 675–684. https://doi.org/10.1016/j.ajhg.2012.03.002 (2012).
Santos, V. S., Souza Araújo, A. A., de Oliveira, J. R., Quintans-Júnior, L. J. & Martins-Filho, P. R. COVID-19 mortality among indigenous people in Brazil: A nationwide register-based study. J. Public Health 43, e250–e251. https://doi.org/10.1093/pubmed/fdaa176 (2021).
Soares, M. C. B. et al. Hospitalizations and deaths of Brazilian children and adolescents with Severe Acute Respiratory Syndrome caused by COVID-19. J. Infect. Dev. Ctries. 16, 1809–1820. https://doi.org/10.3855/jidc.17079 (2022).
Sansone, N. M., Boschiero, M. N., Valencise, F. E., Palamim, C. V. & Marson, F. A. Characterization of demographic data, clinical signs, comorbidities, and outcomes according to the race in hospitalized individuals with COVID-19 in Brazil: An observational study. J. Glob. Health 12, 05027. https://doi.org/10.7189/jogh.12.05027 (2022).
Dos Santos, M. et al. Ethnic/racial disparity in mortality from COVID-19: Data for the year 2020 in Brazil. Spat. Demogr. 11, 1. https://doi.org/10.1007/s40980-022-00112-2 (2023).
Novaes, T. E. R., Lara, D. M. & da Silva, S. G. Severe acute respiratory syndrome (SARS) in the context of the COVID-19 pandemic among Indigenous peoples of Brazil: Epidemiology and risk factors associated with death. J. Racial Ethn. Health Disparities 11, 1908–1917. https://doi.org/10.1007/s40615-023-01660-z (2024).
Cajazeiro, J. M. D., Cardoso, A. M. & Nobre, A. A. Ethnic-racial composition of the population in COVID-19 mortality: A spatial ecological approach to Brazilian health inequalities. Ciênc. Saúde Coletiva 29, e05552024. https://doi.org/10.1590/1413-812320242912.05552024EN (2024).
Leggat-Barr, K., Uchikoshi, F. & Goldman, N. COVID-19 risk factors and mortality among Native Americans. Demogr. Res. 45, 1185–1218. https://doi.org/10.4054/DemRes.2021.45.39 (2021).
Bime, C. et al. Disparities in outcomes of COVID-19 hospitalizations in Native American individuals. Front. Public Health 11, 1220582. https://doi.org/10.3389/fpubh.2023.1220582 (2023).
Hurwitz, I. et al. Disproportionate impact of COVID-19 severity and mortality on hospitalized American Indian/Alaska Native patients. PNAS Nexus 2, pgad259. https://doi.org/10.1093/pnasnexus/pgad259 (2023).
Slutske, W. S. et al. Explaining COVID-19 related mortality disparities in American Indians and Alaska Natives. Sci. Rep. 13, 20974. https://doi.org/10.1038/s41598-023-48260-9 (2023).
Sacuena, E. R. P. et al. Host genetics and the profile of COVID-19 in Indigenous people from the Brazilian Amazon: A pilot study with variants of the ACE1, ACE2 and TMPRSS2 genes. Infect. Genet. Evol. 118, 105564. https://doi.org/10.1016/j.meegid.2024.105564 (2024).
Skoczylas, S., Płoszaj, T. & Zmysłowska, A. Can the MT-CO2 gene surprise us with something? A review of variants considered as pathogenic by identifying conserved sites. Ecol. Genet. Genom. 30, 100216. https://doi.org/10.1016/j.egg.2023.100216 (2024).
Nishigaki, Y. et al. Mitochondrial haplogroup A is a genetic risk factor for atherothrombotic cerebral infarction in Japanese females. Mitochondrion 7, 72–79. https://doi.org/10.1016/j.mito.2006.11.002 (2007).
Nishigaki, Y., Fuku, N. & Tanaka, M. Mitochondrial haplogroups associated with lifestyle-related diseases and longevity in the Japanese population. Geriatr. Gerontol. Int. 10, S221–S235. https://doi.org/10.1111/j.1447-0594.2010.00599.x (2010).
Fuku, N. et al. Mitochondrial haplogroup N9a confers resistance against type 2 diabetes in Asians. Am. J. Hum. Genet. 80, 407–415. https://doi.org/10.1086/512202 (2007).
Rovcanin, B. et al. In silico model of mtDNA mutations effect on secondary and 3D structure of mitochondrial rRNA and tRNA in Leber’s hereditary optic neuropathy. Exp. Eye Res. 201, 108277. https://doi.org/10.1016/j.exer.2020.108277 (2020).
Zaki, E. A. et al. Two common mitochondrial DNA polymorphisms are highly associated with migraine headache and cyclic vomiting syndrome. Cephalalgia 29, 719–728. https://doi.org/10.1111/j.1468-2982.2008.01793.x (2009).
