
Abstract
The viral main protease is a critical drug target due to its role in the SARS-CoV-2 life cycle. In this study, we evaluated 2,000 potential Mpro inhibitors recommended by the FragRep server, with a focus on interactions with the CYS145 residue. Six top candidates were selected for each of the 7JKV and 7TDU Mpro structures, and covalent molecular docking was performed using SeeSAR. We docked twelve ligands to various Mpro sites and performed 100 ns molecular dynamics (MD) simulations on the highest-scoring ligands: lig-14-23, lig-6-32, lig-2-3, lig-0101, lig-7612, lig-811, and lig-837. MD analysis, including Root Mean Square Deviation (RMSD), Radius of Gyration (Rg), Solvent Accessible Surface Area (SASA), and Root Mean Square Fluctuation (RMSF), confirmed the stability of the covalent complexes. Non-covalent ligands, which exhibited higher toxicity profiles, were excluded from the molecular dynamics analysis. The results indicate that lig-7612 and lig-837 form stable interactions with Mpro, suggesting their potential as drug candidates. These inhibitors exhibit pharmacological properties that could influence drug interactions and metabolism in vivo. Further safety and pharmacokinetic assessments are required for therapeutic development.
Similar content being viewed by others

Introduction
In December 2019, humanity faced an unprecedented global health crisis with the emergence of the COVID-19 pandemic. The virus responsible for this crisis, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), joined a lineage of viral threats that included severe acute respiratory syndrome coronavirus 1 (SARS-CoV-1), Middle East respiratory syndrome coronavirus (MERS-CoV), and others. As of January 2023, the world has witnessed over 638 million confirmed COVID-19 cases and the tragic loss of more than 6.7 million lives. This devastating outbreak, characterized by its rapid transmission, highlighted the need for innovative approaches to confront highly adaptable pathogens like SARS-CoV-21,2,3,4.
Within the family of Coronaviridae, SARS-CoV-2 belongs to the Betacoronavirus genus. It is a positive-sense single-stranded RNA virus known for its complex structure and infection mechanism. Despite remarkable progress in vaccine development, the virus’s capacity to mutate continuously poses challenges to the effectiveness of existing vaccines. Thus, the search for effective antiviral drugs and drug targets remains a pressing imperative5,6,7,8,9.
Previous research on related coronaviruses, such as SARS-CoV-1 and MERS-CoV, has illuminated the potential of specific viral proteins as viable drug targets. Among these proteins, the main protease (Mpro), also referred to as 3 C-like protease (3CLpro), stands out as a pivotal enzyme in viral replication. Mpro plays a key role in cleaving polypeptide sequences following glutamine residues, a function that lacks a human protease counterpart. This unique attribute, coupled with its central role in virus replication, positions Mpro as an attractive target for drug development10,11,12,13.
SARS-CoV-2 Mpro boasts a structural complexity characterized by three domains—domains I, II, and III. These domains form a β-barrel structure and α-helical clusters, with the substrate-binding site nestled between domains I and II. This binding site hosts the catalytic dyad essential for hydrolysis, while domain III contributes significantly to enzyme dimerization. Understanding the intricacies of Mpro, including its dimerization and the importance of the N-finger orientation in substrate specificity, underscores the multifaceted nature of its role in viral replication14,15,16,17.
Mpro functions as a cysteine protease and utilizes a unique catalytic dyad consisting of Cys145 and His41, along with an oxyanion hole formed by the main-chain amide NH groups of Gly143, Ser144, and Cys145. The evolution of the oxyanion hole serves to stabilize negatively charged tetrahedral intermediates during the enzymatic reaction pathway facilitated by Mpro. Moreover, catalysis is believed to involve a conserved water molecule, forming hydrogen bonds with His41, His164, and Asp187. These residues, along with Arg40 and Tyr54, constitute a partially negatively charged cluster, hypothesized to act as the third catalytic residue. The Mpro active site accommodates substrates or inhibitors with specific chemical groups at positions P5 through P3’ to occupy substrate-binding subsites S5-S3’. Notably, S1 selectively binds Gln, while S2 prefers medium-sized hydrophobic residues like Leu or Phe. Inhibiting Mpro has been approached through two major strategies: noncovalent inhibitors that interact with the enzyme’s active site via hydrogen bonding and hydrophobic interactions, and alternatively, utilizing electrophilic groups (known as warheads) to exploit the nucleophilic nature of Cys145 thiolate, forming an additional covalent bond with the enzyme to enhance inhibitor binding affinity compared to noncovalent molecules18.
This research paper focuses on covalent drug design targeting SARS-CoV-2 Mpro. Covalent inhibitors have gained prominence due to their potential advantages over non-covalent counterparts. These advantages include heightened selectivity, reduced off-target effects, and enhanced efficacy. Covalent inhibitors leverage the electrophilic nature of covalent bonds to address some of the limitations associated with non-covalent drugs19,20,21,22.
Covalent and non-covalent inhibitors each offer unique advantages and disadvantages in drug design. Non-covalent inhibitors provide flexibility in drug design, are not tied to specific inhibition sites, and form non-permanent interactions with proteins. They allow for ease of identifying binding sites, modulation of inhibition by altering drug structure, lower risk of drug toxicity, and access to large compound libraries. However, they tend to exhibit lower reactivity, limited binding affinity, lower potency, and comparatively lower selectivity23,24,25.
On the other hand, covalent inhibitors can be administered at lower doses, offer higher potency, longer duration of inhibition, and are less sensitive to pharmacokinetic parameters. They provide higher selectivity, higher biochemical efficiency, lower risk of drug resistance, and the ability to target previously undruggable proteins. However, they may result in unforeseen toxicity or hypersensitivity reactions, lead to drug-induced toxicity, carry a risk of generating immunogenic target adducts, and require potent or activated nucleophiles. Covalent inhibitors also depend on accessible nucleophiles and may not be compatible with targets undergoing rapid enzymatic turnover or degradation26,27,28,29.
