
Abstract
Leukemia is the most prevalent form of blood cancer, affecting individuals across all age groups. Early and accurate diagnosis is crucial for effective treatment and improved clinical outcomes. Peripheral blood smear analysis, a key non-invasive diagnostic tool, often suffers from subjective interpretation, inter-observer variability, and a lack of readily available expertise. Although deep learning approaches, particularly Convolutional Neural Networks (CNNs), have demonstrated exceptional performance in binary classification tasks, multiclass classification of leukemia subtypes remains challenging due to limited data availability and morphological similarities between subtypes. This study presents a novel hybrid methodology that combines pre-trained CNN architectures, including VGG16, InceptionV3, and ResNet50, with advanced classification models such as Random Forest (RF), Support Vector Machine (SVM), Extreme Gradient Boosting (XGBoost), and the deep learning-based Multi-Layer Perceptron (MLP). The method leverages publicly available datasets, the Acute Lymphoblastic Leukemia Image Database (ALL-IDB) and the Munich AML Morphology Dataset, to classify healthy cells, lymphoblasts, and myeloblasts. Pre-trained CNNs are employed for feature extraction, while the classifiers refine the predictions for improved accuracy. The proposed approach demonstrated exceptional performance, with the InceptionV3 + SVM combination achieving the highest accuracy of 88%, followed closely by VGG16 + XGBoost at 87%. MLP-based models also achieved strong results, effectively capturing non-linear patterns in the data. In contrast, ResNet50 exhibited limitations, likely due to overfitting caused by the small dataset. The novelty of this work lies in the integration of pre-trained deep learning architectures with hybrid classification techniques, enabling robust multiclass classification in data-constrained scenarios. This innovative approach offers a scalable and precise diagnostic tool, improving the speed and reliability of leukemia subtype identification and providing significant potential to enhance clinical decision-making and patient care.
Similar content being viewed by others
Introduction
Leukemia is the most prevalent form of blood cancer, particularly in children, but it also affects individuals across all age groups. It arises from the uncontrolled proliferation of immature blood cells, which can damage critical body systems such as the immune system, red blood cells (RBCs), and brain tissue1. The development of leukemia involves mutations that lead to the production of leukemic clones, rapidly increasing the number of immature hematopoietic cells known as blasts in the bone marrow. In cases of acute leukemia, more than 20% of the bone marrow may consist of blast cells, resulting in a reduction of normal red blood cells, white cells, and platelets. If left undiagnosed and untreated, the disease can progress rapidly, often resulting in fatal outcomes within a few months2.
In recent years, efforts to enhance the speed and accuracy of leukemia diagnosis have increasingly incorporated advanced technologies such as deep learning. Deep learning algorithms, particularly Convolutional Neural Networks (CNNs), have demonstrated remarkable performance across diverse domains, including medical image processing3. CNNs are highly specialized in detecting visual patterns, making them particularly effective for tasks involving medical imaging4. Despite the widespread success of deep learning for binary classification tasks, such as distinguishing healthy cells from leukemic cells, research into multiclass classification of leukemia subtypes remains relatively limited5. Leukemia can be classified based on abnormalities in white blood cells (WBCs). Acute Lymphoblastic Leukemia (ALL) involves the proliferation of lymphoblasts, while Acute Myeloid Leukemia (AML) involves myeloblasts. ALL is characterized by an abnormal increase in immature lymphocytes, whereas AML is marked by the rapid development of abnormal myelocytes, leading to excessive production of myeloblasts6,7. The identification of these subtypes relies on subtle cytomorphological differences, such as nuclear chromatin texture and cytoplasmic characteristics, which makes manual diagnosis both time-consuming and prone to errors5,8. While cytomorphology plays a critical role in leukemia diagnosis, manual analysis is inherently limited by human error and slow processing times9.
Recent studies have focused on automating leukemia diagnosis using machine learning and deep learning techniques, although they have primarily addressed binary classification. For example, Labati et al. utilized the ALL-IDB dataset for leukemia classification, which is commonly used to distinguish between healthy and leukemic cells10. Similarly, Matek explored the Munich AML Morphology dataset for identifying AML subtypes11. Both datasets have been widely adopted in research due to their high-quality, well-annotated images and applicability in classification tasks10,11. Multiclass classification presents additional challenges, particularly due to the limited availability of annotated datasets, which complicates the accurate identification of multiple leukemia subtypes. Furthermore, differentiating between morphologically similar subtypes, such as lymphoblasts and myeloblasts, often results in lower classification accuracy when using traditional methods12. These limitations highlight the need for a more robust approach capable of handling multiclass classification in a data-constrained environment.
