
Abstract
Inland wetlands are highly diverse, productive ecosystems at the transition between terrestrial and aquatic environments, providing major services to society. They are also under high human pressure and have suffered a progressive decline in total area globally. Here, we describe the genetic diversity and demography of the endangered Lisbon arched-mouth nase Iberochondrostoma olisiponense occurring in the inland wetlands of a major river in southwestern Europe, within a context of extreme habitat changes. Our results highlight the presence of well-structured small population nuclei with evidence of sporadic gene flow at historical and contemporary time scales. Historical reconstructions suggest population isolation consistent with periods of unsuitable habitat conditions severing population connectivity, while small population sizes may be due to limited available habitat coupled to historical as well as recent restricted connectivity among river tributaries. Habitat contraction or loss due to decreased rainfall associated with climate change is expected to impact inland wetland fish regionally, raising additional conservation concerns on species with low genetic diversity and small effective population sizes as I. olisiponense. This study may serve as a proxy for taxa occurring in similar inland wetland ecosystems and may inform conservation efforts and planning regarding the expected drivers of population demographics, distribution and connectivity.
Similar content being viewed by others

Introduction
Inland wetlands are important transition zones between terrestrial and aquatic environments characterized by high productivity and biodiversity1. These include a variety of habitats such as forested and non-forested peatlands, marshes and swamps on alluvial lowlands, among others2. Overall, these ecosystems provide important services such as controlling river discharge volumes and speed, improving water quality, controlling shore erosion, providing food and habitat sources, to name just a few3,4. Indeed, they are historically important areas for human settlement as they provide access to freshwater, productive soils for agriculture and animal production, as well as means for transporting people and goods via river channels5,6. Consequently, natural inland wetlands are currently under intense human-driven pressure globally, which have led to declines in their total area while remaining mostly unprotected7,8.
The location and extent of inland wetlands are intimately dependent on river flow and sediment regimes, as well as on the physiographic/topographic features and vegetation cover of the landscape9. All these features vary considerably in space (e.g. latitude;2) as well as in time (e.g. contemporary vs. geological past). Notably, changes in global sea levels associated with glacial and interglacial cycles have profound impacts on the distribution and extent of inland wetlands10 and, therefore, on their living communities. Nevertheless, anthropogenic pressure during the last centuries has contributed to a concerning reduction in extent and/or alteration of wetlands globally11,12
Inland wetland communities, particularly those occurring in areas closest to river mouths, are expected to be influenced most strongly by the effects of marine regressions and transgressions, hydrological regime shifts (natural and man-made) and human occupation13. These areas are characterized by daily, seasonal and/or permanent flooding and are, thus, highly dynamic in nature6. Tidal freshwater wetlands are a particular case within inland wetlands, as transition zones between marine and freshwaters under daily tidal influence14. However, few studies have focused on understanding the dynamics, population structure and historical demography of inland wetland aquatic species occurring exclusively in these areas, and particularly of fish (e.g.15,16).Towards this end, we focused on a case-study fish species—the Lisbon arched-mouth nase Iberochondrostoma olisiponense—and assessed the levels and distribution of genetic diversity throughout its known range.
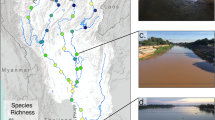
The Lisbon arched-mouth nase is a small-sized leuciscid fish occurring exclusively in inland wetlands of low altitude (generally ~ 10 m altitude, and rarely above 60 m)17, such as marshes and slow-current rivers, including tidal freshwater river sections (18,19; Ribeiro F., pers. observation; Figs. 1 and 2). The species is endemic to the lower section of the Tagus River (Portugal), one of the major rivers in the Iberian Peninsula. The Tagus has a length of ~ 1000 km from its origin in eastern Spain to the mouth in Lisbon, western Portugal, with a catchment area of ~ 81 000 km2 leading to the largest estuary in Europe20. The Lower Tagus Valley is a large alluvial plain bounded by Pleistocene terrace sediments with an extremely low gradient (< 0.24 m/km) and high seasonal flow, and thus susceptible to sea level fluctuations, saline water intrusions and extended periods of freshwater residency5,19,20. The Lisbon arched-mouth nase exhibits a narrow range and low occupancy in the lower section of the Tagus, but may be locally abundant17. Initially thought to occur only in some tributaries, it was later found to live also in the vegetated, low-current side channels in the Tagus mainstream (Fig. 1)17. The Lisbon arched-mouth nase is currently listed as Endangered by the Portuguese Red List Book18 due to the current restricted area of occurrence, including only a handful of sites17,21, and declining population trends18. Thus, this study provides important information on the structure, connectivity and genetic diversity of current population nuclei of the Lisbon arched-mouth nase to inform future conservation plans.

Typical habitat of Lisbon arched-mouth nase Iberochondrostoma olisiponense. Top panels: a freshwater marsh at the Trancão basin (38° 51′ 1.64″ N, 9° 8′ 52.78″ W) in winter (left) and summer (right, depicting a naturalized well). Bottom panels: slow-current tributary, Muge River (left panel, 39° 8′ 20.51″ N, 8°31′26.95″ W), and a tidal freshwater side channel on the Tagus mainstream (right panel, 39° 2′ 51.9″ N 8°48′12.7″ W).

Map of the lower Tagus section, in the western Iberian Peninsula (inset map), showing the 86 sites surveyed with electrofishing (green and black dots) for the Lisbon arched-mouth nase Iberochondrostoma olisiponense. Sites where species´ presence was confirmed are highlighted in green, and sites where samples for genetic analysis were obtained are circled in black contour. Sites where species´ presence was not confirmed are highlighted in black, and sites with historical records are highlighted in blue. Most sites with species´ presence (blue and green) are bounded by the 30 m altitude contour delimiting the inland wetlands in the lower Tagus section (sensu12).
Results
Exploratory data analysis
Out of the initial 89 individuals sampled and processed for DArTseq, three individuals from Muge were filtered out due to missing data > 20%. The filtered dataset, including the two I. lusitanicum specimens, is composed of 86 genotypes and 8,067 binary SNPs and 0.7% missing data. A neighbor-joining tree illustrates the genetic difference between species, and the higher genomic similarity of I. olisiponense across sampling sites except for a single individual from Muge that clusters as basal to the remaining I. olisiponense (Supplemental Figure S1). The Bayesian analysis carried out on the filtered dataset using Structure 2.3 revealed the highest delta K value for K = 3 (Supplemental Figure S2). The coefficient of membership plot identified the two I. lusitanicum individuals as belonging to a separate cluster, and showed an admixed ancestry from I. olisiponense and I. lusitanicum for the same individual from Muge at the base of I. olisiponense cluster in the neighbor-joining tree (Supplemental Figure S2 and S3). The putative hybrid individual and the two I. lusitanicum were removed from further downstream analyses.