Boles, R. G. et al. Are pediatric and adult-onset cyclic vomiting syndrome (CVS) biologically different conditions? Relationship of adult-onset CVS with the migraine and pediatric CVS-associated common mtDNA polymorphisms 16519T and 3010A. Neurogastroenterol. Motil. 21, 936-e72. https://doi.org/10.1111/j.1365-2982.2009.01305.x (2009).
Ye, Z., Xue, A., Huang, Y. & Wu, Q. Children with cyclic vomiting syndrome: Phenotypes, disease burden and mitochondrial DNA analysis. BMC Gastroenterol. 18, 1–8. https://doi.org/10.1186/s12876-018-0836-5 (2018).
Venkatesan, T. et al. Quantitative pedigree analysis and mitochondrial DNA sequence variants in adults with cyclic vomiting syndrome. BMC Gastroenterol. 14, 181. https://doi.org/10.1186/1471-230X-14-181 (2014).
Veenin, K. et al. Association of mitochondrial DNA polymorphisms with pediatric-onset cyclic vomiting syndrome. Front. Pediatr. 10, 876436. https://doi.org/10.3389/fped.2022.876436 (2022).
Luo, Y., Gao, W., Liu, F. & Gao, Y. Mitochondrial nt3010G-nt3970C haplotype is implicated in high-altitude adaptation of Tibetans. Mitochondrial DNA 22, 181–190. https://doi.org/10.3109/19401736.2011.632771 (2011).
Liu, G. et al. Mitochondrial haplogroups and cognitive progression in Parkinson’s disease. Brain 146, 42–49. https://doi.org/10.1093/brain/awac327 (2023).
Jiang, W. et al. Mitochondrial DNA mutations associated with type 2 diabetes mellitus in Chinese Uyghur population. Sci. Rep. 7, 16989. https://doi.org/10.1038/s41598-017-17086-7 (2017).
Zhu, Y., You, J., Xu, C. & Gu, X. Associations of mitochondrial DNA 3777–4679 region mutations with maternally inherited essential hypertensive subjects in China. BMC Med. Genet. 21, 1–9. https://doi.org/10.1186/s12881-020-01045-7 (2020).
Li, C. et al. A preliminary analysis of mitochondrial DNA atlas in the type 2 diabetes patients. Int. J. Diabetes Dev. Ctries. https://doi.org/10.1007/s13410-021-01031-6 (2022).
Russlies, J. et al. Polymorphisms in the mitochondrial genome are associated with bullous pemphigoid in Germans. Front. Immunol. 10, 2200. https://doi.org/10.3389/fimmu.2019.02200 (2019).
Khouri, B. F., de Souza Candido, I. P., Poli-Frederico, R. C. & Bignardi, P. R. Host genetics and COVID-19: Genes underlying the patterns of susceptibility and prognosis. Gene Expr. 22, 222–231 (2023).
Wang, Q. et al. Rare variant contribution to human disease in 281,104 UK Biobank exomes. Nature 597, 527–532. https://doi.org/10.1038/s41586-021-03855-y (2021).
Momozawa, Y. & Mizukami, K. Unique roles of rare variants in the genetics of complex diseases in humans. J. Hum. Genet. 66, 11–23. https://doi.org/10.1038/s10038-020-00845-2 (2021).
Acknowledgements
We are deeply grateful to the research volunteers who generously consented to the use of their biological material for scientific purposes. We also thank the clinical teams responsible for patient care and sample collection during one of the most critical periods for global public health in recent history.
Funding
This work was supported by the Brazilian research funding agencies Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (Grant Nos. 162354/2022-3, 440279/2022-4, 314082/2021-2, 408154/2022-5, 406913/2022-6, and 407864/2023-7) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) (Grant No. 88881.982378/2024-01). Additionally, this study was supported by the Brazilian National Program of Genomics and Precision Health - Genomas Brasil, under the Brazilian Ministry of Health's Departamento de Ciência e Tecnologia da Secretaria de Ciência, Tecnologia e Inovação e do Complexo Econômico- Industrial da Saúde (Decit/SECTICS/MS) (Grant No. 888379/2019).
Author information
Authors and Affiliations
Contributions
G.M.T. designed the study, processed mitochondrial DNA, performed statistical and functional prediction analyses, and wrote and revised the paper. B.O.M. conducted the machine learning analysis and also wrote and revised the paper. N.A.C., R.C.S., M.F.F., G.C.G., and B.O.F. organized the clinical data, contributed to dataset construction, and performed the initial statistical analyses. M.R. performed mtDNA variant calling from whole-genome sequencing data. M.D. provided computational resources, including server access and essential tools, and revised the paper. T.H. secured funding, contributed to the genetic sequencing of samples, and revised the paper. F.S.L.V. provided clinical and genetic data and revised the paper. M.C.B. designed the study, wrote and revised the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tavares, G.M., Missaggia, B.O., Cadore, N.A. et al. Mitochondrial haplogroup A2 is associated with increased COVID-19 mortality in an admixed Brazilian population. Sci Rep 15, 22391 (2025). https://doi.org/10.1038/s41598-025-03578-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-03578-4