In addition to the existing methodologies, recent studies have demonstrated the utility of hybrid approaches that combine deep learning and traditional machine learning for medical imaging tasks. For instance, Iqbal utilized non-deep learning and deep learning methods for recognizing mRNA N4 acetylcytidine (ac4C), highlighting the versatility of combining these techniques for robust feature extraction13. Similarly, Iqbal also explored the application of deep learning to differentiate between diseased and normal cell representations14. Another study by Iqbal showcased the efficient classification of mitochondrial cell images through deep learning approaches15. Moreover, the Drug Investigation Tool proposed by Iqbal et al. (2020) demonstrated the impact of improved correlation techniques in identifying drug effects on cellular images16. These studies underscore the potential of integrating deep learning with traditional machine learning classifiers to enhance the accuracy and robustness of medical image classification systems.
The primary objective of this study is to develop a hybrid method that leverages pre-trained deep learning models for feature extraction and advanced classifiers, including both traditional machine learning models and deep learning-based architectures, for accurate multiclass classification of leukemia subtypes, specifically ALL and AML, from microscopic blood smear images. By integrating CNN architectures such as VGG16, InceptionV3, and ResNet50 with classifiers like Random Forest (RF), Support Vector Machine (SVM), Extreme Gradient Boosting (XGBoost), and the deep learning-based Multi-Layer Perceptron (MLP), the study aims to address the challenges of working with smaller datasets while enhancing classification accuracy and capturing non-linear relationships in complex feature spaces. This study integrates the ALL-IDB and Munich AML Morphology datasets to enhance the model’s generalization across multiple leukemia subtypes, which has been a proven method in similar studies10,11.
Novelty and importance of the study
This study introduces a hybrid approach that combines the feature extraction capabilities of pre-trained deep learning models, such as VGG16, InceptionV3 and ResNet50, with the classifiers including MLP, RF, SVM, and XGBoost. While the application of deep learning in binary classification tasks, particularly for leukemia detection, has been explored in previous studies17,18few have ventured into multiclass classification, which poses unique challenges due to the morphological similarities between leukemia subtypes. The integration of both the ALL-IDB and Munich AML Morphology datasets is a significant aspect of this study, enabling broader generalization across different leukemia subtypes, a capability rarely demonstrated in prior research10,11.
To the best of our knowledge, no prior study has combined these specific CNN architectures (VGG16, InceptionV3, and ResNet50) with a combination of traditional machine learning classifiers, such as RF and SVM, alongside the deep learning-based MLP, to address multiclass leukemia classification using the same datasets. This is particularly significant because multiclass classification is inherently more challenging than binary classification due to the visual similarities between subtypes. Misclassification between subtypes such as lymphoblasts and myeloblasts, which share overlapping cytomorphological characteristics, can hinder accurate diagnosis and effective treatment planning.
This study is pivotal because timely and accurate subtype classification in leukemia is critical for improving patient outcomes. As treatments for leukemia subtypes differ significantly, enhancing diagnostic precision can directly impact survival rates and the effectiveness of therapeutic interventions. By leveraging hybrid models, this research contributes to the development of more reliable, automated diagnostic systems that minimize reliance on manual examination, which is not only prone to human error but also time-intensive8,9. Moreover, with the integration of transfer learning from pre-trained CNNs and the use of classifiers optimized for smaller datasets, this approach demonstrates robust utility even in data-constrained environments, which are common in medical imaging contexts.
This study addresses a critical gap in the existing literature by advancing automated leukemia subtype classification. The findings have the potential to streamline diagnostic processes, reduce diagnostic delays, and significantly improve patient outcomes in clinical practice.
Materials and methods
Automated workflow for leukemia classification
This study aimed to develop an automated system for the processing and analysis of blood smear images to enhance the diagnostic accuracy of ALL and AML. The proposed methodology was designed to assist pathologists by facilitating the segmentation and classification of ALL and AML cells, thereby supporting faster and more precise clinical decision-making.
WBCs are classified into five main subtypes: monocytes, lymphocytes, basophils, eosinophils, and neutrophils, collectively considered healthy cells. For diagnosing ALL, lymphoid WBCs were the primary focus, while myeloid WBCs were analyzed for AML diagnosis. In blood smear images, lymphoid and myeloid cells exhibited distinguishable nuclei, which differed significantly from the surrounding background and other blood cells. The affected cells—lymphoblasts in ALL and myeloblasts in AML—underwent specific morphological changes that could be identified through computational methods.
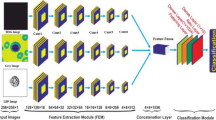
The methodology employed a hybrid approach that combined feature extraction using pre-trained CNNs with classification performed by both traditional machine learning models and deep learning-based classifiers. Semantic segmentation was performed on blood smear images to isolate lymphoid or myeloid cells from the background and other cellular components. Features were then extracted from the segmented regions using pre-trained CNNs (e.g., VGG16, InceptionV3, ResNet50). Finally, classification was conducted using models, including RF, SVM, XGBoost, and a MLP, to categorize cells into healthy WBCs, lymphoblasts, or myeloblasts.
The workflow of the proposed system is summarized in the block diagram (Fig. 1) and included the following steps:
-
1.
Input Data: Blood smear images were sourced from the ALL-IDB and Munich AML Morphology datasets.
