
Abstract
Epidemiological studies show that heart failure often leads to kidney dysfunction, known as cardio-renal syndrome (CRS). Elevated central venous pressure, rather than low cardiac output, strongly correlates with worsening renal function and is increasingly recognized as the cause of CRS. However, the molecular mechanisms behind congestion-mediated worsening of kidney injury remain unclear due to the lack of suitable animal models. Here, we used a novel mouse model of renal congestion and identified injured tubule-specific cell-cell interactions in congested kidneys. We found that Cellular Communication Network Factor 1 (CCN1) played a critical role in this process. Transcriptomic analysis of kidneys with ischemia-reperfusion injury (IRI) and renal congestion showed the upregulation of paracrine chemokine-related pathways. CCN1 was upregulated in the acute phase following kidney injury with renal congestion, and phosphorylated focal adhesion kinase (pFAK), a downstream molecule of CCN1, was present in fibroblasts at injury sites. CCN1 activated FAK, promoting fibroblast and macrophage migration. We further examined the effects of CCN1 deletion in tubular epithelia and found that it reduced pFAK expression and alleviated tissue fibrosis. In conclusion, CCN1 plays a key role in fibroblast migration in congestion-mediated worsening of kidney injury and is a potential therapeutic target to prevent fibrosis.
Similar content being viewed by others


Introduction
The number of patients with end-organ failure, including the kidneys and heart, is increasing in aging societies worldwide1,2. Epidemiological and clinical investigations revealed a vicious cycle between heart and kidney failure in which both disease conditions aggravate each other, and this is called cardio-renal syndrome (CRS)3. Patients with heart failure frequently develop kidney dysfunction, and recent observational findings showed that elevated central venous pressure rather than low cardiac output strongly correlated with worsening renal function during hospitalization4,5. Therefore, renal dysfunction caused by increased renal venous pressure (renal congestion) is being increasingly recognized as the pathophysiology responsible for CRS and also as a strong predictor of the outcomes of HF patients6,7,8,9.
The pathophysiology of renal congestion is multifactorial and has mainly been investigated with a focus on hemodynamic and hormonal responses to elevated renal venous pressure8. The systemic activation of the renin-angiotensin-aldosterone system and sympathetic nervous system in congestive heart failure promotes inflammatory responses in endothelial cells10. In addition, increased renal interstitial pressure caused by congestion reduces renal blood perfusion, which causes local tissue hypoxia, called renal compartment syndrome11. However, due to a lack of appropriate mouse models of renal congestion, the underlying molecular mechanisms have not been sufficiently elucidated12. In addition, since renal congestion is typically accompanied by low cardiac output in HF patients, difficulties are associated with solely investigating the congestion-mediated worsening of renal function.
After acute kidney injury (AKI), regardless of its causes, some patients develop chronic kidney disease (CKD), called AKI to CKD transition13,14,15. Common pathological features of CKD are the loss of nephrons and their replacement with tissue fibrosis. Appropriate cell-cell interactions at the correct timepoints are required for adequate tissue repair after AKI16. However, in the case of severe injury, injured tubular epithelia secrete various pro-fibrotic and pro-inflammatory humoral mediators that accelerate tissue fibrosis and local inflammation, which eventually promotes maladaptive repair17,18,19,20,21,22. Due to the poor prognosis of HF patients with CKD, a preventative approach against congestion-mediated AKI to CKD transition is essential for better outcomes in congestive HF patients. However, injured tubule-specific cell-cell interactions in congestive kidneys remain unclear.
We recently developed a novel mouse model with latent unilateral renal congestion23. We showed that a decrease in renal blood flow and the accumulation of neutrophils within capillaries were responsible for the congestion-mediated worsening of ischemia-reperfusion injury (IRI)23. Therefore, the present study investigated the molecular mechanisms underlying this process using transcriptomics, with a focus on injured tubule-specific cell-cell interactions. We identified injured tubule-derived Cellular Communication Network Factor 1 (CCN1) in the acute phase after IRI with renal congestion and examined its role in the congestion-mediated worsening of IRI and subsequent tissue fibrosis.
Methods
Animal experiments
We generated mice with the CreERT2 cassette in the SLC34a1 locus, which enables the expression of Cre recombinase in the proximal tubules after a tamoxifen injection24. SLC34a1GCE mice were crossed with R26tdTomato reporter mice, in which tdTomato is expressed after the Cre-mediated recombination of the floxed stop cassette to obtain bigenic offspring. Regarding genetic labeling, tamoxifen (Sigma-Aldrich, St. Louis, MO) was dissolved in 3% (vol/vol) ethanol containing corn oil (Sigma-Aldrich). The mixed liquid, which contained 10 mg of tamoxifen in corn oil, was injected orally five times every other day. We generated tubule-specific CCN1 knockout mice (CCN1-KO) by crossing SLC34a1GCE mice and homozygous CCN1 floxed mice (a gift from Prof. Lester F Lau in Illinois Univ, US)25,26. These gene-edited mice and male C57BL/6 mice (purchased from Shimizu, Inc., Kyoto, Japan) were used in experiments and these mice were 10 to 12-week-old male mice.
The surgical contraction of the inferior vena cava (IVC) (IVCC) was performed as previously described23. At the age of 10 to 12 weeks, male mice were anesthetized with isoflurane. After an abdominal median incision, the IVC between the right and left renal veins was carefully exposed. The isolated IVC and a 22-gauge needle were tied using surgical sutures (7 − 0 nylon). The 22-gauge needle was subsequently removed and the wound was closed. Six hundred microliters of saline was injected into the abdominal cavity to maintain body fluids. The same surgical procedure was performed on sham-operated mice, except for IVCC, which were assigned as uninjured mouse kidneys.