Genetic diversity
The final dataset, including only I. olisiponense individuals, is composed of 83 genotypes, and 1,291 binary SNPs with 0.57% of missing data. Overall, genetic diversity indices were low across all sampled locations (Table 1), with the highest genetic diversity values found in Trancão and the lowest in Cabanas and Tagus main stem. The distribution of the genetic diversity in the PCoA (Fig. 3) shows that individuals from Trancão form a cluster apart from those of others locations along the PC1 axis, suggesting marked genetic divergence between downstream and upstream sites. In turn, samples from Muge form a cluster separate from those of the Tagus main stem and Cabanas along the PC2 axis, further suggesting genetic differentiation among the upstream sites. Moreover, the PCoA plot revealed the presence of two individuals from Trancão located between the two main groups of individuals along the PC1 axis.

Principal Coordinate Analysis (PCoA) plot of the Lisbon arched-mouth nase Iberochondrostoma olisiponense samples performed on SNP genotypes. 1—Cabanas (red), 2—Trancão (green), 3—Muge (blue), 4—Tagus mainstem (purple).
Genetic differentiation and population structure
FST values between pairs of sample locations were low yet significant (p < 0.001), except between Cabanas and Tagus. The highest FST values were found in pairwise comparisons including Trancão, followed by those involving Muge (Table 2). These results are consistent with the largest number of private alleles being found in Trancão, followed by Muge (Supplemental Table 1). Conversely, the number of shared alleles was highest between Cabanas and Tagus (Supplemental Table 1).
Structure detected K = 2 as the most probable number of clusters when using the Evanno’s method, whereas the standard method showed K = 4 having the highest likelihood values (Fig. 4). For all K values, Cabanas and Tagus individuals clustered together, whereas those from Muge and Trancão formed distinct clusters at K > 2. The coefficient of membership plots (Fig. 4) for K = 4 showed that two individuals from Trancão had marked admixed ancestries with predominant composition of a fourth cluster not present in high percentage in any of the sampled sites, suggestive of an unsampled source population. Interestingly, these individuals are concordant with those located away from the two main clusters found in the PCoA (Fig. 3). The presence of admixed ancestry from the Tagus/Cabanas cluster in all individuals from Muge and in some from Trancão (Fig. 4) suggests past genetic connectivity across the sampling area.

STRUCTURE analysis of the Lisbon arched-mouth nase Iberochondrostoma olisiponense samples. (a) Bar plot of the posterior probability of the coefficient of membership, each vertical line represents an individual and colours represent the inferred ancestry from K populations. Results for K = 2 to K = 4 are shown; (b) Plot of the Log posterior probability (Ln(P), red line) and Evanno’s method (Delta K, black line) vs K.
The spatially explicit model implemented in geneland with correlated allele frequencies reached convergence of MCMC and the highest likelihood value at K = 4 (Supplemental Figure S3). Individual assignments performed in geneland with K = 4 revealed that samples from Muge and Tagus/Cabanas were distributed in two separate clusters, while all individuals from Trancão except one comprised a third cluster (Fig. 5a–c). A fourth cluster included a single individual from Trancão (Fig. 5d), suggesting the presence of an unsampled population.

GENELAND results for K = 4 using the spatial model with correlated allele frequencies. Plots representing the assignment of pixels to the four clusters. The highest membership values are in light yellow and the contour lines indicate the spatial position of genetic discontinuities between populations. Cluster 1 includes all but one individual from trancão; Cluster 2 includes all individuals from Muge; Cluster 3 includes all individuals from Tagus mainstem and Cabanas; Cluster 4 includes a single individual from Trancão (lower left corner).
In the case of fineRADstructure (Supplemental Figure S4), individuals were clustered into three major groups corresponding to Trancão, Muge and Tagus/Cabanas. However, three individuals from Trancão shared high co-ancestry with two individuals from Tagus in the Tagus/Cabanas group, suggesting recent gene flow between these locations. Furthermore, some individuals in Trancão display high levels of co-ancestry, suggesting a high level of relatedness among them.
Demographic analyses
No outlier SNP locus was detected using Bayescan, thus all SNP loci in the final dataset were used in the demographic analyses. The population effective size results showed reliable confidence intervals except for the Muge sample when using the jackknife method, where an infinite upper interval was obtained. The lowest Ne value was detected in Trancão, although in general all the samples showed low Ne estimates ranging from ~ 100 for Tagus/Cabanas and Muge to ~ 30 for Trancão (Table 3). All estimates had similar values when using different values of Pcrit, suggesting that minor allele frequencies have little influence on the Ne estimates. Historical demographic reconstructions based on gadma highlighted a split between the Trancão and Tagus populations at about 16,000 years ago (Fig. 6). While the obtained divergence time may not be precise, since the mutation rate used was estimated for other taxa, we do not expect it to differ by orders of magnitude and the divergence time should fall within a similar time scale to the one estimated here. The simulations indicate that the population at the time of split was centered in Trancão, followed by an exponential decrease in size until current levels, with some episodes of gene flow both upstream and downstream. The split between Muge and Tagus/Cabanas is estimated to have occurred in very recent historical times, after a period of a small increase in population size (Fig. 6).

GADMA analysis result showing a first population split between Trancão and upstream locations (Muge/ Tagus/Cabanas), and a more recent split between Muge and Tagus/Cabanas. Times is in years before present. Effective population size is depicted in blue with thickness of the lines being proportional to size. Vertical grey arrows indicate events of migration between populations, with line thickness proportional to number of migrators, and direction of arrow indicating direction of gene flow.
Discussion
Our results show marked genetic population structure in the Lisbon arched-mouth nase coupled to small effective population sizes of current population nuclei. Genetic differentiation was detected among all tributaries: the strongest divergence was observed at the site located furthest downstream (i.e., Trancão) while lower but significant genetic differentiation was found between upstream locations (i.e., Muge and Cabanas/Tagus main stem). Individuals from the Cabanas tributary were genetically indistinguishable from those sampled in the Tagus main stem suggesting connectivity between these locations. Indeed, the hypothesis that the species may occur in Cabanas only intermittently has been suggested earlier based on the lack of recruits and on the presence of adults restricted to a small area immediately adjacent to the Tagus main stem17.