-
2.
Preprocessing: Semantic segmentation and data augmentation were applied to enhance image quality and diversity.
-
3.
Feature Extraction: Pre-trained CNNs were used to extract relevant image features from the segmented regions.
-
4.
Classification: Machine learning classifiers, including RF, SVM, XGBoost, and MLP, were utilized for cell categorization.
-
5.
Output: The predicted cell classifications were generated, along with performance evaluation metrics.
This suggested approach was developed to enable precise detection and classification of ALL and AML cells, providing an effective computational tool for advancing leukemia diagnostics. Subsequent sections detail the dataset sources, preprocessing techniques, feature extraction methods, and classification algorithms employed in this study.

Workflow of the proposed system, illustrating the process from data preprocessing to the final classification output.
Dataset integration and preparation
The datasets used to evaluate the suggested approach for ALL classification were sourced from the publicly available Acute Lymphoblastic Leukemia Image Database for Image Processing (ALL-IDB). This database comprises two subsets, ALL-IDB1 and ALL-IDB2, containing microscopic images of blood samples annotated by qualified oncologists. The images were captured using a Canon PowerShot G5 camera attached to an optical laboratory microscope, with magnifications ranging from 300x to 500x. All images were provided in jpg format with a 24-bit color depth10. For this study, the ALL-IDB2 dataset was selected as it includes pre-segmented cells extracted from complete microscopic images, simplifying the processing pipeline. Despite differences in image size, ALL-IDB2 maintains similar grayscale properties to ALL-IDB1, ensuring consistency within the dataset.
For AML, the data were drawn from the Munich AML Morphology Dataset, which contains expert-labeled single-cell images from peripheral blood smears of 100 AML-diagnosed patients and 100 non-malignant cases. These images were collected at the Munich University Hospital between 2014 and 2017 using an M8 digital microscope/scanner (Precipoint GmbH, Freising, Germany) at 100x optical magnification with oil immersion. Experienced professionals categorized both pathological and non-pathological leukocytes based on morphological guidelines derived from clinical practice11.
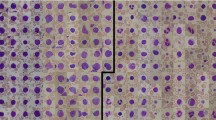
To improve the robustness and generalization capabilities of the proposed system, the study integrated the ALL-IDB2 and Munich AML Morphology datasets. This unified dataset contained a total of 390 images, with 130 images for each of the three classes: healthy cells, lymphoblasts, and myeloblasts. The balanced dataset ensures equitable representation of each class, which is critical for minimizing bias and enhancing classification accuracy. Representative images of the three cell types are shown in Fig. 2, illustrating their distinct morphological features.

Morphological characteristics of (a) healthy WBCs (130 images), (b) lymphoblasts (130 images), and (c) myeloblasts (130 images) are shown. These images are used for model training and evaluation.
Sample pre-processing
To address the limited number of microscopic blood sample images available in the datasets, data augmentation techniques were applied to artificially expand the training set. Augmentations included rotations of 60° and 90°, horizontal flips, vertical flips, and random shifts within the range of (1.0, 1.0). These transformations were chosen to simulate variations in cell orientation and positioning that occur naturally during sample preparation or microscopic imaging. By increasing dataset diversity, these augmentations reduced the risk of overfitting and improved the model’s generalization capabilities. Examples of these augmentation techniques applied to an original training image are shown in Fig. 3.
Before augmentation, all input images were resized to 256 × 256 pixels and normalized to ensure uniformity and compatibility with the U-Net model’s input requirements. This preprocessing step standardized the dataset and improved training efficiency.
Semantic segmentation was employed to isolate WBCs from background artifacts and other cellular components. Background removal plays a critical role in bioimage classification tasks by reducing noise, enhancing focus on relevant features, and mitigating bias caused by variable or cluttered backgrounds19.
The segmentation was performed using a U-Net architecture20. The U-Net model consisted of convolutional layers with 3 × 3 filters for feature extraction, dropout layers with a rate of 0.5 to prevent overfitting, max-pooling layers for down-sampling, and transpose convolutional layers for up-sampling21. Concatenation layers combined features from different levels of the network, enabling the integration of both low- and high-level features22. The output of the model was a single-channel segmentation map representing the isolated WBCs.The augmented dataset served as input for the segmentation step, ensuring diverse and representative samples for training the U-Net model. Figure 4 demonstrates the segmentation process, where the U-Net architecture successfully isolated WBCs from the background.

Augmented data used in training.

Semantic Segmentation of White Blood Cells. (a) Original raw image. (b) Segmented image with background removal.
Pre-trained networks for feature selection
Several pre-trained CNNs, including VGG-1623, InceptionV324, and ResNet-5025, were employed in this study for feature extraction. These networks, trained on large datasets such as ImageNet, are well-suited for image classification tasks due to their robust architectures and widespread use. In this study, the feature extraction process can be represented mathematically in Eq. (1) as:
where:
-
\(X=\left\{ {{x_1},{x_2}, \ldots ,{x_n}} \right\}\) is the dataset, with \({x_i} \in {{\mathbb{R}}^{h \times w \times c}}\), representing an image of height h, width w, and c channels (e.g., c = 3 for RGB),
-
n is the number of the input images.