The mechanism of action of original covalent drugs remained unclear for a long time. Aspirin, a covalent drug approved by the World Health Organization, illustrates the concept. Aspirin exhibits anti-inflammatory and analgesic properties by irreversibly acetylating cyclooxygenase enzymes. These enzymes are essential catalysts in the formation of pro-inflammatory prostaglandins. Covalent inhibitors are divided into two categories: reversible and irreversible, depending on their binding energy. Irreversible covalent bonds have higher binding energy30,31.
Various inter- and intramolecular chemical reactions can break covalent bonds, including water, changes in environmental pH values, and specific enzymes. Covalent drugs feature mildly reactive functional groups that form covalent bonds with protein targets, enhancing their affinity beyond non-covalent interactions. Early covalent drugs, such as aspirin and penicillin, were discovered serendipitously and targeted active sites to inhibit enzymatic activity32,33.
In this study, we employ computational tools to design covalent drugs aimed at combating SARS-CoV-2 through fragment replacement in potent compounds.
Materials and methods
Receptor structure Preparation
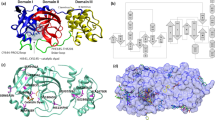
To improve the reliability of docking outcomes and better capture the conformational variability of the Mpro binding site, we first screened a list of SARS-CoV-2 main protease crystal structures available in the RCSB (Research Collaboratory for Structural Bioinformatics) website. Two protein structures of the SARS-CoV-2 Mpro, with the Protein Data Bank (PDB) accession codes 7JKV and 7TDU, were obtained from the RCSB website (Fig. 1). Preparing the protein structures involved several steps: adding hydrogen atoms, completing missing side-chains, establishing potential disulfide bridges, and eliminating water molecules. Small ligands situated within the protein’s active site and dimethyl sulfoxide (DMS) from the crystallization buffer were excluded. Subsequently, the resultant protein structure underwent further energy minimization using Chimera 1.17.1 software. Visualization of both protein structures was accomplished using Chimera 1.17.1 software and Discovery Studio 2021.

The structure of the 7JKV and 7TDU proteins with two ligands attached. The amino acids His-41 and Cys-145 are shown in space-filling models.
Ligand selection
In this study, the covalent hybrid inhibitors (BBH-1, BBH-2, NBH-2, YH-53, 5 h, WU-04, and S-217622) were used by reviewing the literature on hepatitis C and SARS-CoV-1 protease inhibitors. These inhibitors exhibit potent antiviral properties against SARS-CoV-2, with YH-53, 5 h, WU-04 and S-217,622 showing remarkable efficacy. X-ray structure analyses elucidate their mechanisms of action, revealing interactions such as covalent bonding and polar interactions with target proteins. The compounds show promising results in in vitro and in vivo studies, positioning them as potential candidates for advanced antiviral development18,34,35,36.
For drug design, we used the FragRep web server, a pivotal tool in structure-based ligand design through fragment replacement. FragRep, with its user-friendly interface, employs a fragment replacement strategy that considers both geometric requirements and compatibility with the local protein environment. The server’s systematic approach ensures the selection of fragments that not only meet structural feasibility criteria, but also fit seamlessly into the local protein environment. The FragRep Web Server, freely available at http://xundrug.cn/fragrep. From the output of the FragRep server, which included suggestions for at least 2000 inhibitors targeting the 7JKV and 7TDU structures, we selectively identified compounds with the most favorable binding affinities. Default parameters of SeeSAR were maintained throughout the docking process. The resulting file was saved in .PDB format and evaluated using SeeSAR37.
Site prediction and druggability analysis of SARS-CoV-2 Mpro
The prepared protein structures were exported as PDB files and analyzed using the “Binding Site” tool within the BioSolveIT Suite. Site predictions and druggability analysis were performed using the Difference of Gaussian (DoG) site pocket finder. DoGSite detects potential binding pockets and sub-pockets of the protein and analyzes their geometric and physico-chemical properties, estimating druggability with a support vector machine (SVM).
Covalent docking on SARS-CoV-2 Mpro
After ligand selection by Fragrep, covalent docking was performed using SeeSAR software, a platform that enables the identification of covalent binding sites and various key points. Upon loading the protein structure and defining the ligand, SeeSAR detects potential ligands that covalently bind to the structure, recognizes attachment points, and aligns them with the binding site by selecting the covalent ligand.
SeeSAR provides an advanced framework called CovXplorer, which allows users to evaluate docking poses for binding affinity and advance potential candidates to subsequent steps. CovXplorer also interprets and converts warheads into their ligand-binding form, integrating over 30 established covalent warheads into the workflow while allowing for the easy addition of new ones. The software visualizes binding modes within SeeSAR, presenting comprehensive molecular insights such as ligand parameters, molecular torsions, lipophilic and overall ligand efficiencies, intra- and intermolecular collisions, and specific atomic interactions within the complex38.
Predicting pharmacokinetic characteristics
After an acceptable docking score was obtained, the pharmacokinetic specifications, physicochemical attributes and drug-like properties of high binding affinity inhibitors were evaluated using SwissADME (http://www.swissadme.ch/) and pkCSM server (https://biosig.lab.uq.edu.au/pkcsm/prediction). These platforms are designed to facilitate the calculation of critical parameters for single or multiple molecules and provide a user-friendly interface for easy input and interpretation of results.4041
Toxicity assessment
Toxicity assessment was conducted using the eMOLTOX webtool (http://xundrug.cn/moltox), analyzing various components within the drug to predict potential toxicities. This assessment aids in determining the drug’s safety for human use alongside its therapeutic effectiveness. The eMolTox web tool serves as a tool for anticipating potential toxicity linked to a specific molecule, leveraging extensive data from in vitro and in vivo experiments in toxicology for model development. It employs Mondrian conformal prediction to gauge the confidence level of these predictions41.