Our results further suggest the presence of additional unsampled population nuclei of the Lisbon arched-mouth nase. This was most evident in Trancão where some admixed individuals had genetic ancestry not found in the remaining sampled sites (Figs. 4 and 5). This may be due to the presence of additional populations undetected during our field survey or currently extinct. Indeed, historical records of species´ presence include sites located along the lower section of the sampled area21, of which some were not surveyed here17 (e.g., lower section of Rio Maior—Vala da Azambuja) due to recurrent and continuous pollution events in these sites19. The potential for additional population nuclei and a wider spatial distribution of the Lisbon arched-mouth nase is also supported by recent records of this species in the tidal oligohaline and mesohaline sections of the Tagus mainstem, ~ 40 to 50 km upstream from the confluence with Trancão18,19. These sections have regular occurrences of other freshwater fishes, such as the cyprinid Luciobarbus bocagei and the leuciscid Pseudochondrostoma polylepis (22,23and Costa J.L., pers. communication). Indeed, some relatives within the family Leuciscidae occurring in other large estuaries in similar environments exhibit tolerance to water salinities up to 18%o (e.g.,24,25,26,27), suggesting a similar tolerance for I. olisiponense.
While current genetic connectivity appears restricted among the population nuclei detected in the Lisbon arched-mouth nase, we found evidence of sporadic gene flow at historical and contemporary time scales. Specifically, pulses of past gene flow between the diverged populations were reconstructed by GADMA (Fig. 6), and admixed individuals were found in Trancão (Fig. 4, and Supplemental Figure S4). These observations suggest that there were periods of suitable hydrological conditions in the past that allowed for long- distance movement of individuals along the lower Tagus basin. Indeed, connectivity along the Tagus main stem may have occurred during sporadic periods of high fluvial activity (i.e., high freshwater inflow) recorded in the last 10 000 yr BP28. Presently, low salinity conditions (< 10 ppt) occur during winter and last about for up to 3 weeks at CoastNet Station located ~ 4 km downstream from the Trancão river mouth. This condition creates a wide oligohaline-mesohaline section in the upper Tagus estuary, which could allow some degree of connectivity between freshwater populations. Moreover, the CoastNet Station located ~ 11 km upstream of the Trancão outlet, has low salinity values (< 10 ppt) for more than 5 months per year (data from 2019 to 2023; http://geoportal.coastnet.pt/).
Since the estimated time of speciation of the Lisbon arched-mouth nase, ~ 12.5–7.9 Myr before present (BP) in the late Miocene29, the lower Tagus basin has undergone major reworkings of its morphology and environmental conditions. Notably, the region has alternated between freshwater and brackish/saltwater conditions due to marine regressions and transgressions during glacial and interglacial periods30,31,32,33. Changes in relative sea level and sediment supply in the last 30 kyr BP have shaped the lower Tagus basin from a deep incised channel within a broad fluvial plain during the lowstand period, to the development of extensive marsh areas and a broad estuarine–marine central basin upon rapid sea-level rise34. The effects of these shifts through time may vary among species, depending on their specific habitat requirements. For instance, the Lisbon arched-mouth nase is strongly associated with lowlands of riverine habitats with slow current, soft substrate and abundant riparian and aquatic vegetation, including seasonal wetlands17. Although salinity tolerance was never tested for the Lisbon arched-mouth nase, it is likely that it does not tolerate salinities larger than 15 ppt (see above for details on other leuscicids). Such habitat requirements may have resulted in habitat tracking during sea level fluctuations, as well as in recent habitat loss/contraction driven by human-mediated pressure in the Lisbon arched-mouth nase, shaping its current genetic diversity distribution.
The current pattern of genetic differentiation in the Lisbon arched-mouth nase may comply with expectations of isolation-by-distance, with more distant populations exhibiting large genetic distances. However, the timings of population divergence reconstructed here (Fig. 6) suggest that processes occurring at evolutionary time-scales may be the main drivers of the observed genetic structure. For instance, differentiation of the most downstream population (Trancão) from those further upstream appears to have occurred first, and to date far back into the past compared to the much more recent split of the two upstream population nuclei (Fig. 6). Within the geological context of the lower Tagus section, gene flow among tributaries via the Tagus main stem was likely restricted until current sea levels were reached (< 4 000 yr BP) concomitant with floodplain and levee development in the mid- and upper- basin sections33,35. Limited among-tributary connectivity was likely felt during low-stand periods, when the Tagus was then braided river system with a steep gradient fed by fast flowing water carrying an abundant supply of coarse sediments33 and thus unsuitable to the Lisbon arched-mouth nase. In turn, salt/brackish water conditions may have extended into the lower reaches of some of the Tagus River tributaries during high-stand periods, also limiting among-tributary connectivity (e.g., Muge31,36,37).
The recent divergence of upstream populations suggests more contemporary drivers of genetic discontinuity among tributaries. Water usage has grown dramatically in the last century in the lower Tagus section and major changes in river flow dynamics have occurred associated with increase damming, river regulation and water transfer20,38,39. Seasonal floods in the lower Tagus basin during winter have decreased dramatically in frequency and intensity since 1970, associated with human-driven changes in the river channels and, more recently, construction of several reservoirs20,38,39. In small tributaries, such as Muge, former wetlands have been drained for agricultural practices or transformed into rice paddies, and former river channels have been cleared of riparian vegetation with water flow controlled through a system of small levees and/or gates (Ribeiro F., Veríssimo A., pers. observations). Such pronounced habitat changes coupled to regulation of flow regimes may contribute to a recent decrease in gene flow between tributaries and/or the river main stem.
The low occupancy and restricted distribution of the Lisbon arched-mouth nase, which occurs exclusively on alluvial floodplain and tidal freshwater habitats, may be the result of a historically limited range of suitable habitats. These lowlands are characterized by sandy and mud substrate with slow current, and abundant aquatic vegetation with some riparian trees such as willows (Salix sp.)10. Contraction and expansion of these habitats associated with global climate shifts and sea level changes occurred repeatedly during an extended time period33,35, including the age of many contemporary taxa. Such conditions could have severely limited population growth and genetic diversity in inland wetland taxa occupying riverine lowland regions near estuaries. On the other hand, it may be expected that populations may have persisted mainly in areas upstream of the recurrent brackish/saltwater influence of marine transgressions. Thus, population nuclei of the Lisbon arched-mouth nase persisting in pockets of suitable habitats may have served as the source of recruits for new populations and of migrants connecting populations in different tributaries, such as Maior and Magos13, upon re-establishment of more broadly distributed suitable environmental conditions.