-
\(\Phi (X;\theta )\) is the feature extraction function, where θ are the parameters of the pre-trained CNN.
-
Z is the features matrix, with each row representing a d-dimensional feature vector extracted from the corresponding input image.
For feature extraction, VGG-16, a series-based CNN, processes each layer sequentially, taking input from the previous layer. In contrast, InceptionV3 uses a Directed Acyclic Graph (DAG) structure for more complex pathways between layers. ResNet-50, a residual network, leverages skip connections to address the vanishing gradient problem and maintain accuracy in deep networks. The extracted feature matrix Z enables effective representation of input data for downstream tasks such as classification. Here, d corresponds to the number of features extracted from the final or penultimate layer of the network.
Classifiers for leukemia image classification
The study employs a range of classifiers, including traditional machine learning models like RF, SVM, and XGBoost, as well as the deep learning-based MLP, to classify leukemia images. These classifiers were selected due to their complementary strengths in handling diverse data characteristics. RF is known for its robustness and ability to generalize effectively on small datasets26SVM excels in high-dimensional feature spaces27XGBoost provides scalability and effective regularization to mitigate overfitting28and MLP captures complex non-linear relationships in the feature space29.
Feature vectors extracted from the fully connected layers of pre-trained CNNs were utilized as inputs to these classifiers for final classification. The extracted features (Z) served as input to the classifiers, while classifier-specific parameters were denoted as θ. The classifiers’ performance was evaluated using precision, recall, F1-score, and accuracy to comprehensively assess their effectiveness in distinguishing between healthy cells, lymphoblasts, and myeloblasts.
Deep learning-based multi-layer perceptron (MLP)
The MLP classifier was implemented to model non-linear relationships in the feature space. Its architecture consisted of an input layer of dimension d, a single hidden layer with 128 neurons, and an output layer with three neurons corresponding to the three classes: healthy, lymphoblasts, and myeloblasts. The prediction function for MLP can be expressed in Eq. (2) as:
where:
-
\({W_i}\) and \({W_h}\) : Weight matrices for the input and hidden layers, respectively,
-
\({b_i}\) and \({b_h}\) : Bias terms,
-
\(\sigma \left( \cdot \right)\): Rectified Linear Unit(ReLu) activation function.
The final probabilities were computed using the softmax function. Optimization was performed using the Adam optimizer with a learning rate of 0.001. Categorical cross-entropy was used as the loss function, and dropout layers with a rate of 0.5 were incorporated to mitigate overfitting. The model was trained for 50 epochs with a batch size of 32, and hyperparameters were selected based on validation performance30 .
Random forest (RF)
RF was selected for its robustness and ability to generalize effectively across diverse datasets. It consists of an ensemble of T decision trees, with predictions aggregated through majority voting. The final prediction function is defined in Eq. (3) as:
where:
-
\({h_t}(Z)\) : Prediction from the t-th tree.
-
T: Total number of trees in the ensemble.
For this study, T was set to 100, and the maximum tree depth was left unrestricted to allow full tree growth. The minimum number of samples required to split a node was set to 2, and the minimum number of samples required for a leaf node was set to 128. Hyperparameter optimization, including T, was conducted using RandomizedSearchCV method to ensure optimal performance28,31.
Support vector machines (SVM)
Support Vector Machines (SVMs) were employed for their effectiveness in high-dimensional spaces and their ability to handle non-linear relationships using kernel functions. The SVM classifier separates data points by constructing a hyperplane defined in Eq. (4) as:
where:
-
W: weight vector,
-
b: Bias term,
-
\({\text{sign}}( \cdot )\): determines the class label,
Extreme gradient boosting (XGBoost)
XGBoost was chosen for its scalability and ability to handle large datasets effectively while preventing overfitting through regularization and subsampling techniques. The XGBoost model aggregates predictions from K trees, defined in Eq. (5) as:
where:
-
\({f_k}\left( {{\rm Z};T} \right)\) : Prediction from k-th tree,
-
\(\eta\) : Learning rate,
-
K: Total number of boosting rounds.
In this study, K was set to 100 (ηestimators) and the learning rate (η) was set to 0.328. The maximum tree depth was set to 6, and the subsample parameter, controlling the fraction of samples used for training each tree, was set to 1.0. Additionally, L2 regularization was applied to reduce overfitting risks28. Hyperparameter tuning was performed using RandomizedSearchCV to achieve optimal results31.
Model evaluation and performance metrics
The classifiers’ performance was evaluated using precision, recall, F1-score, and accuracy. These metrics provided a comprehensive assessment of the classifiers’ ability to distinguish between healthy cells, lymphoblasts, and myeloblasts32.