In some experiments, bilateral IRI surgery was simultaneously performed as previously described24,27. Both kidneys were exposed and mice were subjected to ischemia by clamping the renal pedicle with a non-traumatic micro aneurysm clamp (Roboz Surgical Instrument Co., Rockville, MD, USA), which was removed after 20 min. During the surgical procedure, mice were kept on a heat pad to maintain their body temperature and the intestines were covered with cotton gauze soaked with warm saline.
All the mice were randomly allocated into control groups or models, however, investigators were not blind to surgical or treatment groups. Mice and rats were housed under pathogen‑free conditions (temperature, 25˚C; 12‑h light/dark cycle) and had free access to normal chow and water. All experiments were conducted following approval from the Kyoto Prefectural University of Medicine Animal Experiment Committee (Approval number: #2024-107−1) and in accordance with facility guidelines, the Guidelines for Proper Conduct of Animal Experiments of the Japanese Ministry of Education, Culture, Sports, Science and Technology, and ARRIVE guidelines.
Doppler echography, echocardiography, and blood pressure measurements
Doppler echography and blood pressure measurements were performed as previously described23,28. Doppler echography of the renal veins was conducted using the 2100 Imaging System (VisualSonics Inc., Toronto, Canada). During echography, the heart rate of mice was kept between 400 and 500 beats/min by adjusting the depth of anesthesia with isoflurane (3.0–5.0% for induction; 1.0–2.0% for maintenance). Heart rate and blood pressure were measured using the non-invasive tail cuff method (BP-98 A, Softron Co., Ltd., Tokyo, Japan) before IVC coarctation surgery and 1 day after surgery.
Tissue Preparation and histology
Mice were anesthetized and sacrificed, and the kidneys were removed at the indicated time points. The kidneys were cut into pieces for further analyses. To obtain frozen sections, the kidneys were fixed with 4% paraformaldehyde (Wako Pure Chemical Industries, Ltd., Osaka, Japan) on ice for 1 h, incubated in 30% (vol/vol) sucrose in PBS at 4 °C overnight, embedded in optimum cutting temperature compound (Sakura Finetek Japan Co., Ltd., Tokyo, Japan), and cut into 8-µm-thick sections. To obtain paraffin sections, the kidneys were fixed with 4% paraformaldehyde and embedded in paraffin by Applied Medical Research Laboratory (Osaka, Japan). Paraffin-embedded tissues were cut into 4-µm-thick sections. Periodic acid-Schiff (PAS) staining and Picro-Sirius Red staining were performed according to standard procedures.
The tubular injury score was evaluated using PAS-stained specimens. According to previously described methods17,27,29the degree of tubular injury was scored semi-quantitatively in 5 randomly selected images from 25 consecutive non-overlapping cortical or outer medullary fields of kidney sections stained with PAS under high magnification (n = 5) using the following scoring system: 0, 0%; 1, 1–10%; 2, 11–25%; 3, 26–50%; 4, 51–75%; and 5, 76–100%. Tubular injury was judged by tubular atrophy, tubular dilation, protein casts, necrotic cells, and brush border loss.
The interstitial fibrosis score was evaluated using Picro-Sirius Red-stained specimens. The degree of fibrosis was scored semi-quantitatively in 5 randomly selected images from 25 consecutive non-overlapping cortical or outer medullary fields of kidney sections stained with Sirus-red under high magnification. Interstitial fibrosis (defined as a red-stained area on Picro-Sirius Red staining) was quantified using the following scores: 0, 0%; 1, 1–10%; 2, 11–25%; 3, 26–50%; 4, 51–75%; and 5, 76–100%17,29.
Immunofluorescence analysis
In immunofluorescence staining, frozen sections were rehydrated and permeabilized with 0.5% Triton X-100 in PBS for 5 min. Cells were rehydrated, fixed with 4% paraformaldehyde, and permeabilized with 0.5% Triton X-100 in PBS for 5 min. Samples were then blocked with 3% bovine serum albumin (BSA) (Nacalai Tesque, Kyoto, Japan) in PBS and sequentially incubated with the primary antibodies shown in Supplementary Table S2 at 4 °C overnight, followed by an incubation with the dye-conjugate secondary antibodies shown in Supplementary Table S2 at room temperature for 1 h. Nuclear counterstaining was performed using DAPI or DRAQ5 (DR05500; BioStatus, Leicestershire, UK; 1:200), followed by mounting in Prolong-Gold (Thermo Fisher Scientific, Waltham, MA). Images were obtained by confocal microscopy (FV1000, Olympus).
Immunohistochemical analysis
After deparaffinization and antigen retrieval, endogenous peroxidase was quenched with 3.0% H2O2 in methanol for 20 min. Blockade was performed using 3.0% BSA in PBS for 30 min. Regarding nitrotyrosine staining, blocking was conducted using 2.0% fish gelatin in PBS instead of 3.0% BSA in PBS. Sections were then incubated with the primary antibodies listed in Supplemental Table S2. Frozen sections were used for F4/80 staining instead of paraffin sections. As described in the immunofluorescence analysis, frozen sections were rehydrated and permeabilized with 0.5% Triton X-100 in PBS for 5 min. Sections were then blocked with 3% BSA (Nacalai Tesque, Kyoto, Japan) in PBS and sequentially incubated with the primary antibodies listed in Supplementary Table S2 at 4 °C overnight. All sections were incubated with the secondary antibodies shown in Supplemental Table S2. Diaminobenzidine chromogenic substrate (K3468, Agilent Technologies, Santa Clara, CA) was used for color visualization followed by counterstaining with hematoxylin. All sections were observed using BZ-X800 (Keyence Corporation, Osaka, Japan).