Molecular dynamics (MD) simulations on SARS-CoV-2 Mpro complexes
In this study, molecular dynamics (MD) simulations were used to investigate atomic behavior, evaluate structural stability, and analyze conformational changes at the atomic level. The top-ranked compounds (MD simulation did not include non-covalent docking compounds due to their unfavorable toxicity characteristics compared to covalently docked ligands.) and the crystal structure of the Mpro enzyme were subjected to MD simulations using GROMACS v5.0 with the GROMOS96 54a7 force field and the SPC216 water model, with a time step of 1 fs for 100 ns and with cutoff distances of 12 Å for van der Waals interactions. The systems were neutralized with 0.15 M NaCl and solvated using the TIP3P water model. The systems were then embedded in a rectangular water box with an edge distance of 10 Å.
The systems were minimized in 50,000 steps, followed by equilibration for 100 ps under constant number [N], constant volume [V], and constant temperature [T] (NVT). The Berendsen thermostat algorithm was used during NVT equilibration to maintain a constant volume for 100 ps and a constant temperature of 310 K. Finally, the production step was performed under constant number [N], constant pressure [P], and constant temperature [T] (NPT). The NPT equilibration was performed at a constant pressure of 1 bar for 100 ps, controlled by the Parrinello-Rahman barostat42. The Particle Mesh Ewald approximation with a 1 nm cutoff was used to calculate the long-range electrostatic interactions, including Coulomb and van der Waals interactions. After equilibration, a 100 ns simulation run was performed with default parameters, saving coordinates at every 2 fs time frame43,44.
Built-in ‘gmx’ commands within GROMACS were used to calculate parameters including Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuation (RMSF) to assess stability and residual fluctuations, Radius of Gyration (Rg) to assess compactness, Hydrogen Bonding analysis for neighboring interactions, and Solvent Accessible Surface Area (SASA) calculation.
MM/GBSA binding free energy analysis and Residue-Specific contributions
The gmx_MMPBSA program (version 1.4.2) was employed to estimate the MM/GBSA (Molecular Mechanics/Generalized Born Surface Area) binding free energy of the complex system during the final 10 ns of molecular dynamics (MD) simulation. The MM/GBSA binding energy is determined by aggregating various interactions and is represented as:
The terms G_complex, G_receptor, and G_ligand represent the Gibbs free energy of the ligand-protein complex, protein, and unbound ligand, respectively. ΔH corresponds to the enthalpy of binding, while -TΔS accounts for the conformational entropy change after ligand binding.
Here, ΔEMM = (ΔEbond + ΔEangle + ΔEdihedral) + ΔEele + ΔEvdW, represents the molecular mechanical energy changes in the gas phase, encompassing alterations in internal energies (ΔE_int for bond, angle, and dihedral energies), electrostatic energies (ΔE_ele), and van der Waals energies (ΔE_vdW).
Also, ΔG_sol is determined by the summation of polar contribution (electrostatic solvation energy calculated using the Generalized Born model) and non-polar contribution (ΔE_surf estimated using solvent-accessible surface area) between the solute and the continuum solvent45,46.
Furthermore, the investigation into the free energy contribution per residue was conducted to elucidate the involvement of specific amino acid residues and the ligand in the interaction.
Results
Inhibitor selection for Mpro
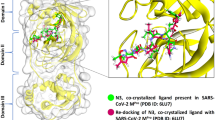
We screened a set of 2000 Mpro inhibitors suggested by the FragRep server (Fig. 2; Supplementary Table S1). Several of these compounds showed interactions with the catalytic residue CYS145. Candidate compounds were selected based on relatively high binding scores and favorable interactions within the S1, S2, S3, and S4 subsites of Mpro, as observed by visual inspection.

FragRep’s selected inhibitor candidates for 7JKV and 7TDU.
In addition, reference affinity ranges reported for known covalent and non-covalent Mpro inhibitors were considered. Ligands with estimated binding affinities below approximately 1,000,000 nM for non-covalent interactions and below 10,000,000 nM for covalent interactions were included based on previously studied compounds such as boceprevir, narlaprevir, and PF-00835231.
Using these criteria, we selected six candidate compounds each for the 7JKV and 7TDU Mpro structures for further analysis.
Covalent molecular Docking of the ligands with Mpro
To date, there is no approved drug or vaccine specifically targeting the SARS-CoV-2 main protease. Molecular docking serves as an exploratory method, utilizing existing antiviral drugs to assess their potential efficacy against SARS-CoV-2. Notably, the current strategy for designing small-molecule inhibitors of SARS-CoV-2 Mpro has resulted in the development of PF-07321332 (nirmatrelvir, Paxlovid™, Pfizer Inc.), an orally bioavailable compound. This reversible covalent peptidomimetic inhibitor leverages the nucleophilic properties of the catalytic Cys145 thiolate and incorporates structural elements from boceprevir, GC-376, and previous inhibitors of SARS-CoV-1 Mpro. However, the ongoing design of novel Mpro inhibitors, encompassing both covalent and noncovalent compounds, remains imperative in the fight against COVID-19 and for preparedness against potential future coronavirus outbreaks. A refined inhibitor design approach should take into account not only conventional hydrogen bonds but also unconventional intermolecular interactions, including C-H···O(N) and X-H···π contacts (where X = O, N, S, or C62), involving Mpro residues. Thus, the significance of hydrogen atoms, constituting approximately half of the total atom count in both small molecules and proteins, cannot be underestimated18.