The consistently small effective population size of upstream populations of the Lisbon arched-mouth nase may reflect such historically variable but generally limited availability of suitable habitats, coupled with restricted or absent connectivity among tributaries. Such conditions may have limited population size increase and expansion into the mid-section of the lower Tagus basin over the last 16 000 yr BP due to a wide marine intrusion5,35,40. Favorable conditions re-established in the more recent past (~ 4000 yr BP) may have expanded the area of suitable Lisbon arched-mouth nase habitats to those observed nowadays. During this period, fluvial connectivity among tributaries may have been restored, potentially leading to a slight increase in population size in upstream locations (i.e., Muge and Tagus/Cabanas; Fig. 6). Despite this, the extent of suitable wetland habitats in the upper section of the lower Tagus basin may have been limited by a combination of human-driven land use changes (e.g. deforestation and land reclamation for agriculture and pasture), coupled to a decrease in rainfall particularly in the last 3000 y BP5. On the other hand, continuous contraction of suitable habitat in downstream populations (e.g., Trancão) may have promoted a continuous population size decrease. Indeed, the area occupied by floodplains in the lowermost section of Tagus basin has been decreasing since the past ~ 10,000 y BP, associated with the sea-level rise and invasion of saltwater into the estuary and of brackish waters into areas of previous freshwater floodplains33. Currently, the Trancão population nucleus is located in an area of high human occupation and intensive agricultural practices, with high water demand particularly during the dry season. This often leads to severe reductions of the local wetland area with aquatic species being confined to a few man-made wells that were previously used for irrigation and are naturalizing due to lack of maintenance (Fig. 1; Santos D., Gante H., Ribeiro F., Veríssimo A., pers. observations). These semi-natural wells have dirt walls, with 3–5 m deep and have a diameter of ~ 6 m, being encircled by giant cane (Arundo donax) and halophytic plant species (Bolboschoenus sp. and Typha sp.) (Santos D., Gante H., Ribeiro F., Veríssimo A., pers. observations).
The results shown here have important conservation implications. All population nuclei detected for the Lisbon arched-mouth nase showed low genetic diversity and correspondingly small effective population sizes (Tables 1 and 3), highlighting critical conservation concerns for the species. Small populations are more vulnerable to demographic, environmental, and genetic stochasticity compared to large populations, which elevates their extinction risk41. Also, low occupancy and population isolation reduce the probability of demographic rescue, which in turn impedes gene flow, increases genetic drift and ultimately heightens the risk of local extinction42. Habitat specialists occurring at low densities and with small geographical ranges display the highest risk43. This raises concerns regarding the adaptive potential and resilience of the Lisbon arched-mouth nase populations to future environmental changes. Indeed, future climate scenarios predict non-negligible impacts in rainfall regimes for the region; specifically, both longer dry seasons and more frequent droughts are expected due to rainfall reduction during spring and fall and, thus, to decreased annual precipitation (6% per decade till 2050)44,45. These more frequent and prolonged droughts could lead to local extinctions and/or strong population bottleneck effects due to wetland desiccation and habitat contraction/loss. It remains to be determined whether other inland wetland species inhabiting the lower Tagus exhibit similar vulnerability as reported in the Lisbon arched-mouth nase. However, such patterns may be expected for habitat specialist species of inland wetlands, which should be prioritized for research and conservation efforts.
While inland wetland communities may be adapted to the natural temporal variability of their ecosystems, this is not necessarily true for the most recent past where the pace and mode of variability are not natural but human-driven7,11,12. In the last century, the Lisbon arched-mouth nase has likely contracted both its distribution and abundance, with some populations seemingly driven to extinction in recent decades17,21. Indeed, the lower Tagus drainage has been subjected to an increasing set of human-induced pressures, such as land reclamation for urban and agricultural uses, pollution events, invasive species, and water extraction for human and agriculture consumption19,20,46. Such human-driven pressures are typical of inland wetland ecosystems2,11,47, underscoring the urgent need to understand the genetic diversity, population structure and connectivity of existing populations of lowland and tidal freshwater species to inform conservation prioritization and planning. Of particular urgency is more comprehensive inventory and characterization of inland wetlands and their associated biodiversity. For instance, about 30 inland wetlands were identified in the lower Tagus section recently for which there are no data on the resident fish community19. These efforts may uncover yet unknown population nuclei of species of conservation concern, such as the Lisbon arched-mouth nase.
In summary, we show genetic differentiation among small population nuclei in an endangered fish species with an exclusive distribution on alluvial floodplains and tidal freshwaters. Historical demographic reconstructions suggest that population isolation is consistent with periods of unsuitable habitat conditions while small population sizes may be due to limited available habitat coupled to restricted connectivity among tributaries. These conditions may result from the effect of marine transgressions and regressions on hydrological flow regimes and salinity, and more recently from human intervention on the landscape. This study may serve as a reference for taxa in similar inland wetland ecosystems, and may inform conservation efforts and planning given the predicted trends of population demographics, distribution and connectivity.
Methods
Sampling
The original sampling survey was conducted throughout the lower Tagus River basin for a total of 86 sites, including the main river stem and 19 tributaries, using electrofishing11. This study and its experimental protocols were approved by the Portuguese nature conservation authority (Instituto da Conservação da Natureza e das Florestas) under permit number 410/2016/CAPT, and were carried out according to relevant guidelines and regulations. Sites with reported species´ presence (including historical records) were largely confined within lowland wetland areas (mostly < 30 m in altitude17; Fig. 2). No large barriers (e.g., dams or weirs) are present along the sampling area, besides some century-old water mills of small height (generally less than 1 m high). The Lisbon arched-mouth nase was sampled from six sites (Fig. 2) distributed across three tributaries and the Tagus main stem: Cabanas (one site, n = 22), Muge (two sites, n = 25) and Trancão (one site, n = 22), and the Tagus main stem (two sites, n = 18). Two Portuguese arched-mouth nase individuals, Iberochondrostoma lusitanicum, originating from the Muge river were also genotyped to assess putative introgression with the Lisbon arched-mouth nase, detected in previous surveys48. Fish were anesthetized with MS222 and a small clip (~ 0.5 cm2) of the pelvic fin or lower caudal lobe was sampled and stored in 96% ethanol. Upon sampling, fish were allowed to recover from anesthesia before being returned live to their original location. This study is reported in accordance with ARRIVE guidelines.