A confusion matrix was utilized to quantify the performance of the models, detailing the number of True Positives (TP), False Negatives (FN), True Negatives (TN), and False Positives (FP) for each class32. These values were then used to compute the following metrics:
-
1.
Precision:
Precision measures the proportion of correctly classified positive samples to the total number of predicted positive samples. It is defined in Eq. (6) as:
-
2.
Recall:
Recall, also known as sensitivity, calculates the proportion of correctly classified positive samples to the total number of actual positive samples. It is defined in Eq. (7) as:
-
3.
F1-Score:
The F1-score is the harmonic mean of precision and recall, providing a balanced measure of the model’s performance, particularly in the presence of class imbalance. It is defined in Eq. (8) as:
-
4.
Accuracy:
Accuracy represents the overall correctness of the model and is defined in Eq. (9) as:
These metrics provided insights into the strengths and weaknesses of each classifier. Precision and recall were particularly critical for evaluating the classifiers’ ability to identify lymphoblasts and myeloblasts, as false negatives in these categories could have significant clinical implications32. The confusion matrix was computed for each classifier to analyze their performance across individual classes.
Results
The classification performance of different combinations of pre-trained CNNs and classifiers, including both traditional machine learning models and the deep learning-based MLP, is summarized in Table 1. Three CNN architectures, VGG16, InceptionV3, and ResNet50, were employed for feature extraction, followed by classification using RF, SVM, XGBoost, and MLP. The models were evaluated for their ability to classify leukemia cells into three categories: healthy, lymphoblast, and myeloblast.
The classification performance, as evaluated through accuracy, precision, recall, and F1-score, is presented in Table 1. The VGG16-based models demonstrated high classification performance across most classifier combinations. The VGG16 + XGBoost combination achieved the highest accuracy for this feature extractor at 87%, with precision (0.87), recall (0.86), and F1-score (0.85). The VGG16 + MLP combination performed well, achieving an accuracy of 82%, while the VGG16 + RF combination achieved an accuracy of 81%. The VGG16 + SVM combination exhibited lower performance, achieving an accuracy of 74%.
The InceptionV3-based models showed strong performance, with the InceptionV3 + SVM combination achieving the highest overall accuracy of 88%, alongside precision (0.86), recall (0.87), and F1-score (0.87). The InceptionV3 + MLP combination achieved an accuracy of 79%, while the combinations with RF and XGBoost achieved accuracies of 78% and 77%, respectively.
In contrast, the ResNet50-based models exhibited lower performance. The ResNet50 + RF combination achieved the highest accuracy for this feature extractor at 76%, followed by the ResNet50 + MLP combination at 75%. The combinations of ResNet50 with SVM and XGBoost yielded accuracies of 70%. These results highlight challenges in feature extraction using ResNet50 for this dataset.
Confusion matrices provide additional insights into the classification performance of selected model combinations by highlighting the distribution of true and predicted labels (Fig. 5). For the VGG16 + XGBoost combination, the confusion matrix revealed high classification accuracy across all categories, with 124 healthy cells, 112 lymphoblast cells, and 108 myeloblast cells correctly classified. Misclassifications were minimal, with 6 healthy cells misclassified as lymphoblasts and 10 lymphoblast cells misclassified as myeloblasts. The InceptionV3 + SVM combination also showed consistent accuracy, with 126 healthy cells, 114 lymphoblast cells, and 109 myeloblast cells correctly classified. Misclassifications included 8 lymphoblast cells predicted as myeloblasts, reflecting a strong overall performance. The ResNet50 + RF combination exhibited higher misclassification rates. The confusion matrix revealed that 118 healthy cells were correctly classified, but significant errors were observed for lymphoblast and myeloblast cells. Specifically, 18 lymphoblast cells were misclassified as healthy, and 16 myeloblast cells were misclassified as lymphoblasts. These results suggest that ResNet50 struggles with feature extraction, leading to confusion between these categories.
Across all combinations, myeloblast cells showed higher misclassification rates compared to healthy and lymphoblast cells. This trend indicates that the morphological features of myeloblast cells may overlap with those of other categories, posing challenges for accurate classification.



Confusion Matrices for Feature Extractors and Classifiers. (a) Confusion matrices for VGG16 with MLP, RF, SVM, and XGBoost. (b) Confusion matrices for InceptionV3 with MLP, RF, SVM, and XGBoost. (c) Confusion matrices for ResNet50 with MLP, RF, SVM, and XGBoost.
The classification performance varied significantly based on the feature extractor and classifier combinations. The VGG16 + XGBoost and InceptionV3 + SVM combinations demonstrated high accuracy and minimal misclassifications, achieving 87% and 88% accuracy, respectively. The VGG16 + MLP combination also performed well, with an accuracy of 82%. In contrast, ResNet50-based models consistently underperformed, with the highest accuracy being 76% (ResNet50 + RF).
The confusion matrix analysis reinforced these findings, highlighting the minimal misclassifications for VGG16 + XGBoost and InceptionV3 + SVM. Conversely, ResNet50-based models exhibited higher error rates, particularly for lymphoblast and myeloblast cells. These results emphasize the importance of selecting appropriate feature extractors and classifiers for leukemia classification.