The results of the binding affinity assessment for the selected ligands, drawn from a comprehensive literature review, are presented in supplementary table S1. These ligands, namely BBH-1, BBH-2, NBH-2, YH-53 (also known as 5 h), WU-04, S-217,622, and CCF0058981, demonstrate varying levels of affinity towards their respective targets. BBH-1, derived from Boceprevir, displayed a binding affinity ranging around 24032.113176 nM, while BBH-2 and NBH-2 exhibited affinities around 298984.956851 nM and 65862221.643120 nM, respectively, both linked to Boceprevir and Narlaprevir. YH-53 (or 5 h), associated with PF-00835231, showcased an affinity near 199432.029821 nM (supplementary table S1).
The designed ligands were subjected to covalent molecular docking with Mpro. Using a generated grid box, 12 selected ligands were docked to different sites within Mpro. The obtained docking results for all 12 ligands are shown in Fig. 3.


Docking of Covalent Inhibitors with 7JKV and 7TDU. Visualize the 3D structure of both the ligand and the protein, as well as the presence and status of hydrogen and van der Waals bonds, using Discovery Studio software.
Our designed covalent ligands (lig-F837, lig-F802, lig-F807, lig-F811, lig-F7612, and lig-F101) exhibit similarities and distinct features in their binding characteristics when compared to PF-00835231. Similar to PF-00835231, all of our ligands form covalent bonds with Cys145 with comparable bond distances (1.79–1.88 Å). However, several of the ligands (lig-F811, lig-F7612, and lig-F101) demonstrate dual covalent interactions with Cys145, which may provide more stable anchoring.
The hydrogen bonding patterns show some differences. PF-00835231 interacts with His163, Glu166, and Phe140; however, our ligands consistently form hydrogen bonds with His163 (1.77–1.82 Å) and Glu166 (1.79–2.13 Å) but not Phe140. Some ligands (lig-F802, lig-F811, and lig-F101) show particularly short contacts with Gln189 (1.56–1.70 Å), suggesting stronger electrostatic interactions than those observed in PF-00835231.
The hydrophobic interactions appear more varied. PF-00835231 uses an isobutyl group at P2 to stack with His41 and other residues. In contrast, our ligands primarily interact with His164 (2.00–2.06 Å) and Gln189. Several of our ligands (lig-F837 and lig-F802) demonstrate potential halogen bonding with Gln189 (1.50–1.56 Å), which has not been reported for PF-00835231.
Additional polar interactions with Asn142 and Glu146, along with consistent dual contacts with Glu166, suggest that our compounds may achieve comparable or improved binding through alternative interaction networks.
These differences in interaction patterns suggest that our ligands may have alternative binding mechanisms that are worth investigating further, especially with regard to resistance profiles and selectivity. The presence of various interaction types, such as covalent, hydrogen bonding, hydrophobic, and potential halogen bonding, across our ligand series indicates that they may serve as effective Mpro inhibitors47.
MD simulation analysis
The MD simulation study aimed to validate the molecular docking results obtained using the static crystal structure of SARS-CoV-2 Mpro. By employing molecular dynamic simulation, the dynamic behavior of the receptor and ligand interactions was explored to ascertain stable binding conformations. lig-14-23, lig-6-32, lig-2-3, lig-0101, lig-7612, lig-811, lig-837, identified with higher covalent docking among the screened compounds, were subjected to a 100 ns molecular dynamic simulation in conjunction with SARS-CoV-2 Mpro. Utilizing the simple point charge (SPC) water model, the simulation allowed an analysis of parameters such as Root Mean Square Deviation (RMSD), Hydrogen bonds, radius of gyration (Rg) Solvent Accessible Surface Area (SASA), and Root Mean Square Fluctuation (RMSF).
RMSD analysis
The RMSD analysis shown the stability of the protein backbone in complex with the specific ligand throughout the dynamic simulation. A lower RMSD value across the trajectory implied greater stability of the protein-ligand complex, while a higher RMSD value indicated relatively lower stability. RMSD analysis revealed that the mean RMSD values for all 100 ns simulations were approximately 1.9 Å. Among the ligands, lig-6-32 exhibits the lowest RMSD, showed stability practically in its entire trajectory, not exceeding the average value of 1.4 Å. Subsequently, lig-811 displays the next lowest RMSD, followed by lig-14-23 and lig-7612. Lig-837 demonstrates a slightly higher RMSD, while lig-0101 possesses the highest RMSD. The RMSD fluctuates over time, indicating that the complexes are not static but are constantly moving. However, the fluctuations are relatively small for all four ligands, suggesting that the complexes are relatively stable. However, the fluctuations are relatively small for all four ligands, suggesting that the complexes are relatively stable (Fig. 4a).

(a) RMSD Plot of the time-dependent Protein-ligand for lig-14-23, lig-6-32, lig-2-3, lig-0101, lig7612, lig-811 and lig-837 ligands interacting with the binding pocket of the 7jkv and 7TDU structures of SARS-CoV-2 Mpro enzyme. (b) ) RMSF Plot of the time-dependent Protein-ligand for lig-14-23, lig-6-32, lig-2-3, lig-0101, lig7612, lig. 811 and lig-837 ligands interacting with the binding pocket of the 7jkv and 7TDU structures of SARS-CoV-2 Mpro enzyme.
RMSF analysis
The flexibility of amino acids within the active site of the receptors concerning the selected ligands was investigated over a 100 ns duration. Figure 4b illustrates the RMSF plot for lig-14-23, lig-6-32, lig-2-3, lig-0101, lig-7612, lig-811, and lig-837 ligand complexes with receptors. While the RMSF plots exhibit overall similarity; however, the plots for the lig-14-23 and lig-0101 complexes display numerous residual fluctuations during their interactions with the receptors.