Data collection and laboratory procedures
Genomic DNA (gDNA) was extracted using a high-salt protocol modified for low yield samples following49. The final elution of gDNA was performed in 25 µl of nuclease-free water. The gDNA extractions were checked for quality by agarose gel electrophoresis, and quantified using Qubit (Invitrogen). Individual gDNA extracts were standardized to 400 ng in 20 µl and shipped to Diversity Arrays Technology (University of Canberra, Australia) for sequencing a reduced representation of the genome from double-digested restriction fragments using a DArTseq protocol. The resulting library was sequenced in an Illumina HiSeq 2500 as single-end reads of 77 nucleotides (nt), and genotyped using the proprietary DArTSoft14 pipeline. Around 30% of the samples were run as internal replicates to provide confidence levels in genotype calls.
Data analysis
Only loci with reproducibility higher than 99% were retained. Moreover, additional SNP quality control filters were applied to retain loci with < 5% missing data, keeping individuals with > 80% of SNP loci genotyped, and keeping loci with Minor Allele Frequencies (MAF) > 1%. The presence of private alleles between sites was assessed with the function gl.report.pa of the dartR 1.1.11 R package50. No monomorphic loci were detected after filtering. A global Hardy–Weinberg Equilibrium (HWE) and Linkage disequilibrium (LD) test was performed by GENEPOP 4.051 using 10,000 dememorization steps of Markov chains, 20 batches and 5,000 iterations per batch. Loci not conforming to HWE expectations were excluded from further analyses.
Exploratory data analysis on the final filtered dataset aimed at assessing overall structure among multilocus genotypes, as well as identifying putative hybrid individuals. This was performed by Principal Coordinate Analysis (PCoA) and by constructing a neighbor-joining tree using dartR 1.1.11 R package50. Genetic diversity indices, namely observed heterozygosity (Ho), gene diversity (He), allelic richness (Ar), and inbreeding coefficient (FIS) were calculated using the hierfstat R package 1.2.252. Input datafiles for the different analyses were obtained by conversion of the original genlight object with the multilocus genotypes into a Structure format file (with the function gl2structure of the dartR package), and then converted into various formats using PGDSpider v2.0.6.053.
Genetic differentiation was evaluated by means of pairwise FST values among sample collections, computed with the function gl.fst.pop of the dartR 1.1.11 R package50 and 1000 bootstraps across loci to generate confidence intervals and p-values. STRUCTURE v2.3.454 was run to assess overall population structure patterns based on inferences on the most likely number of K clusters (i.e., the probability of the data given K genetic clusters of individuals), and to estimate the probability of membership of each individual to each cluster using a Markov Chain Monte Carlo (MCMC) method. Structure 2.3 was run using sampling location information55 to improve inferences when there are small genetic differences between sample collections (as was the case here). We ran ten independent replicates for each value of K, ranging 1–5, using the admixture model with correlated allele frequencies54,56. Each run consisted of 100,000 MCMC steps with a burn-in period of 10,000 steps. For these analyses we used an approach described by55: the lambda value has been estimated by running the Structure 2.3 software for K = 2–4 allowing updates for lambda value. The optimum lambda value obtained was used to perform longer MCMC runs as described above. This strategy allowed modeling the frequency spectrum of SNPs which may be skewed towards rare alleles. The convergence of the MCMC runs was evaluated considering the extent of variation of the log probability among runs. The most likely number of clusters was inferred from both the standard method (i.e., plotting ln Pr (X|K) vs K) and the Delta K statistic57 based on a rate of change in the log probability of the data over consecutive K values. The results were averaged over multiple runs using the clummp software58 and plotted using the distruct program59.
Additionally, the spatially explicit Bayesian clustering program geneland 3.2.460 was used to further investigate genetic structure and areas of genetic discontinuity. geneland considers individual multi-locus genotype data searching for the best fit to HWE and linkage disequilibrium, but also incorporates spatial data (i.e., individual geographical coordinates) to test the assumption that populations are spatially organized. geneland was run assuming a correlated allele frequency model with spatial information, thereby accounting for putative spatial correlation of genotypes. Any genetic boundaries detected are assumed to separate K random mating subpopulations, subdividing the space similarly to the Poisson-Voronoi tessellation60,61. Ten replicate runs were performed for each K value, ranging 1–5, using 500,000 MCMC steps, a thinning interval of 500 generations, a maximum rate of Poisson process fixed to 400, and spatial coordinates uncertainty of 2.5 km. The prior for allele frequencies was modeled assuming a Dirichlet distribution62. The posterior probability of subpopulation membership was computed for each pixel of the spatial domain (400 × 400 pixels), using a burn-in of 500 iterations.
The fineRADstructure package v0.263 was used with default settings to quantify the ancestry sources in each population. FineRADstructure utilizes a fineSTRUCTURE MCMC clustering algorithm64 to infer a co-ancestry matrix, which is a summary of nearest neighbor haplotype relationships across the dataset63.
Prior to demographic analyses, non-neutral loci were screened using Bayescan version 2.165,66,67 with following run parameters: -n 5000 -thin 10 -nbp 20 -pilot 5000 -burn 50,000 -pr_odds 100. MCMC chain convergence and effective sample size were checked using the functions in the R package coda68, and putative outliers were plotted and assessed using the built-in R function plot_bayescan. Demographic inferences on the size of population nuclei of the Lisbon arched-mouth nase were made by calculating the contemporary effective population size based on the LD method (NeLD) using NeEstimator v.2.0169. Among several demographic and genetic methods available to estimate temporal and contemporary Ne, the linkage disequilibrium (LD) method70 performs best, even for small population sizes71. This method was run assuming different values of Pcrit to assess the impact of minor allele frequencies on the Ne estimates obtained.
Furthermore, GADMA72 was used to model the demographic history of the sampled populations. Like other demographic inference methods, GADMA is based on the comparison of allele frequency spectra (ASF) between a hypothetical model and the data. The difference being that GADMA automatically searches for the best model, in an unsupervised way, being only necessary to define a set of parameter constraints. Such a model assumes two sequential population splits in which we define a maximum complexity of two moments of demographic change before and after the second split and one moment before the first split. These included parameters such as population size and its variation, segregation time, and migration rates. Timings of demographic change were estimated based on a mutation rate estimated for fish ddRAD loci of 6.6 × 10–8 per site per generation73, and a generation time of 2 years. SNPs were considered to be unlinked. Model evaluation was performed with Moments74 using a down projected ASF dimensions (Trancao: 25, Tagus/Cabanas: 46, Muge: 27), which according to a test performed with easySFS (https://github.com/isaacovercast/easySFS) keeps at least 90% of the segregation sites. We performed three independent searchers using the local optimizer powell and the best model was chosen based on Akaike’s information criterion.