Discussion
This study evaluated a hybrid approach for leukemia cell classification, combining pre-trained CNN feature extractors with both traditional machine learning classifiers and the deep learning-based Multi-Layer Perceptron (MLP) to address challenges such as limited datasets and the complexity of multiclass classification.
The findings highlight the potential of hybrid methods to bridge the gap between deep learning and traditional classifiers, achieving state-of-the-art performance for leukemia diagnosis. Among the models tested, VGG16 + XGBoost and InceptionV3 + SVM emerged as the most effective, achieving accuracies of 87.00% and 88.00%, respectively, with high precision, recall, and F1-scores. These results underscore the importance of pairing robust feature extractors with effective classifiers to optimize classification performance.
The VGG16 + XGBoost combination demonstrated balanced precision and recall scores of 0.86 and an F1-score of 0.85, indicating a robust ability to minimize both false positives and false negatives. XGBoost’s regularization capabilities and ability to handle structured datasets contributed significantly to its superior performance. Minimal misclassifications were observed between morphologically similar classes, such as lymphoblasts and healthy cells, as evidenced by the confusion matrix (Fig. 5a). These findings align with existing literature, where gradient boosting has been highlighted as an effective method for structured classification tasks33.
The InceptionV3 + SVM model achieved the highest overall accuracy at 88.00%, supported by strong precision and recall scores. InceptionV3’s hierarchical feature extraction capabilities complemented SVM’s kernel-based classification, allowing for optimal decision boundaries in high-dimensional feature spaces. This combination demonstrated consistent true positive rates across all classes, with minimal errors (Fig. 5b). The superior recall of 0.87 suggests that the model is highly effective at identifying true positives, which is critical for medical applications. These results corroborate findings from Umar, who also identified InceptionV3 + SVM as a robust combination for leukemia classification, achieving a similar accuracy of 87.05% 33.
In contrast, ResNet50-based models consistently underperformed across all classifier combinations. The highest accuracy achieved with ResNet50 was 76.00% when paired with RF. The confusion matrices for ResNet50-based models (Fig. 5c) revealed higher misclassification rates, particularly for lymphoblast and myeloblast cells. The deeper architecture of ResNet50, while advantageous for larger datasets, likely overfit the limited dataset used in this study, emphasizing the need for data-driven architecture selection when working with constrained datasets.
The sequential CNN model demonstrated the challenges of working with limited data. As shown in Fig. 4, the training accuracy approached perfection, while the validation accuracy plateaued at 0.4–0.5, indicating significant overfitting. This gap reflects the model’s inability to generalize beyond the training data, a common limitation in deep learning models applied to small datasets. While data augmentation techniques were employed, they were insufficient to fully mitigate this issue, highlighting the importance of regularization strategies, such as dropout, weight decay, or adversarial training, to improve model robustness.
Comparison with prior studies
The findings of this study align closely with and extend existing literature (Table 2). Umar et al. (2022) reported an accuracy of 87.05% for the InceptionV3 + SVM combination, which is comparable to the 88.00% achieved here. However, the introduction of XGBoost in this study significantly improved the performance of VGG16-based models, increasing accuracy from 81.00% (VGG16 + SVM) to 87.00%. This result highlights the importance of carefully selecting classifiers to complement feature extractors, particularly when dealing with small datasets.
Pałczyński explored lightweight architectures like MobileNetV2 combined with RF and XGBoost, achieving accuracies of 76.90% and 75.30%, respectively. These results, while promising, were lower than those achieved by VGG16 and InceptionV3 in this study, suggesting that deeper architectures are better suited for extracting the complex morphological features necessary for leukemia classification tasks.
Limitations
Despite the promising results, this study faced several limitations. The primary limitation was the small dataset size, which restricted the generalizability of deeper architectures like ResNet50. While data augmentation techniques were applied to increase dataset diversity, they could not fully compensate for the lack of real-world variability. The morphological similarities between lymphoblasts and healthy cells further complicated the classification task, resulting in occasional misclassifications even in the best-performing models.
Additionally, the computational complexity of deeper architectures, such as InceptionV3, limited their suitability for real-time clinical applications. While these models performed well in classification tasks, their high training and inference times underscore the need for lightweight and efficient alternatives for deployment in resource-constrained environments.
Future directions
Future research should focus on addressing the limitations identified in this study. Expanding datasets through collaborative efforts or synthetic data generation using GANs could provide the diversity and volume necessary for robust training. Incorporating multimodal data, such as genomic or proteomic information, could further enhance classification performance and provide a more comprehensive diagnostic framework.
Exploring advanced architectures like EfficientNet and Vision Transformers could provide improved feature extraction capabilities while maintaining parameter efficiency. Lightweight versions of high-performing models, such as VGG16 + XGBoost, should be developed using techniques like model quantization or pruning, enabling real-time applicability without compromising accuracy.