Rg analysis
Figure 5a illustrates the radius of gyration (Rg) plot for lig-14-23, lig-6-32, lig-2-3, lig-0101, lig-7612, lig-811, and lig-837 ligands engaging with the binding pocket of the SARS-CoV-2 Mpro enzyme’s 7JKV and 7TDU structures. Rg serves as a metric for assessing the compactness of macromolecules and functions as a crucial equilibrium criterion for the system. The lig-7612 (black) and lig-0101 (gray) demonstrate an increase in Rg compared to other conformations. This increase continues until 50 ns and 90 ns of simulation time, respectively, after which the Rg values stabilize and reach convergence (Fig. 5a).

(a) Rg Plot of the time-dependent Protein-ligand for lig-14-23, lig-6-32, lig-2-3, lig-0101, lig7612, lig-811 and lig-837 ligands interacting with the binding pocket of the 7jkv and 7TDU structures of SARS-CoV-2 Mpro enzyme (b) SASA Plot of the time-dependent Protein-ligand for lig-14-23, lig-6-32, lig-2-3, lig-0101, lig7612, lig-811 and lig-837 ligands interacting with the binding pocket of the 7JKV and 7TDU structures of SARS-CoV-2 Mpro enzyme.
SASA analysis
Solvent Accessible Surface Area (SASA) refers to the surface area of a protein that interacts with its surrounding solvent molecules. Throughout the 100 ns molecular dynamics (MD) simulations, the average SASA values for lig-14-23, lig-6-32, lig-2-3, lig-0101, lig-7612, lig-811, lig-837 complexe with 7jkv and 7tdu, were monitored. Notably, no significant changes in SASA values were observed as a consequence of ligand binding. Following the initial period, SASA values exhibited fluctuations around a consistent baseline. This led to the assumption that the 100 ns simulation duration was adequate for sampling equilibrated systems. It is noteworthy that the lig-837 ligand exhibited the highest SASA value among the ligands studied (Fig. 5b).
Hydrogen bonds
Hydrogen bonds play a pivotal role in protein-ligand interactions, exerting influence over drug affinity, specificity, adsorption, and metabolism. Consequently, the number of hydrogen bonds within the protein-ligand complex was scrutinized throughout the 100 ns trajectory. Notably, lig-7612 and lig-837 exhibited the highest count of hydrogen bonds over the 100 ns duration, signifying a robust and sustained interaction with the receptors (Fig. 6).

Hydrogen bonds Plot of the time-dependent Protein-ligand for lig-14-23, lig-6-32, lig-2-3, lig-0101, lig7612, lig-811 and lig-837 ligands interacting with the binding pocket of the 7jkv and 7TDU structures of SARS-CoV-2 Mpro enzyme.
Free energy landscape analysis
The GROMACS gmx sham tool was employed to conduct analysis of the Free Energy Landscape (FEL) in order to evaluate conformational changes in the protein. A Gibbs free energy landscape analysis was executed utilizing the two principal components, namely RMSD and Rg. In the resulting Gibbs Energy landscape analysis (Fig. 7 and Supplementary Figure S1), color gradations range from blue to red, with blue denoting the highest stability state of the protein and red indicating a lower stability state. The graphical representation illustrates that the binding interaction reaches its most stable state at an RMSD of approximately 0.15 nm for lig-837. This point signifies the optimal binding orientation of the drug and protein target. Similarly, for lig-7621, the binding interaction its most stable state at an RMSD of approximately 0.18 nm.
From the outcomes of the free energy contribution per residue analysis (supplementary Figure S1 and S2), it can be inferred that among the amino acid residues within the 7jkv receptor, Met165 exhibited the highest energy contribution value, followed by Gln189. Moreover, in lig-837, Met165 demonstrated the highest energy contribution value, followed by Met49. In the case of lig-7612, Glu166, His163, and Met165 displayed the greatest energy contribution values.

Free Energy Landscape of 7jkv, lig-837 and lig-7612.
Pharmacokinetic properties analysis
We evaluated the pharmacokinetic properties of several ligands for 7JKV using various parameters. Ligands F802, F807, F811, F837, F101, and F7612 were assessed for their molecular weight, logP (lipophilicity), topological polar surface area (TPSA), molecular formula, rotatable bonds, hydrogen bond acceptors and donors, molar refractivity, gastrointestinal (GI) absorption, blood-brain barrier (BBB) permeability, P-glycoprotein (P-gp) substrate status, inhibition potential for various cytochrome P450 (CYP) isoforms, skin permeation, synthetic accessibility, and toxicity measures such as AMES toxicity, maximum tolerated dose, hERG inhibition, and toxicities in aquatic organisms like minnow and Tetrahymena Pyriformis. Among these ligands, F807 exhibited the highest affinity, with a molecular weight of 645.756, a logP value of 4.57438, and a TPSA of 181.87 Ų. It showed low GI absorption and no BBB permeability or P-gp substrate characteristics. F101 displayed the lowest skin permeation potential (-7.84 cm/s), whereas F7612 presented the highest synthetic accessibility with a score of 6.95 (Supplementary Table S2).
Among the evaluated ligands of 7TDU, 2–3 showed remarkable pharmacokinetic properties (Supplementary Table S3). Ligand 2–3 shows higher affinity than the others, indicating a potentially stronger binding interaction. However, 2–3 shows better properties in terms of logP and rotatable bonds, but relatively lower intestinal absorption compared to 5–8. All ligands have interactions with CYP enzymes and are identified as P-glycoprotein substrates. Ligand 2–3, with its higher affinity and certain favorable pharmacokinetic properties, may be more promising for further consideration despite its comparatively lower intestinal absorption.