Data availability
Genotypes called for the 83 individuals for the Lisbon arched-mouth nase Iberochondrostoma olisiponense used in the analyses are available for download as file “Iolisiponensis_SNPgenotypes_genepop.txt” in genepop format at the link https://data.mendeley.com/preview/r4b5mdzjt9?a=d696afc8-5d2a-4e1c-962f-185d851977d7. Raw read sequences were deposited in GenBank under accession no. PRJNA1207780.
References
Keddy, P. A. et al. Wet and wonderful: The world’s largest wetlands are conservation priorities. Bioscience 59(1), 39–51. https://doi.org/10.1525/BIO.2009.59.1.8 (2009).
Davidson, N. C. & Finlayson, C. M. Extent, regional distribution and changes in area of different classes of wetland. Mar. Freshw. Res. 69(10), 1525–1533. https://doi.org/10.1071/MF17377 (2018).
De Groot, R., Stuip, M., Finlayson, C. & Davidson, N. Valuing wetlands: Guidance for valuing the benefits derived from wetland ecosystem services, Ramsar Technical Report No. 3/CBD Technical Series No. 27. ’, Montreal, Canada (2006).
Finlayson, C., D’Cruz, R. & Davidson, N. Ecosystems and human well-being: Wetlands and water. Synthesis. Millennium Ecosystem Assessment (2005).
Azevêdo, T. M. et al. Floodplain sediments of the tagus river, portugal: Assessing avulsion, channel migration and human impact. In Sedimentary Processes Environments and Basins 535–554 (Wiley, 2007). https://doi.org/10.1002/9781444304411.ch21.
Odum, W. E. Comparative ecology of tidal freshwater and salt marshes. Ann. Rev. Ecul. Sysl 19, 147–176 (1988).
Davidson, N. C., Fluet-Chouinard, E. & Finlayson, C. M. Global extent and distribution of wetlands: Trends and issues. Mar. Freshw. Res. 69(4), 620–627. https://doi.org/10.1071/MF17019 (2018).
Reis, V. et al. A global assessment of inland wetland conservation status. Bioscience 67(6), 523. https://doi.org/10.1093/biosci/bix045 (2017).
Ward, J. V. & Stanford, J. A. The serial discontinuity concept: Extending the model to floodplain rivers. Regul. Rivers Res. Manag. 10(2–4), 159–168. https://doi.org/10.1002/RRR.3450100211 (1995).
Kaplan, J. O. Wetlands at the last glacial maximum: Distribution and methane emissions. Geophys. Res. Lett. https://doi.org/10.1029/2001GL013366 (2002).
Davidson, N. C. How much wetland has the world lost? Long-term and recent trends in global wetland area. Mar. Freshw. Res. 65, 934–941. https://doi.org/10.1071/MF14173 (2014).
Fluet-Chouinard, E. et al. Extensive global wetland loss over the past three centuries. Kees Klein Goldewijk 614, 26. https://doi.org/10.1038/s41586-022-05572-6 (2023).
Čížková, H. et al. Actual state of European wetlands and their possible future in the context of global climate change. Aquat. Sci. 75(1), 3–26. https://doi.org/10.1007/S00027-011-0233-4/TABLES/2 (2013).
Baldwin, A. H., Barendregt, A. & Whigham, D. F. Tidal Freshwater Wetlands—An Introduction to the Ecosystem (2009).
Baerwald, M., Bien, V., Feyrer, F. & May, B. Genetic analysis reveals two distinct Sacramento splittail (Pogonichthys macrolepidotus) populations. Conserv. Genet. 8, 159–167 (2007).
Feyrer, F. et al. Metapopulation structure of a semi-anadromous fish in a dynamic environment. Can. J. Fish. Aquat. Sci. 72(5), 709–721. https://doi.org/10.1139/cjfas-2014-0433 (2015).
Verissimo, A. et al. Distribution and demography of the critically endangered Lisbon arched-mouth nase, Iberochondrostoma olisiponense. Fish. Mediterr. Environ. 2018, 1–13. https://doi.org/10.29094/fishmed.2018.002 (2018).
Magalhães, M. et al. Livro Vermelho dos Peixes Dulciaquícolas e Diádromos de Portugal Continental. (2023).
Ribeiro, D. ‘Inland Wetlands in the Lower Tagus: Land Uses, Habitat Condition and Fish Communities (University of Lisbon, 2023).
Gante, H. F., Santos, C. D. & Alves, M. J. A new species of Chondrostoma Agassiz, 1832 (Cypriniformes: Cyprinidae) with sexual dimorphism from the lower Rio Tejo Basin, Portugal. Zootaxa 1616, 23–35 (2007).
Cabral, H. N., Costa, M. J. & Salgado, J. P. Does the Tagus estuary fish community reflect environmental changes?. Clim. Res. 18(1–2), 119–126. https://doi.org/10.3354/CR018119 (2001).
Costa, M. J., Vasconcelos, R., Costa, J. L. & Cabral, H. N. River flow influence on the fish community of the Tagus estuary (Portugal). Hydrobiologia 587(1), 113–123. https://doi.org/10.1007/S10750-007-0690-X/FIGURES/6 (2007).
Feyrer, F., Hobbs, J. & Sommer, T. Salinity inhabited by age-0 splittail (Pogonichthys macrolepidotus) as determined by direct field observation and retrospective analyses with otolith chemistry. San Franc. Estuary Watershed Sci. https://doi.org/10.15447/SFEWS.2010V8ISS2ART2 (2010).
Ostrand, K. G. & Wilde, G. R. Temperature, dissolved oxygen, and salinity tolerances of five prairie stream fishes and their role in explaining fish assemblage patterns. Trans. Am. Fish. Soc. 130, 742–749. https://doi.org/10.1577/1548-8659(2001)130%3c0742:TDOAST%3e2.0.CO;2 (2001).
Whiterod, N. R. & Walker, K. F. Will rising salinity in the Murray-Darling Basin affect common carp (Cyprinus carpio L.)?. Mar. Freshwater Res. 57, 817–823. https://doi.org/10.1071/MF06021 (2006).
Bianco, P. G. & Nordlie, F. The salinity tolerance of Pseudophoxinus stymphalicus (Cyprinidae) and Valencia letourneuxi (Valenciidae) from western Greece suggests a revision of the ecological categories of freshwater fishes. Ital. J. Zool. 3(75), 285–293. https://doi.org/10.1080/11250000801931753 (2008).