Additionally, integrating explainable AI techniques into the models could enhance their interpretability, providing insights into predictions and increasing trust among clinicians. Ensemble approaches, which aggregate predictions from multiple models, could further optimize performance, particularly for challenging multiclass classification tasks. These advancements could position hybrid models as a cornerstone of precision medicine, bridging the gap between AI research and clinical practice.
Implications for clinical practice
The findings of this study have significant implications for medical imaging, particularly in leukemia diagnosis. The success of VGG16 + XGBoost and InceptionV3 + SVM demonstrates the potential of hybrid approaches to achieve robust performance in multiclass classification tasks. These models combine the strengths of deep learning and traditional machine learning, offering a scalable and efficient framework for medical image classification.
By balancing feature extraction capabilities and classification robustness, hybrid approaches provide a reliable foundation for future advancements in medical imaging. The continued exploration of advanced architectures, synthetic data generation, and explainable AI techniques will enable the development of clinically applicable tools, improving diagnostic accuracy and patient outcomes in hematological disorders.
Conclusion
This study demonstrated the effectiveness of hybrid approaches combining pre-trained CNN feature extractors with traditional machine learning classifiers for leukemia cell classification. By leveraging the strengths of deep learning for feature extraction and traditional classifiers for robust decision-making, the models achieved state-of-the-art performance despite the constraints of a small dataset. Among the evaluated models, VGG16 + XGBoost and InceptionV3 + SVM emerged as the top performers, achieving accuracies of 87.00% and 88.00%, respectively. These results underscore the potential of hybrid models to address the challenges of multiclass classification in medical imaging, particularly when datasets are limited. The success of these combinations highlights the importance of carefully pairing feature extractors and classifiers to optimize performance.
While this study achieved promising results, it also highlighted significant challenges, including overfitting in deep learning models, the limited size of annotated datasets, and the inherent morphological similarities between certain leukemia subtypes. Addressing these limitations will require the development of larger, more diverse datasets, as well as the application of advanced architectures such as EfficientNet or Vision Transformers. Future research should also explore synthetic data generation, explainable AI techniques, and lightweight model optimization to enhance generalizability, interpretability, and clinical applicability. These findings contribute to the growing body of evidence supporting hybrid approaches in medical image classification and provide a strong foundation for future advancements in leukemia diagnosis and beyond.
Data availability
The Acute Lymphoblastic Leukemia Image Database (ALL-IDB) and Munich AML Morphology Dataset repository [https://www.cancerimagingarchive.net/collection/aml-cytomorphology_lmu/] are publicly available for academic and non-commercial use. The dataset includes a collection of images used for the processing and diagnosis of acute lymphoblastic leukemia (ALL). Researchers and practitioners can access the ALL-IDB for various image processing applications by visiting the following link: [https://scotti.di.unimi.it/all/]. The dataset is fully annotated and includes high-resolution images of bone marrow smears, facilitating leukemia detection and classification tasks. More details on the dataset, including licensing information, can be found in the original publication: Labati, R., Piuri, V., & Scotti, F. (2011). All-IDB: The acute lymphoblastic leukemia image database for image processing. 2011 18th IEEE International Conference on Image Processing (ICIP), 2045–2048. https://doi.org/10.1109/ICIP.2011.6115881.
References
Vasighizaker, A., Sharma, A. & Dehzangi, A. A novel one-class classification approach to accurately predict disease-gene association in acute myeloid leukemia cancer. PLoS One. 14, e0226115 (2019).
Mughal, B., Sharif, M., Muhammad, N. & Saba, T. A novel classification scheme to decline the mortality rate among women due to breast tumor. Microsc Res. Tech. 81, 171–180 (2018).
Lewis, J. E., Cooper, L. A. D., Jaye, D. L. & Pozdnyakova, O. Automated Deep Learning-Based Diagnosis and Molecular Characterization of Acute Myeloid Leukemia using Flow Cytometry (2023). https://doi.org/10.1101/2023.09.18.558289.
Bharati, P. & Pramanik, A. Deep learning Techniques—R-CNN to mask R-CNN: A Survey 657–668 (2020). https://doi.org/10.1007/978-981-13-9042-5_56.
Carmona-Rivera, C. & Kaplan, M. J. Neutrophil biology. In Encyclopedia of Cell Biology 750–758 (Elsevier, 2016). https://doi.org/10.1016/B978-0-12-394447-4.30112-2.
Bibi, N., Sikandar, M., Ud Din, I., Almogren, A. & Ali, S. IoMT-based automated detection and classification of leukemia using deep learning. J. Healthc. Eng. 2020, 1–12 (2020).
Yu, W. et al. Automatic classification of leukocytes using deep neural network. In IEEE 12th International Conference on ASIC (ASICON) 1041–1044 (IEEE, 2017). https://doi.org/10.1109/ASICON.2017.8252657.