Separation of tdTomato-positive proximal tubular epithelia using FACS
The separation of tdTomato-positive proximal tubular epithelia using FACS was performed as previously described30,31,32. The kidney cortex was minced and a single-cell suspension was generated via research-grade Liberase TL (5401020001: Sigma-Aldrich) and 60 units/ml of DNAse Inhibitor (D5025:Sigma-Aldrich) at 37 °C for 30 min. Cells were washed with PBS two times, filtered through 70- and 40-µm cell strainers, resuspended in PBS and 10% fetal bovine serum (FBS) with 500:1 DAPI (2 mg/mL), and subjected to FACS using MA900 (SONY). Dead cells (DAPI-positive cells) were excluded during FACS. tdTomato-positive and DAPI-negative cells were collected in DMEM and 10% FBS.
Cell culture
Normal rat kidney epithelial cells (NRK52E) and normal rat kidney fibroblasts (NRK49F) were obtained from the JCRB Cell Bank. NRK52E and NRK49F were cultured in DMEM (Fujifilm Wako, Osaka, Japan) containing 1% penicillin and streptomycin (Invitrogen, Carlsbad, CA) and 5% FBS (Invitrogen) at 37 °C in a humidified 5% CO2 and 95% air atmosphere.
At the indicated time points, human recombinant CCN1 (PeproTech, Rocky Hill, NJ, USA) was applied for 10–30 min in the Western Blot analysis, for 3–24 h in qPCR; TGFβ (PeproTech) was applied for 3 h in qPCR; the focal adhesion kinase (FAK) inhibitor GSK2256098 (Selleck Biotech, Houston, TX) for 10 min in the Western Blot analysis; H2O2 for 24 h in qPCR; or vehicle was added to the medium.
Rat bone marrow-derived macrophage (BMM) isolation
A primary culture of BMM was obtained as previously described33,34. In brief, Wister rats were anesthetized and sacrificed, and the femurs and tibias were removed. Both ends of these bones were cut, and bone marrow was washed out by PBS. To differentiate to macrophages, bone marrow cells were cultured in complete medium (DMEM/F12 (Thermo Fisher Scientific, Waltham, MA) supplemented with 10% FBS,1% penicillin/streptomycin, and 20% of L-929 cell-conditioned medium for 5 days after which the cells were used for analysis.
Cell proliferation assay
Cells were seeded at a density of 1 × 103 cells/well in 96-well plates. The number of cells was counted using an ADAM-MC automatic cell counter (Digital Bio, Japan) that functions using the propidium iodide staining method for dead cells. After overnight growth, the medium was replaced with fresh medium or changed to medium containing CCN1 or H2O2 (Fujifilm Wako, Osaka, Japan). Twenty-four hours later, the number of viable cells in each well was measured using the Premix WST-1 Cell Proliferation Assay System (Takara Bio, Shiga, Japan) according to the manufacturer’s instructions.
Wound healing and single-cell migration assay
Cells were seeded on a 6-well plate and cultured until 90–95% confluent. Artificial scratch gaps (wounds) were made in each cell monolayer using a 1000-µl pipette tip, cells were then immediately washed by PBS two times, and medium was replaced with fresh culture medium. Digital images of the wounded area were acquired at 0 and 24 h. Free Java-based Image J image processing software (Wayne Rasband, National Institutes of Health, Bethesda MD, USA) was used to calculate cell migration by measuring the wounded area at 0 and 24 h.
In the single-cell migration assay on NRK49F and BMM, cells were seeded on a 6-well plate. A few hours later, only NRK49F, not BMM, were treated with mitomycin C (Fujifilm Wako, Osaka, Japan) at 3 µg/ml for 1 h to inhibit cell proliferation. Thereafter, cells were immediately washed by PBS two times and culture medium was replaced with fresh culture medium containing CCN1. Live cell imaging of individual cell tracking was monitored by the JuLI™ Stage Cell History Recorder (NanoEntek, Korea) every 10 min for 24 h and migration data were collected. Tracking data were analyzed using the Tracking Tool software (Gradientech). The mean accumulated distance of migrated NRK49F and BMM was analyzed.
Hypoxia assay
Hypoxia was induced as previously described35. NRK52E were seeded on a 6-well plate and cultured in DMEM. To induce hypoxia, cells were cultured in a gas barrier pouch bag for 24 h using the nBIONIX Hypoxic Cell Culture Kit (AR Brown Co., Ltd., Tokyo, Japan).
Western blot analysis
Total cell extracts were obtained using lysing Buffer 17 (895943: R&D Systems, Inc., Minneapolis, MN, USA) at room temperature. After sonication, proteins were denatured by heating at 95 °C for 5 min and separated by SDS-PAGE. Proteins were then transferred onto polyvinylidene difluoride membranes (Immobilon-P IPVH00010: Millipore, MA, USA). After blocking with 5% non-fat milk or 3% BSA in TBS/0.1% Tween20 at room temperature for 1 h, the membrane was incubated with the corresponding primary antibody (Supplementary Table S1) at 4 °C overnight. After washing with TBS/0.1% Tween20, secondary antibodies were added (Supplementary Table S1). Chemiluminescence was detected using an ECL select Western blot detection reagent (RPN2235: GE Healthcare UK Ltd., Amersham Place, England) or Clarity Max Western ECL substrate (1705062: Bio-Rad Laboratories, Inc., Hercules, CA, USA). Signal intensities were evaluated using Image J software (National Institutes of Health, Bethesda, MD).
RNA extraction and real-time quantitative PCR
Total RNA was extracted from the cortex of kidneys, FACS-sorted cells, NRK49F, NRK52E, or BMM using NucleoSpin® RNA (Macherey-Nagel, Duren, Germany). Two hundred nanograms of total RNA was reverse transcribed to synthesize cDNA using a PrimeScripttm RT Master Mix (RR036A: Takara Bio Inc., Shiga, Japan). The real-time detection of PCR products was performed using KAPA SYBR FAST qPCR Master Mix (2×) Universal (KK4602:Kapa Biosystems, Wilmington, MA) and a Thermal Cycler Dice Real Time System (Takara Bio Inc., Shiga, Japan). Gene expression was quantified using GAPDH and β-actin as an internal control. Primers are listed in Supplementary Table S3.