Benito, G., Sopeña, A., Sánchez-Moya, Y., Machado, M. J. & Pérez-González, A. Palaeoflood record of the Tagus River (Central Spain) during the Late Pleistocene and Holocene. Quat. Sci. Rev. 22, 1737–1756. https://doi.org/10.1016/S0277-3791(03)00133-1 (2003).
Gante, H. F., Santos, C. D. & Alves, M. J. Phylogenetic relationships of the newly described species Chondrostoma olisiponensis (Teleostei: Cyprinidae). J. Fish. Biol. 76(4), 965–974. https://doi.org/10.1111/J.1095-8649.2010.02536.X (2010).
Pais, J. The neogene of the lower Tagus Basin (Portugal). Span. J. Palaeontol. 19(2), 229. https://doi.org/10.7203/sjp.19.2.20534 (2004).
Van Der Schriek, T., Passmore, D. G., Rolaõ, J. & Stevenson, A. C. Estuarine-fluvial floodplain formation in the Holocene Lower Tagus valley (Central Portugal) and implications for Quaternary fluvial system evolution. Quat. Sci. Rev. 26, 2937–2957. https://doi.org/10.1016/j.quascirev.2007.07.020 (2007).
Almeida, I. M. et al. Holocene paleoenvironmental evolution of the Lisbon downtown area as recorded in the Esteiro da Baixa sediments—First results. J. Coast. Res. 56, 574–578 (2009).
Vis, G. J., Kasse, C. & Vandenberghe, J. Late Pleistocene and Holocene palaeogeography of the Lower Tagus Valley (Portugal): Effects of relative sea level, valley morphology and sediment supply. Quat. Sci. Rev. 27(17–18), 1682–1709. https://doi.org/10.1016/j.quascirev.2008.07.003 (2008).
Vis, G. J. & Kasse, C. Late Quaternary valley-fill succession of the Lower Tagus Valley, Portugal. Sediment. Geol. 221(1–4), 19–39. https://doi.org/10.1016/j.sedgeo.2009.07.010 (2009).
Azevêdo, T. M. & Gonçalves, M. A. Geochemistry of core sediments from the Middle Tagus alluvial plain (Portugal) since the last glacial: using background determination methods to outline environmental changes. Environ. Earth Sci. 59(1), 191–204. https://doi.org/10.1007/s12665-009-0016-6 (2009).
Lentacker, A. Fish remains from Portugal: Preliminary analysis of the Mesolithic shell-midden sites of Cabeço da Amoreira and Cabeço da Arruda. Annales du Musée Royal de l’Afrique Centrale, Sciences Zoologiques 274, 263–271 (1994).
Lentacker, A. Preliminary results of the fauna of Cabeço de Amoreira and Cabeço de Arruda (Muge, Portugal). Trabalhos de Antropologia e Etnologia 26(1–4), 9–26 (1986).
Rodrigues, A. O Rio Tejo, Clube do Colecionador dos Correios. (CTT—Correios de Portugal, 2018).
Fernandes, M. R., Aguiar, F. C., Martins, M. J., Rivaes, R. & Ferreira, M. T. Long-term human-generated alterations of Tagus River: Effects of hydrological regulation and land-use changes in distinct river zones. Catena (Amst) 188, 104466. https://doi.org/10.1016/J.CATENA.2020.104466 (2020).
Sabater, S. et al. The Iberian rivers’. In Rivers of Europe 181–224 (Elsevier, 2022).
Gonçalves, V. S., Sousa, A. C., Texugo, A. & Ramos-Pereira, A. In the Sorraia river valley (Coruche, Portugal): Settlement dynamics of ancient peasant societies on the left bank of the Lower Tagus river (5500 to 1800 B.C.E.). Cuadernos de Prehistoria y Arqueologia de la Universidad de Granada 31, 95–158. https://doi.org/10.30827/CPAG.v31i0.21118 (2021).
Simberloff, D. The proximate causes of extinction. In Patterns and Processes in the History of Life 259–276 (Springer, 1986). https://doi.org/10.1007/978-3-642-70831-2_14.
Crisfield, V. E., Guillaume Blanchet, F., Raudsepp-Hearne, C. & Gravel, D. How and why species are rare: Towards an understanding of the ecological causes of rarity. Ecography 2024(2), e07037. https://doi.org/10.1111/ECOG.07037 (2024).
Purvis, A., Gittleman, J. L., Cowlishaw, G. & Mace, G. M. Predicting extinction risk in declining species. Proc. R Soc. Lond. B Biol. Sci. 267(1456), 1947–1952. https://doi.org/10.1098/RSPB.2000.1234 (2000).
Guerreiro, S. B., Kilsby, C. G. & Fowler, H. J. Rainfall in Iberian transnational basins: A drier future for the Douro, Tagus and Guadiana?. Clim. Change 135(3–4), 467–480. https://doi.org/10.1007/S10584-015-1575-Z/FIGURES/5 (2016).
Pambianchi, G., Gentilucci, M., Sondermann, M. N. & Proença De Oliveira, R. Climate Adaptation Needs to Reduce Water Scarcity Vulnerability in the Tagus River Basin (2022). https://doi.org/10.3390/w14162527.
Anastácio, P. M. et al. Non-native freshwater fauna in Portugal: A review. Sci. Total Environ. 650, 1923–1934. https://doi.org/10.1016/j.scitotenv.2018.09.251 (2019).
Šmejkal, M. et al. Wetland fish in peril: A synergy between habitat loss and biological invasions drives the extinction of neglected native fauna. Biol. Conserv. 302(August), 2025. https://doi.org/10.1016/j.biocon.2024.110948 (2024).
Sousa-Santos, C. et al. Evolutionary history and population genetics of a cyprinid fish (Iberochondrostoma olisiponensis) endangered by introgression from a more abundant relative. Conserv. Genet. 15(3), 665–677. https://doi.org/10.1007/S10592-014-0568-1/METRICS (2014).
Boileau, N. et al. A complex mode of aggressive mimicry in a scale-eating cichlid fish. Biol. Lett. https://doi.org/10.1098/rsbl.2015.0521 (2015).
Gruber, B., Unmack, P. J., Berry, O. F. & Georges, A. dartr: An r package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Mol. Ecol. Resour. 18(3), 691–699. https://doi.org/10.1111/1755-0998.12745 (2018).
Rousset, F. genepop’007: A complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 8(1), 103–106. https://doi.org/10.1111/J.1471-8286.2007.01931.X (2008).
Goudet, J. HIERFSTAT, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 5(1), 184–186. https://doi.org/10.1111/j.1471-8286.2004.00828.x (2005).