Toxicity prediction for covalent ligands
The toxicity profiles of covalent ligand including: F802, F807, F811, F837, F101, and F7612 reveal diverse and complex actions across various physiological systems and biological pathways. These compounds exhibit cytotoxic effects on different cell lines, indicating potential adverse impacts on organs such as the kidney, heart, and liver. Additionally, they interact with specific signaling pathways, including the farnesoid-X-receptor (FXR) and androgen receptor (AR) pathways, with potential implications for liver function, endocrine, and central nervous systems. Furthermore, the activation of the heat shock response signaling pathway and DNA damage induction through H2AX agonism suggest broader cellular effects. The structural features of F101 raise alerts for potential toxicological concerns (Supplementary Table S4).
Discussion
Mpro plays a crucial role in the SARS-CoV-2 replication cycle by cleaving the translated polyprotein chain at 11 distinct regions labeled A-K. Inhibiting Mpro activity effectively halts viral replication. Through sequence analysis of peptides cleaved by Mpro, a consistent pattern emerges. Cleavage by Mpro consistently occurs between a glutamine residue (P1) and either a serine, alanine, or asparagine residue (P1’). Furthermore, the critical glutamine residue in the substrate peptide is consistently preceded by a bulky nonpolar residue such as valine, leucine, or phenylalanine (P2). Several residues within the active site, including Thr24, Thr26, Asn142, Asn143, His163, His164, Glu166, Gln189, and Thr190, form hydrogen bonds with the peptide. These residues act as anchors, facilitating substrate peptide recognition and resulting proper alignment and orientation within the active site. This arrangement results close contact between the target glutamine residue and the catalytic Cys145 residue. Literature suggests that residues within the P4-P1’ range are crucial for peptide recognition and anchoring. The catalytic reaction is facilitated by a Cys-His dyad. The His41 residue activates the nucleophilic character of Cys145, leading to the covalent attachment of the peptide substrate to the enzyme. This reaction intermediate is subsequently hydrolyzed, releasing the reaction product from the active site. Mpro has emerged as a validated and prominent antiviral drug target, with Mpro inhibitors demonstrating potent antiviral activity in cell cultures and animal models. Notably, Pfizer has advanced two Mpro inhibitors, PF-07304814 and PF-07321332, to phase I clinical trials.4950515253
In this study, we first evaluated a pool of 2000 Mpro inhibitors recommended by the FragRep server, focusing on their interactions with CYS145 of Mpro. We identified top candidates based on their favorable interactions within the S1, S2, S3, and S4 binding sites of Mpro. We then selected six top-ranked candidates for each of the 7JKV and 7TDU structures for further analysis. Following this selection process, the selected ligands were subjected to covalent molecular docking with Mpro. In this process, 12 selected ligands were docked to different sites within Mpro using the SeeSAR software, maintaining default parameters. MD simulations were then performed to validate the stability of the ligand-Mpro complexes. Ligands with higher docking scores, namely lig-14-23, lig-6-32, lig-2-3, lig-0101, lig-7612, lig-811, and lig-837, underwent 100 ns simulations in conjunction with SARS-CoV-2 Mpro. The simulations allowed the analysis of various parameters, including Root Mean Square Deviation (RMSD), Radius of Gyration (Rg), Solvent Accessible Surface Area (SASA), and Root Mean Square Fluctuation (RMSF). Analysis of the MD simulation results revealed the stability of the protein backbone in complex with the specific ligands throughout the dynamic simulation. In particular, lig-6-32 exhibited the lowest RMSD, indicating stability throughout its trajectory. This relatively low deviation indicates that lig-6-32 maintains a consistent binding conformation within the active site. This consistent binding may enhance inhibitory efficacy by preventing frequent dissociation or repositioning. In addition, lig-811, lig-14-23, and lig-7612 also exhibited low RMSD values, suggesting relatively stable complexes. We calculated the Rg value to evaluate changes in Mpro’s structural compactness upon ligand binding. Throughout the simulation period, most of the ligand-Mpro complexes maintained stable Rg values, which indicates that their overall structural integrity was preserved. Rg analysis revealed lower, more consistent Rg values for lig-7612 and lig-0101, suggesting a more compact and potentially stable structure. Hydrogen bond analysis highlighted lig-7612 and lig-837 as having the highest number of hydrogen bonds over the duration of the simulation, indicating robust and sustained interactions with the receptors. These stable hydrogen bonding patterns could improve ligand retention within the binding site. This would prolong the inhibitory action and increase the binding affinity. RMSF analysis revealed residual fluctuations during ligand-receptor interactions, particularly notable in lig-14-23 and lig-0101 complexes. These higher fluctuations, particularly in residues adjacent to the active site, may indicate local flexibility that could aid in better accommodation or result in reduced binding precision. This could potentially influence inhibitory performance.
In addition, the free energy landscape (FEL) analysis evaluated the conformational stability of the ligand-Mpro complexes. The results showed that lig-837 and lig-7621 exhibited optimal stability at specific RMSD values. These energy minima suggest that the ligands adopt favorable binding modes during simulation, indicating a potential for sustained interaction with the target site.
Conclusion
Our findings underscore the potential of these ligands as inhibitors against SARS-CoV-2, which may aid in further therapeutic development. By elucidating the structural and dynamic properties of ligand-Mpro complexes, our study contributes to ongoing efforts to identify effective treatments for COVID-19 and future coronavirus outbreaks. In addition, the pharmacokinetic properties and toxicity predictions of the identified ligands may offer hepatotoxicity, genotoxicity and immunotoxicity risks at toxic doses, so the need for further research and in vitro and in vivo studies essential to effectively target SARS-CoV-2 and address the COVID-19 pandemic.
Data availability
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
References
Hattori, S. et al. A small molecule compound with an Indole moiety inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat Commun 12, (2021).
Zhu, N. et al. A novel coronavirus from patients with pneumonia in china, 2019. N. Engl. J. Med. https://doi.org/10.1056/nejmoa2001017 (2020).
Cui, J., Li, F. & Shi, Z. L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 17 181–192. Preprint athttps://doi.org/10.1038/s41579-018-0118-9 (2019).