RNA sequencing (RNA-seq)
RNA samples were provided to the NGS core facility of the Genome Information Research Center at the Research Institute for Microbial Diseases of Osaka University for library construction and sequencing. Library preparation was performed using a TruSeq-stranded mRNA sample prep. kit (Illumina, San Diego, CA) according to the manufacturer’s instructions. Sequencing was conducted on an Illumina NovaSeq6000 platform in the 100-bp single-end mode. Sequenced reads were mapped to mouse reference genome sequences (GRCm38) using STAR v 2.7.10a (https://github.com/alexdobin/STAR/) and uniquely mapped reads were counted by the featureCounts function in the Subread package (https://subread.sourceforge.net).
Bioinformatic analysis
Data were analyzed using R software version 4.3.2 (https://www.R-project.org/). The edgeR package was used for a differential expression analysis36. Genes with p value < 0.05 and an absolute log2 fold change > 1 were considered to be significant differentially expressed genes. The fgsea package was used for a gene set enrichment analysis. We used pheatmap package to generate the heatmap.
Statistical analysis
Results are expressed as the mean ± SEM. Each experiment was performed using at least 5 mice per group. Quantification was conducted using at least 5 high-power field images for each kidney section. Statistical analyses were performed by the paired t-test for comparisons between the congested side and contralateral side, by the unpaired t-test for comparisons of two variables, and by a one-way ANOVA and Dunnett’s post hoc test for comparisons of multiple variables. P-values < 0.05 were considered to be significant.
Results
Renal congestion exacerbates IRI and subsequent tissue fibrosis
To elucidate the impact of renal congestion on the transition of AKI to CKD, we investigated the effects of IRI in the unilateral renal congestion model where IVC between the left and right renal veins was constricted, causing renal congestion only in the left kidney23. We performed IVCC, simultaneously introduced renal IRI in both kidneys by 20 min of clamping (Fig. 1a), and sacrificed mice 1, 3 or 7 days after surgery (Fig. 1b). Systolic blood pressure did not significantly differ after IVCC (Fig. 1c). Ultrasonography of the kidneys revealed dilatation of the left renal vein after IVCC (Fig. 1d and e). Doppler ultrasonography demonstrated that the peak velocity of renal venous flow was markedly reduced and its pulse wave disappeared in congestive kidneys (Fig. 1d and f). A histological analysis of PAS staining of the kidney 1 day after surgery and a semi-quantitative analysis revealed extensive tubular injury in IRI kidneys with or without renal congestion (Cong-IRI and CLK-IRI, respectively, Fig. 1g and h). Immunohistochemistry for kidney injury molecule-1 (KIM1) and a macrophage marker (F4/80) showed stronger positive staining for KIM1 and the accumulation of macrophages in both Cong-IRI and CLK-IRI (Fig. 1g, Supplementary Fig S1a). A histological analysis of Sirius red staining 7 days after surgery revealed tissue fibrosis in Cong-IRI and CLK-IRI; however, it was more prominent in Cong-IRI (Fig. 1i). A semi-quantitative analysis showed tissue fibrosis in both the cortex and medulla of IRI kidneys, and its exacerbation by renal congestion (Fig. 1j). qPCR analyses over time revealed that the up-regulated expression of a proximal tubular injury marker (Havcr1) by IRI was sustained in Cong-IRI (Fig. 1k). The up-regulated expression of fibrosis markers (Col1a1 and Fn1) at later time points was more pronounced in Cong-IRI than in CLK-IRI (Fig. 1k). No significant differences were observed in the expression of markers for tubular integrity (Lrp2), macrophages (Cd68), or neutrophils (Ly6g) between Cong-IRI and CLK-IRI on day 1 (Fig. 1k, Supplementary Figure S1b).

Renal congestion exacerbates ischemia-reperfusion injury and subsequent fibrosis. (a). Scheme of unilateral inferior vena cava (IVC) constriction between renal veins (IVCC) and bilateral IRI surgery. (b). Experimental scheme. IVCC and bilateral IRI surgery were performed on 10-week-old male mice, which were sacrificed 1, 3, and 7 days later. (c) Systolic blood pressure measurements before and after IVCC. (d) Two-dimensional and Doppler echography of renal veins before and after IVCC. Yellow lines and red arrowheads indicate the border of the kidney and renal veins, respectively. (e) Diameters from renal vein measurements. (f) Velocity from renal vein measurements. N = 5 for CLK or Cong mice, n = 3 for CLK-IRI or Cong-IRI mice in (c, e, and f). (g) Images of periodic acid Schiff (PAS) staining and immunostaining of kidney sections for KIM-1 1 day after surgery. (h) Semiquantitative analysis of scores for tubular injury 1 day after surgery. (i) Images of Picro-Sirius red staining 7 days after surgery. (j) Semiquantitative analysis of fibrosis scores. N = 6 for CLK or Cong mice, n = 9 for CLK-IRI or Cong-IRI mice in (h, and j). (k) qPCR of RNA from whole kidneys for the representative markers for tubular injury (Havcr1) and fibrosis (Col1a1 and Fn1). N = 5–10 for each group. All mRNA values are expressed as relative expression to that in uninjured kidneys. In all groups, data are means ± SEM. Statistical analyses were performed using the paired t-test between CLK and Cong, and a one-way ANOVA and Dunnett’s post hoc test for comparisons of multiple variables in (c, e, f, h, j and k). *P < 0.05. Bars = 40 μm in PAS, KIM1, and Sirius Red staining.