Morgan, E. A. Surgical pathology of hematopoietic neoplasms. In Pathobiology of Human Disease 3607–3627 (Elsevier, 2014). https://doi.org/10.1016/B978-0-12-386456-7.06908-2.
Douglas, C., Tkachuk & Jack, V. Hirschmann. Approach to the microscopic evaluation of blood and bone marrow. Wintrobe Atlas Clin. Hematolog (2007).
Labati, R. D., Piuri, V., Scotti, F., All & -IDB. The acute lymphoblastic leukemia image database for image processing. In 18th IEEE International Conference on Image Processing 2045–2048 (IEEE, 2011). https://doi.org/10.1109/ICIP.2011.6115881.
Matek Christian, S. et al. A single-cell morphological dataset of leukocytes from AML patients and non-malignant controls (AML-Cytomorphology_LMU). In The Cancer Imaging Archive (TCIA)[Internet] (2019).
Abhishek, A., Jha, R. K., Sinha, R. & Jha, K. Automated classification of acute leukemia on a heterogeneous dataset using machine learning and deep learning techniques. Biomed. Signal. Process. Control. 72, 103341 (2022).
Iqbal, M. S. et al. Recognition of mRNA N4 acetylcytidine (ac4C) by using Non-Deep vs. Deep learning. Appl. Sci. 12, 1344 (2022).
Iqbal, M. S., Ahmad, I., Bin, L., Khan, S. & Rodrigues, J. J. P. C. Deep learning recognition of diseased and normal cell representation. Trans. Emerg. Telecommun. Technol. 32, 1456 (2021).
Iqbal, M. S. et al. Efficient cell classification of mitochondrial images by using deep learning. J. Opt. 48, 113–122 (2019).
Iqbal, M. S., Ahmad, I., Asif, M., Kim, S. & Mehmood, R. M. Drug investigation tool: identifying the effect of drug on cell image by using improved correlation. Softw. Pract. Exp. 51, 260–270 (2021).
Pałczyński, K. et al. IoT application of transfer learning in hybrid artificial intelligence systems for acute lymphoblastic leukemia classification. Sensors 21, 8025 (2021).
Abir, W. H., Uddin, M. F., Khanam, F. R. & Khan, M. M. Explainable AI in diagnosing and anticipating leukemia using transfer learning method (2023). https://doi.org/10.1155/2022/5140148.
Takeda, K., Sakai, T. & Mitate, E. Background removal for debiasing computer-aided cytological diagnosis. Int. J. Comput. Assist. Radiol. Surg. 19, 2165–2174 (2024).
Ronneberger, O., Fischer, P. & Brox, T. U-Net: Convolutional Networks for Biomedical Image Segmentation (2015).
Nitish Srivastava, G. et al. Dropout: a simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. (2014).
Krizhevsky, A., Sutskever, I. & Hinton, G. E. ImageNet classification with deep convolutional neural networks. Commun. ACM. 60, 84–90 (2017).
Simonyan, K. & Zisserman, A. Very Deep Convolutional Networks for Large-Scale Image Recognition (2014).
Szegedy, C., Vanhoucke, V., Ioffe, S., Shlens, J. & Wojna, Z. Rethinking the inception architecture for computer vision. in IEEE Conference on Computer Vision and Pattern Recognition (CVPR) 2818–2826 (IEEE, 2016). https://doi.org/10.1109/CVPR.2016.308.
He, K., Zhang, X., Ren, S. & Sun, J. Deep Residual Learning for Image Recognition (2015).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 (2001).
Cortes, C. & Vapnik, V. Support-Vector Networks 297 273–297 (1995).
Chen, T. & Guestrin, C. XGBoost. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining 785–794 (ACM, 2016). https://doi.org/10.1145/2939672.2939785.
Christopher, M. Bishop. Pattern Recognition and Machine Learning (Springer, 2006).
Kingma, D. P. & Ba, J. Adam: a method for stochastic optimization (2014).
James Bergstra & Yoshua Bengio. Random search for hyper-parameter optimization. J. Mach. Learn. Res. 12, 281–305 (2012).
Powers, D. M. W. Evaluation: from precision, recall and F-measure to ROC, informedness, markedness and correlation (2020).
Umar, H., Muhammad, Y. & Arif, U. R. Transfer learning for the medical diagnosis of acute leukemia cancer. in 3 rd International Conference on Computational Sciences and Technologies, Pakistan (2023).
Author information
Authors and Affiliations
Contributions
Sorayya Malek, JunJie Tang, Bryan Liew contributed equally to this work, focusing on study design, analysis, and validation, under the supervision and guidance of Sazzli Kasim. Song Cheen and Xue Ning Kiew assisted in manuscript writing. Raja Ezman Raja Shariff and Norashikin Saidon worked as proof-readers for the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kasim, S., Malek, S., Tang, J. et al. Multiclass leukemia cell classification using hybrid deep learning and machine learning with CNN-based feature extraction. Sci Rep 15, 23782 (2025). https://doi.org/10.1038/s41598-025-05585-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-05585-x