Bayani, F. et al. An overview of the vaccine platforms to combat COVID-19 with a focus on the subunit vaccines. Progress Biophys. Mol. Biol. Preprint at https://doi.org/10.1016/j.pbiomolbio.2023.02.004 (2023).
Pillaiyar, T., Manickam, M., Namasivayam, V., Hayashi, Y. & Jung, S. H. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: Peptidomimetics and small molecule chemotherapy. J. Med. Chem. 59 6595–6628. Preprint at https://doi.org/10.1021/acs.jmedchem.5b01461 (2016).
Woo, P. C. Y. et al. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports Bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavi. J. Virol. 86, 3995–4008 (2012).
Huang, C. et al. A new generation Mpro inhibitor with potent activity against SARS-CoV-2 Omicron variants. Signal. Transduct. Target. Ther. https://doi.org/10.1038/s41392-023-01392-w (2023).
Bayani, F., Safaei Hashkavaei, N., Karamian, M. R., Uskoković, V. & Sefidbakht, Y. In Silico design of a multi-epitope vaccine against the Spike and the nucleocapsid proteins of the Omicron variant of SARS-CoV-2. J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2023.2170470 (2023).
Rezaei, S., Sefidbakht, Y. & Uskoković, V. Tracking the pipeline: immunoinformatics and the COVID-19 vaccine design. Brief. Bioinform. https://doi.org/10.1093/bib/bbab241 (2021).
Unoh, Y. et al. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19. J. Med. Chem. https://doi.org/10.1021/acs.jmedchem.2c00117 (2022).
Konno, S. et al. 3CL protease inhibitors with an electrophilic arylketone moiety as Anti-SARS-CoV-2 agents. J. Med. Chem. https://doi.org/10.1021/acs.jmedchem.1c00665 (2022).
Verma, D. K., Kapoor, S., Das, S. & Thakur, K. G. Potential inhibitors of SARS-CoV-2 main protease (Mpro) identified from the library of FDA-Approved drugs using molecular Docking studies. Biomedicines https://doi.org/10.3390/biomedicines11010085 (2023).
Han, S. H. et al. Structure-Based optimization of ML300-Derived, noncovalent inhibitors targeting the severe acute respiratory syndrome coronavirus 3CL protease (SARS-CoV-2 3CLpro). J. Med. Chem. 65, 2880–2904 (2022).
Cho, E. et al. Dynamic profiling of β-Coronavirus 3CL MproProtease Ligand-Binding sites. J. Chem. Inf. Model. https://doi.org/10.1021/acs.jcim.1c00449 (2021).
Hsu, W. C. et al. Critical assessment of important regions in the subunit association and catalytic action of the severe acute respiratory syndrome coronavirus main protease. J. Biol. Chem. https://doi.org/10.1074/jbc.M502556200 (2005).
Macip, G. et al. Haste makes waste: A critical review of docking-based virtual screening in drug repurposing for SARS-CoV-2 main protease (M-pro) inhibition. Med. Res.Rev. 42 744–769. Preprint at https://doi.org/10.1002/med.21862 (2022).
Hegyi, A. & Ziebuhr, J. Conservation of substrate specificities among coronavirus main proteases. J. Gen. Virol. 83, 595–599 (2002).
Kneller, D. W. et al. Covalent narlaprevir- and boceprevir-derived hybrid inhibitors of SARS-CoV-2 main protease. Nat Commun 13, (2022).
Wu, Q. & Huang, S. Y. HCovDock: an efficient Docking method for modeling covalent protein-ligand interactions. Brief. Bioinform. https://doi.org/10.1093/bib/bbac559 (2023).
Song, Q., Zeng, L., Zheng, Q., Liu, S. & SCARdock: A web server and manually curated resource for discovering covalent ligands. ACS Omega. https://doi.org/10.1021/acsomega.2c08147 (2022).
Ghosh, A. K., Samanta, I., Mondal, A. & Liu, W. R. Covalent Inhibition in Drug Discovery. ChemMedChem Preprint at https://doi.org/10.1002/cmdc.201900107 (2019).
Sutanto, F., Konstantinidou, M. & Dömling, A. Covalent inhibitors: A rational approach to drug discovery. RSC Medicinal Chemistry 11 876–884 .Preprint at https://doi.org/10.1039/d0md00154f (2020).
Kahne, D. & Still, W. C. Hydrolysis of a peptide bond in neutral water. J. Am. Chem. Soc. https://doi.org/10.1021/ja00230a041 (1988).
Huang, F., Han, X., Xiao, X. & Zhou, J. Covalent warheads targeting cysteine residue: The promising approach in drug development. Molecules Preprint at https://doi.org/10.3390/molecules27227728 (2022).
Oksenberg, D. et al. GBT440 increases haemoglobin oxygen affinity, reduces sickling and prolongs RBC half-life in a murine model of sickle cell disease. Br. J. Haematol. https://doi.org/10.1111/bjh.14214 (2016).
Schaefer, D. & Cheng, X. Recent advances in covalent drug discovery. Pharmaceuticals Preprint athttps://doi.org/10.3390/ph16050663 (2023).
Kim, H., Hwang, Y. S., Kim, M. & Park, S. B. Recent advances in the development of covalent inhibitors. RSC Med. Chemistry Preprint at https://doi.org/10.1039/d1md00068c (2021).
Zhou, S., Chan, E., Duan, W., Huang, M. & Chen, Y. Z. Drug bioactivation, covalent binding to target proteins and toxicity relevance. Drug Metab. Rev. Preprint at https://doi.org/10.1081/DMR-200028812 (2005).
Uetrecht, J. & Naisbitt, D. J. Idiosyncratic adverse drug reactions: Current concepts. Pharmacol. Rev. Preprint at https://doi.org/10.1124/pr.113.007450 (2013).