Renal congestion promotes the sustained up-regulation of CCN1 in IRI kidneys
To investigate changes in molecular pathways, we examined differences in the transcriptome of kidney tissue between CLK-IRI and Cong-IRI by RNA-seq (Fig. 2a). We annotated gene expression using the Kyoto Encyclopedia of Genes and Genomes pathway analysis, and among the top 10 significantly enriched up-regulated pathways, paracrine chemokine-related pathways (ECM receptor interaction, cytokine-cytokine receptor interaction, and chemokine signaling pathway) were identified in Cong-IRI (Fig. 2b). Based on the up-regulation of chemokine/cytokine pathways in RNA-seq, we performed a time-course analysis of the expression of major humoral mediators. The gene expression of Ccn1 (encoding CCN1), Ccn2 (encoding CTGF), and Tnfa was up-regulated at early time points in IRI kidneys, while that of Ccl2, Tgfb, and Pdgfb was up-regulated at later time points (Fig. 2c). Based on the sustained up-regulation of Ccn1 at later time points in Cong-IRI and the findings of single-cell based ligand-receptor interaction analyses in a previous study37we focused on the role of CCN1 in the congestion-mediated worsening of tissue fibrosis after IRI.

Transcriptomics of the IRI kidney with renal congestion. (a) Clusters of differentially expressed genes (DEGs) in the renal tissues of IRI kidneys with and without renal congestion. Each column represents a mouse sample and each row represents a gene. Red indicates up-regulation, and blue shows down-regulation. Each group has 3 samples. (b) The Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis of DEGs. The top 10 up-regulated pathways in IRI kidneys with renal congestion are displayed. (c) qPCR of representative markers of pro-inflammatory cytokines (Ccl2 and Tnf) and pro-fibrotic cytokines (Ccn1, Tgfb1, Ccn2, and Pdgfb) in whole kidneys. N = 5–10 for each group. All mRNA values are expressed as relative expression to that in uninjured kidneys. In all groups, data are means ± SEM. Statistical analyses were performed using the paired t-test between CLK and Cong, and a one-way ANOVA and Dunnett’s post hoc test for comparisons of multiple variables in (c). *P < 0.05.
Oxidative stress promote the up-regulation of CCN1 in tubular epithelial cells
IRI is a dual-phased tissue injury, e.g., a hypoxic phase followed by reperfusion-induced oxidative stress38. Therefore, we performed an in vitro analysis using the rat tubular epithelial cell line NRK52E to investigate whether the secretion of CCN1 following renal tubular injury is promoted by oxidative stress or hypoxia. In NRK52E exposed to oxidative stress using H2O2, the expression of oxidative stress markers (Hmox1, Sod1, Sod2, and Cat), Havcr1, and Ccn1 was significantly up-regulated in a dose-dependent manner (Fig. 3a). We also examined the effects of oxidative stress in congestive kidneys. Hmox1 and Sod1 gene expression was higher in Cong-IRI than in CLK-IRI (Fig. 3b). Immunostaining for nitrotyrosine, a marker of oxidative stress, revealed a marked increase in its expression in Cong-IRI (Fig. 3c). After a 24-hour exposure to hypoxia, the expression of Gapdh, one of the hypoxia-inducible factor target genes, and Havcr1 were up-regulated (Fig. 3d). Ccn1 was tended to be up-regulated, but there was not statistical significance (Fig. 3d).

Oxidative stress induces the secretion of CCN1 from tubular epithelial cells. (a) qPCR of Ccn1, the tubular injury marker (Havcr1), and oxidative stress markers (Homx1, Cat, Sod1 and Sod2) in NRK52E treated with H2O2 for 24 h (n = 3 each). All mRNA values are expressed as relative expression to that in untreated cells. (b) qPCR of oxidative stress markers (Homx1, Cat, Sod1, and Sod2) in kidney tissue. N = 6 for CLK or Cong mice, n = 10 for CLK-IRI or Cong-IRI mice. All mRNA values are expressed as relative expression to that in uninjured kidneys. (c) Immunostaining of kidney sections for nitrotyrosine (NT) in wild-type mice. (d) qPCR of Ccn1, Gapdh, and Havcr1 in NRK52E cultured under hypoxic conditions for 24 h (n = 3 each). In all groups, data are means ± SEM. Statistical analyses were performed using the paired t-test between CLK and Cong, the unpaired t-test for comparisons of two variables, and a one-way ANOVA and Dunnett’s post hoc test for comparisons of multiple variables in (a, b, and d). *P < 0.05. Bars = 40 μm in (c).
CCN1 accelerates fibroblast migration and proliferation through FAK signaling
Based on the up-regulation of CCN1 in Cong-IRI, we comprehensively examined the effects of CCN1 on various cell populations. Previous studies showed that the migration of detached fibroblasts played an essential pathophysiological role in the congestion-mediated worsening of kidney fibrosis39,40; therefore, we examined fibroblast migration using NRK49F, rat kidney fibroblasts. A single-cell migration analysis showed that CCN1 promoted the migration of NRK49F (Fig. 4a, Supplementary movie S1, S2). CCN1 also enhanced the proliferation of NRK49F in a dose-dependent manner (Fig. 4b). Regarding extracellular matrix (ECM) synthesis and transdifferentiation to myofibroblasts, CCN1 did not affect the gene expression of Acta2, Col4a1, or Fn1 (Fig. 4c). Regarding downstream signaling molecules, a Western blot analysis revealed that the phosphorylation of FAK and its downstream molecules, such as Akt and Erk, was increased by CCN1 in NRK49F (Fig. 4d). Immunostaining for pFAK and phalloidin showed marked increases in pFAK expression in CCN1-treated NRK49F (Fig. 4e). We also examined the expression of pFAK in vivo. In an immunofluorescence analysis of kidneys, PDGFRβ + interstitial fibroblasts were positive for pFAK + cells in Cong-IRI (Fig. 4f). pFAK + cells accumulated around KIM1 + injured tubular epithelial cells in Cong-IRI (Fig. 4g).