Lischer, H. E. L. & Excoffier, L. PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28(2), 298–299. https://doi.org/10.1093/BIOINFORMATICS/BTR642 (2012).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155(2), 945–959. https://doi.org/10.1111/j.1471-8286.2007.01758.x (2000).
Hubisz, M. J., Falush, D., Stephens, M. & Pritchard, J. K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 9(5), 1322–1332. https://doi.org/10.1111/J.1755-0998.2009.02591.X (2009).
Falush, D., Stephens, M. & Pritchard, J. K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 164(4), 1567–1587. https://doi.org/10.1111/j.1471-8286.2007.01758.x (2003).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 14(8), 2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x (2005).
Jakobsson, M. & Rosenberg, N. A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23(14), 1801–1806. https://doi.org/10.1093/BIOINFORMATICS/BTM233 (2007).
Rosenberg, N. A. distruct: A program for the graphical display of population structure. Mol. Ecol. Notes 4(1), 137–138. https://doi.org/10.1046/J.1471-8286.2003.00566.X (2004).
Guillot, G., Mortier, F. & Estoup, A. GENELAND: A computer package for landscape genetics. Mol. Ecol. Notes 5(3), 712–715. https://doi.org/10.1111/j.1471-8286.2005.01031.x (2005).
Manel, S. et al. A new individual-based spatial approach for identifying genetic discontinuities in natural populations. Mol. Ecol. 16(10), 2031–2043. https://doi.org/10.1111/J.1365-294X.2007.03293.X (2007).
Balding, D. J. Likelihood-based inference for genetic correlation coefficients. Theor. Popul. Biol. 63(3), 221–230. https://doi.org/10.1016/S0040-5809(03)00007-8 (2003).
Malinsky, M., Trucchi, E., Lawson, D. J. & Falush, D. RADpainter and fineRADstructure: Population Inference from RADseq Data. Mol. Biol. Evol. 35(5), 1284–1290. https://doi.org/10.1093/MOLBEV/MSY023 (2018).
Lawson, D. J., Hellenthal, G., Myers, S. & Falush, D. Inference of population structure using dense haplotype data. PLoS Genet. 8(1), 1002453. https://doi.org/10.1371/journal.pgen.1002453 (2012).
Foll, M. & Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 180(2), 977–993. https://doi.org/10.1534/genetics.108.092221 (2008).
Foll, M., Fischer, M. C., Heckel, G. & Excoffier, L. Estimating population structure from AFLP amplification intensity. Mol. Ecol. 19(21), 4638–4647. https://doi.org/10.1111/J.1365-294X.2010.04820.X (2010).
Fischer, M. C., Foll, M., Excoffier, L. & Heckel, G. Enhanced AFLP genome scans detect local adaptation in high-altitude populations of a small rodent (Microtus arvalis). Mol. Ecol. 20(7), 1450–1462. https://doi.org/10.1111/J.1365-294X.2011.05015.X (2011).
Plummer, M., Best, N., Cowles, K. & Vines, K. ‘CODA: Convergence Diagnosis and Output Analysis for MCMC’, R News. Accessed 08 October 08 2024. Available: https://journal.r-project.org/articles/RN-2006-002/RN-2006-002.pdf
Do, C. et al. NeEstimator v2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 14(1), 209–214. https://doi.org/10.1111/1755-0998.12157 (2014).
Hill, W. G. Estimation of effective population size from data on linkage disequilibrium1. Genet. Res. (Camb.) 38(3), 209–216. https://doi.org/10.1017/S0016672300020553 (1981).
Gilbert, K. J. & Whitlock, M. C. Evaluating methods for estimating local effective population size with and without migration. Evolution (N Y) 69(8), 2154–2166. https://doi.org/10.1111/EVO.12713 (2015).
Noskova, E., Ulyantsev, V., Koepfli, K. P., Obrien, S. J. & Dobrynin, P. GADMA: Genetic algorithm for inferring demographic history of multiple populations from allele frequency spectrum data’. Gigascience https://doi.org/10.1093/gigascience/giaa005 (2020).
Recknagel, H., Elmer, K. R. & Meyer, A. A hybrid genetic linkage map of two ecologically and morphologically divergent midas cichlid fishes (Amphilophus spp.) obtained by massively parallel DNA sequencing (ddRADSeq). G3 Genes Genomes Genet. 3(1), 65–74. https://doi.org/10.1534/G3.112.003897/-/DC1 (2013).
Jouganous, J., Long, W., Ragsdale, A. P. & Gravel, S. Inferring the joint demographic history of multiple populations: Beyond the diffusion approximation. Genetics 206(3), 1549–1567. https://doi.org/10.1534/GENETICS.117.200493 (2017).
Acknowledgements
This work was partly funded by the Mohammed Bin Zayed Species Conservation Fund #152510795, and by FEDER Funds through the Operational Competitiveness Factors Program COMPETE and by Portuguese funds through the Foundation for Science and Technology (FCT) within the scope of projects ENVMETAGENOMICS (PTDC/BIA-CBI/31644/2017) and FRISK (PTDC/AAG-MAA/0350/2014). AV was funded by FCT (DOIs:https://doi.org/10.54499/DL57/2016/CP1440/CP1646/CT0001; https://doi.org/10.54499/2022.02937.CEECIND/CP1730/CT0011). Data on current salinity values in the lower Tagus basin are freely available by the Infrastructure CoastNet (http://geoportal.coastnet.pt). The project CoastNet was supported by (PINFRA/22128/2016)”, funded by Foundation for Science and Technology (FCT) and the European Regional Development Fund (FEDER), through LISBOA2020 and ALENTEJO2020 regional operational programs, in the framework of the National Roadmap of Research Infrastructures of strategic relevance. The authors would like to thank Filipa Santos for her help with laboratory work, and Nicolas Boileau for his feedback on DNA extraction methods of low yield samples.
Author information
Authors and Affiliations
Contributions
FR, HG, CDS, MJA, AV obtained the funding; CDS, MJA, HG, FR and AV planned the study and conducted the fieldwork; AV performed the laboratory work; GR, MC, HG and AV conducted the data analysis; GR, HG and AV wrote the manuscript with significant contributions from CDS and FR; all authors have reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Riccioni, G., Curto, M., Santos, C.D. et al. Genetic structure and historical demography of inland wetland fish using the endangered Lisbon arched-mouth nase as a case-study. Sci Rep 15, 22998 (2025). https://doi.org/10.1038/s41598-025-05280-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-05280-x