Lei, J., Zhou, Y., Xie, D. & Zhang, Y. Mechanistic insights into a classic Wonder drug-aspirin. J. Am. Chem. Soc. https://doi.org/10.1021/ja5112964 (2015).
Vane, J. R. & Botting, R. M. The mechanism of action of aspirin. in Thrombosis Res. (2003). https://doi.org/10.1016/S0049-3848(03)00379-7
Ghosh, A. K., Samanta, I., Mondal, A. & Liu, W. R. Covalent Inhibition in Drug Discovery. ChemMedChem 14 889–906. Preprint at https://doi.org/10.1002/cmdc.201900107 (2019).
Bauer, R. A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today Preprint at https://doi.org/10.1016/j.drudis.2015.05.005 (2015).
Yue Yang Yue Yang School of Medicine & 716000, Y. U. Y. C. M. by Y. Y. Y.-D. L.-B. Z. X.-Y. B. Z.-Y. F.-B. H.-L. L. Progress in Research on Inhibitors Targeting SARS-CoV-2 Main Protease (Mpro). The American Chemical Society (ACS).
Lin, C. et al. Structural basis for the Inhibition of coronaviral main proteases by ensitrelvir. Structure 31, (2023).
Hou, N. et al. Development of highly potent noncovalent inhibitors of SARS-CoV-2 3CLpro. ACS Cent. Sci 9, (2023).
Shan, J., Pan, X., Wang, X., Xiao, X. & Ji, C. FragRep: A web server for Structure-Based drug design by fragment replacement. J Chem. Inf. Model 60, (2020).
[Internet]. Covalent docking software update: SeeSAR 11.2 and CovXplorer Workflow [Internet]. Preprint at (2021).
Daina, A., Michielin, O., Zoete, V. & SwissADME A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. https://doi.org/10.1038/srep42717 (2017).
Pires, D. E. V., Blundell, T. L., Ascher, D. B. & pkCSM Predicting small-molecule Pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. https://doi.org/10.1021/acs.jmedchem.5b00104 (2015).
Ji, C., Svensson, F., Zoufir, A. & Bender, A. eMolTox: Prediction of molecular toxicity with confidence. Bioinformatics 34, (2018).
Andersen, H. C. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. https://doi.org/10.1063/1.439486 (1980).
Singh, H., Raja, A., Prakash, A., Medhi, B. & Gmx_qk An automated Protein/Protein-Ligand complex simulation workflow bridged to MM/PBSA, based on Gromacs and Zenity-Dependent GUI for beginners in MD simulation study. J Chem. Inf. Model 63, (2023).
Eric Irrgang, M., Davis, C. & Kasson, P. M. Gmxapi: A GROMACS-native Python interface for molecular dynamics with ensemble and plugin support. PLoS Comput. Biol 18, (2022).
Valdés-Tresanco, M. S., Valdés-Tresanco, M. E., Valiente, P. A. & Moreno, E. Gmx_MMPBSA: A new tool to perform End-State free energy calculations with GROMACS. J. Chem. Theory Comput. https://doi.org/10.1021/acs.jctc.1c00645 (2021).
Valdés-Tresanco, M. S., Valdés-Tresanco, M. E., Valiente, P. A. & Moreno, E. Gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J Chem. Theory Comput 17, (2021).
Chen, W. et al. Review of preclinical data of PF-07304814 and its active metabolite derivatives against SARS-CoV-2 infection. Frontiers in Pharmacology 13. Preprint at https://doi.org/10.3389/fphar.2022.1035969(2022).
Ma, C. et al. Discovery of Di- and trihaloacetamides as covalent SARS-CoV-2 main protease inhibitors with high target specificity. J. Am. Chem. Soc. https://doi.org/10.1021/jacs.1c08060 (2021).
Fernandes, H. S., Sousa, S. F. & Cerqueira, N. M. F. S. A. New insights into the catalytic mechanism of the SARS-CoV-2 main protease: an ONIOM QM/MM approach. Mol. Divers. https://doi.org/10.1007/s11030-021-10259-7 (2022).
MH, H. E. A. & AI-driven covalent drug design strategies targeting main protease (Mpro) against SARS-CoV-2: structural Insights and molecular mechanisms. Preprint at https://doi.org/10.1080/07391102.2024.2317979(2024).
Ghasemlou, A., Uskoković, V. & Sefidbakht, Y. Exploration of potential inhibitors for SARS-CoV‐2 Mpro considering its mutants via structure‐based drug design, molecular docking, MD simulations, MM/PBSA, and DFT calculations. Biotechnol. Appl. Biochem. 70, 439–457 (2023).
Ghassemlou, A., Sefidbakht, Y. & Rahmandoust, M. The Main Protease of SARS COV-2 and Its Specific Inhibitors. in COVID-19 121–147 (Springer, 2021). https://doi.org/10.1007/978-981-16-3108-5_4
Author information
Authors and Affiliations
Contributions
The authors confirm their contributions to the paper as follows: Yahya Sefidbakht and Fatemeh Bayani were responsible for the study conception and design. Fatemeh Bayani and Mohammad Hossein Haghir Ebrahim Abadi carried out data collection. The analysis and interpretation of the results were performed by Mohammad Hossein Haghir Ebrahim Abadi and Fatemeh Bayani. The draft manuscript was prepared by Mohammad Hossein Haghir Ebrahim Abadi, Fatemeh Bayani, and Yahya Sefidbakht providing supervision and project administration. All authors reviewed the results and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Abadi, M.H.H.E., Bayani, F. & Sefidbakht, Y. Identification of potential COVID-19 Mpro inhibitors through covalent drug docking, molecular dynamics simulation, and MMGBSA calculation. Sci Rep 15, 20500 (2025). https://doi.org/10.1038/s41598-025-05375-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-05375-5