CCN1 accelerates the migration and proliferation of fibroblasts through focal adhesion kinase signaling. (a) Analysis of the migration distance of NRK49F treated with or without CCN1 (2 µg/ml) for 24 h by the Tracking Tool software (Gradientech). (b) Cell proliferation assay data indicating that CCN1 accelerated the proliferation of NRK49F (n = 6 each). (c) qPCR of representative markers for myofibroblasts (Acta2) and the extracellular matrix (Col4a1 and Fn1) in NRK49F treated with CCN1(2 µg/ml) or TGFβ (10 ng/ml) for 24 h (n = 3 each). (d) Western blot analysis of pFAK, FAK, pERK, ERK, pAKT, AKT, and GAPDH in NRK49F with CCN1 (2 µg/ml) (n = 3 each). The optical density of the bands for pFAK, pAKT, and pERK were normalized against those for FAK, AKT, and ERK. (e) Co-immunofluorescence images of pFAK and Phalloidin in NRK49F treated with CCN1. (f) Co-immunofluorescence images of pFAK and PDGFRb in wild-type mice. (g) Co-immunofluorescence images of pFAK and Kim1 in wild-type mice. In all groups, data are means ± SEM. Statistical analyses were performed by the unpaired t-test for comparisons of two variables, and by an ANOVA and Dunnett’s post hoc test for comparisons of multiple variables in (a, b, c, and d). *P < 0.05. Bars = 20 μm in (e, f, and g).
Regarding the effects of the pharmacological inhibition of FAK on CCN1 signaling, Western blotting showed that the FAK inhibitor reduced the CCN1-mediated phosphorylation of FAK and ERK in NRK49F (Fig. 5a). Furthermore, the FAK inhibitor reduced the CCN1-induced migration of NRK49F (Fig. 5b). An immunofluorescence analysis revealed the down-regulation of pFAK and phalloidin in NRK49F treated with the FAK inhibitor (Fig. 5c).

The FAK inhibitor reduced the CCN1-mediated migration of fibroblasts. (a) Western blot analysis of pFAK, FAK, pERK, ERK, pAKT, AKT, and GAPDH in NRK49F with or without CCN1 (2 µg/ml) and/or the FAK inhibitor (10 µM) (n = 3 each). The optical density of the bands for pFAK, pAKT, and pERK were normalized against FAK, AKT, and ERK. (b) Analysis of the migration distance of NRK49F treated with or without CCN1 (2 µg/ml) and/or the FAK inhibitor (10 µM) for 24 h by the Tracking Tool software (Gradientech). (c) Co-immunofluorescence images of pFAK and Phalloidin in NRK49F treated with CCN1 (2 µg/ml) and/or the FAK inhibitor (10 µM) for 10 min. In all groups, data are means ± SEM. Statistical analyses were performed by an ANOVA and Dunnett’s post hoc test for comparisons of multiple variables in (a, and b). *P < 0.05. Bars = 20 μm in (c).
CCN1 promotes macrophage, not epithelial cell migration
We examined the effects of CCN1 on tubular epithelial cells using NRK52E. A Western blot analysis revealed that the phosphorylation of AKT and ERK was increased by CCN1 in NRK52E (Fig. 6e). In contrast to fibroblasts, CCN1 did not accelerate migration (Fig. 6a and b). However, CCN1 increased the proliferation of NRK52E in a dose-dependent manner (Fig. 6c). CCN1 did not affect the gene expression of mesenchymal markers (Twist1, Snail, Zeb1, and Zeb2), suggesting that CCN1 did not promote epithelial-mesenchymal transition (EMT) (Fig. 6d).

CCN1 promotes the migration of macrophages, but not epithelial cells. (a) Representative images of the scratch migration assay on NRK52E treated CCN1. (b) Quantification of the migrated area of NRK52E (n = 6 each). (c) Cell proliferation assay data indicating that CCN1 accelerated the proliferation of NRK52E (n = 6 each). (d) qPCR of representative markers for epithelial-mesenchymal transition (Twist1, Snail, Zeb1, Zeb2) in NRK52E treated with CCN1 (2 µg/ml) or TGFβ (10 ng/ml) for 3 h (n = 3 each). (e) Western blot analysis of pERK, ERK, pAKT, AKT, and GAPDH in NRK52E treated with CCN1 (2 µg/ml) (n = 3 each). The optical density of the bands for pAKT and pERK were normalized against those for AKT and ERK. (f) Transparent and immunofluorescence (F4/80) images of bone marrow-derived macrophages (BMM). (g) Analysis of the BMM migration distance for 24 h after the CCN1 (1, 2, and 5 µg/ml) treatment for 24 h by the Tracking Tool software (Gradientech). (h) qPCR of representative markers for macrophage polarization (Tnfa, Ym1) in BMM treated with CCN1 (2 µg/ml) for 24 h (n = 3 each). (i) Western blot analysis of pFAK, FAK, pERK, ERK, and GAPDH in BMM treated with CCN1 (2 µg/ml) (n = 3 each). The optical density of the bands for pFAK and pERK were normalized against those for FAK and ERK. (j) Co-immunofluorescence images of pFAK and F4/80 in wild-type mice. In all groups, data are means ± SEM. Statistical analyses were performed by the unpaired t-test for comparisons of two variables and by an ANOVA and Dunnett’s post hoc test for comparisons of multiple variables in (b, c, d, e, g, h, and i). *P < 0.05. Bars = 20 μm in (j).
We also examined the effects of CCN1 on macrophage phenotypes. A primary culture of rat bone BMM was obtained, and we confirmed that all cells were positive for F4/80 (Fig. 6f). A single-cell migration analysis showed that CCN1 accelerated the migration of BMM in a dose-dependent manner (Fig. 6g, Supplementary movie S3, S4). Regarding macrophage polarization, CCN1 did not affect the gene expression of the M1 or M2 marker (Tnfa and Ym1, respectively, Fig. 6h). A Western blot analysis revealed that the phosphorylation of FAK and ERK was increased by CCN1 in BMM (Fig. 6i). In vivo, an immunofluorescence analysis showed that F4/80 + cells (macrophages) were positive for pFAK + in Cong-IRI (Fig. 6j). Overall, CCN1 promoted the migration of fibroblasts and macrophages, but not tubular epithelial cells through CCN1-FAK signaling.
Genetic deletion of CCN1 in tubular epithelial cells ameliorated the congestion-mediated worsening of AKI and subsequent tissue fibrosis
To elucidate the specific role of injured tubule-derived CCN1 in the progression of tissue fibrosis in congestive kidneys, we performed in vivo experiments using CCN1-KO mice. To directly confirm the deletion of CCN1 in proximal tubular epithelial cells, we generated trigenic mice carrying a heterozygous SLC34a1GCE allele, heterozygous R26tdTomato allele, with or without a homozygous floxed CCN1 gene. After the induction of exclusive recombination in proximal tubules by multiple oral doses of tamoxifen, mice were sacrificed 1 day after IRI (Supplementary Figure S2a, S2b). We collected tdTomato + tubular epithelia by FACS and RNA was extracted (Supplementary Figure S2c). RT-PCR showed that the gene expression of Havcr1 was up-regulated after IRI in both WT and CCN1-KO mice, whereas CCN1 was up-regulated in WT mice only, indicating that its expression in injured tubular epithelial cells was successfully disrupted in CCN1-KO mice (Supplementary Figure S2d).
We then examined the effects of CCN1-KO on the congestion-mediated progression of tissue fibrosis. After the induction of exclusive recombination in the proximal tubules by multiple oral doses of tamoxifen, mice were sacrificed 1 or 7 days after IRI +/- IVCC (Fig. 7a). One day after surgery, a histological analysis revealed extensive tissue damage in both WT and CCN1-KO kidneys, although it was slightly less severe in CCN1-KO mice (Fig. 7b and c). Immunohistochemistry for KIM1 and F4/80 showed no significant change between WT and CCN1-KO mice (Fig. 7b). qPCR of Havcr1, Ccl2, and Cd68 showed no significant differences between WT and CCN1-KO mice (Fig. 7d) An immunofluorescence analysis revealed the presence of pFAK + cells around KIM1 + injured tubules in Cong-IRI kidneys of WT mice, and there were fewer of these cells in CCN1-KO mice (Fig. 7e). Interstitial pFAK + cells were positive for PDGFRβ or F4/80 in the Cong-IRI kidneys of WT mice, and there were fewer of these cells in CCN1-KO kidneys (Fig. 7f and g). These results suggest that tubule-specific CCN1-KO inhibited the accumulation of fibroblasts or macrophages around injured tubules.

The PT-specific knockout of CCN1 inhibits the accumulation of fibroblasts and macrophages at injured tubules in congestive kidneys. (a) IVCC and bilateral IRI surgery were performed on 10-week-old male mice, which were then sacrificed 1 and 7 days later. (b) Images of periodic acid Schiff (PAS) staining and immunostaining for KIM-1 and F4/80 in kidney tissue obtained 1 day after surgery. (c) Semiquantitative analysis of scores for tubular injury 1 day after surgery. N = 9 for WT mice and n = 11 for CCN1-KO mice (d) qPCR analysis of Ccn1, Havcr1, Ccl2, and Cd68 in kidney tissues 1 day after surgery. N = 9 for WT mice and n = 11 for CCN1-KO mice. All mRNA values are expressed as relative expression to that in uninjured kidneys. (e), (f), (g) Co-immunofluorescence images of PDGFRβ, KIM1, F4/80, and pFAK. In all groups, data are means ± SEM. Statistical analyses were performed using the paired t-test between CLK and Cong in (c and d). *P < 0.05. Bars = 40 μm in PAS staining and KIM1 staining and 20 μm in F4/80 staining in (b). Bars = 20 μm in (e, f, and g).
Seven days after surgery, Sirius Res staining and its semi-quantitative analysis showed that the congestion-mediated worsening of tissue fibrosis was no longer detectable in CCN1-KO mice (Fig. 8a and b). qPCR analyses revealed the congestion-mediated up-regulation of the mRNA expression of markers for tubular injury, tissue fibrosis, and macrophages in WT mice, whereas these differences were canceled in CCN1-KO mice (Fig. 8c). Collectively, these results suggest that injured tubule-derived CCN1 contributed to the progression of the congestion-mediated worsening of kidney fibrosis after injury.

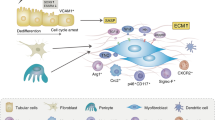
The PT-specific knockout of CCN1 inhibits congestion-mediated fibrosis after IRI. (a) Images of Picro-Sirius red staining in the kidneys 7 days after surgery. (b) Semiquantitative analysis of fibrosis scores. N = 9 for WT mice and n = 13 for CCN1-KO mice. (c) qPCR of representative markers for a pro-inflammatory cytokine (Ccl2), pro-fibrotic cytokines (Tgfb1 and Ccn2), fibrosis (Col1a1, Col3a1, and Fn1), mesenchymal stem cells (Vim), tubular injury (Havcr1), myofibroblasts (Acta2), and macrophages (Cd68) in kidney tissues 7 days after surgery. N = 10 for WT mice and n = 13 for CCN1-KO mice. All mRNA values are expressed as relative expression to that in uninjured kidneys. (d) Scheme for the proposed mechanisms by which injured tubule-derived CCN1 exacerbates renal congestion-mediated acute kidney injury and fibrosis. In all groups, data are means ± SEM. Statistical analyses were performed using the paired t-test between CLK and Cong in (b, and c). *P < 0.05. Bars = 40 μm in (a).
Discussion
Emerging clinical evidence sheds light on the importance of renal congestion in the development of renal dysfunction in HF patients4,5,41,42. However, due to a lack of appropriate murine renal congestion models, experimental studies mainly focused on hemodynamic changes in congestive kidneys using large animals, such as rats and dogs12and research on the molecular mechanisms underlying renal congestion is limited. The major advantage of the present study is that the unilateral renal congestion mouse model we established recently enabled us to investigate the molecular pathogenesis of the congestion-mediated worsening of renal injury using a combination of transcriptomics and genetically engineered animals. In the present study, using a newly developed mouse unilateral renal congestion model, we comprehensively analyzed three important pathophysiological relationships: kidney injury with increased venous pressure, AKI to CKD transition, and cell-cell interactions in congestive kidneys. While renal fibrosis is accelerated in the chronic phase after injury in congestive kidneys, we found that CCN1 was activated early in congestive kidneys and also that fibroblasts and macrophages accumulated around injured tubules, resulting in accelerated fibrosis. Taken together with the results of the in vitro analysis, the congestion-mediated promotion of oxidative stress after injury appeared to induce the up-regulation of CCN1 in epithelial cells, thereby contributing to tissue fibrosis after injury (Fig. 8d).
Based on previous findings, hemodynamic and pathological changes occur in renal congestion, which may be major contributors to the congestion-mediated worsening of IRI6,9,10. Regarding hemodynamic changes, the renal arteriovenous pressure gradient is important for maintaining renal blood perfusion9; therefore, increased venous pressure reduces the arterio-venous gradient, resulting in less blood perfusion. A hemodynamic examination using isolated dog kidneys revealed that similar to a decrease in arterial pressure, with an increase in renal venous pressure, renal blood flow and urine output decrease, which eventually leads to renal dysfunction43,44. In the present study, blood flow was reduced in congestive kidneys after IRI, which may have exacerbated tissue hypoxia and oxidative stress, resulting in the worsening of tissue injury and subsequent tissue fibrosis.
Transcriptomics of renal tissues in our experiments showed increased focal adhesion, cytokine-cytokine receptors, and chemokine signaling activation, indicating that paracrine effects between various cell populations contribute to the pathophysiology of renal congestion. Previous ligand-receptor interaction analyses from single-cell RNA-seq in the acute phase of IRI revealed a stronger interaction between CCN1 from tubular epithelia and its downstream signaling molecules in fibroblasts37. Consistent with these findings, serial measurements of the humoral factors exerting paracrine effects in the present study showed that CCN1 and CCN2 were elevated at earlier time points after injury, while the well-known pro-fibrotic factor TGFβ and PDGF increased in a later phase, suggesting that CCN1 plays a different role from these molecules on the worsening of IRI in congestive kidneys.
CCN1 binds to integrins and activates its downstream signaling molecules, including FAK and ERK, in various cells45,46. In the present study, we comprehensively investigated the effects of CCN1 on fibroblasts, macrophages, and tubular epithelial cells, which are major contributors to the development of renal fibrosis after tissue injury. We verified that CCN1 induced the migration and proliferation of fibroblasts in vitro, which were canceled by the FAK inhibitor. Furthermore, in vivo, pFAK-positive fibroblasts clustered around injured tubules in congested kidneys, and this was ameliorated in tubular epithelial-specific CCN1-KO mice. However, unlike the TGFβ stimulation, CCN1 did not facilitate the transdifferentiation from fibroblasts to myofibroblasts, which are major contributors to the production of ECM in fibrotic tissue. Previous studies showed that an increase in venous pressure in congestive kidneys induced the dilatation of peritubular capillaries, resulting in the detachment of pericytes39,40. Pericytes are a subpopulation of interstitial mesenchymal cells that attach to capillaries, and in the case of tissue injury, they detach, activate, and eventually produce ECM through transdifferentiation to myofibroblasts47,48,49. More recently, we have demonstrated that conditioned medium from injured tubules promotes strong chemotaxis, and it was canceled by CCN1-KO in tubular epithelia and by FAK inhibition in fibroblasts in vitro50. Taken together with the up-regulation of CCN1, the detachment of pericytes as well as interstitial fibroblast migration may be accelerated in congestive kidneys with IRI.
Regarding the factors regulating the excretion of CCN1 from injured tubules, based on previous findings showing that the up-regulation of CCN1 only occurred in the acute phase after IRI51,52,53,54we focused on the dual processes of IRI: the initial phase of tissue ischemia followed by increased oxidative stress during the reperfusion phase38. Our in vitro experiments showed that oxidative stress up-regulated the expression of CCN1 in tubular epithelial cells in a dose-dependent manner. In addition, our in vivo experiments showed that IRI-induced oxidative stress was further exacerbated by renal congestion. Previous our intravital imaging and the analysis of blood perfusion by laser doppler demonstrated that blood flow velocity was reduced in IRI kidney, and it was further decreased by renal congestion23. The results of these in vivo and in vitro experiments suggest that renal congestion contributed to the exacerbation of ischemia and promotion of oxidative stress from tissue hypoperfusion, resulting in the induction of CCN1 from injured tubules.
Regarding the time-course analysis of CCN1 expression in kidney tissue, although CCN1 was predominantly upregulated at earlier time points in the IRI kidney, its expression was sustained at later time points in the Cong-IRI kidney. There are two possible explanations for this sustained upregulation of CCN1 after injury. First, based on the upregulation of Havcr1 expression in the Cong-IRI kidney at later time points, continued CCN1 secretion may result from residual tubular damage. If the injury is severe, some damage may persist in the tubules, leading to prolonged CCN1 expression. Second, CCN1 may also be secreted by fibroblasts. Indeed, our recent renal single-cell RNA sequencing (scRNA-seq) analysis using a public database suggests that fibroblasts may also express and secrete CCN150. In cases of severe injury, the number of fibroblasts in fibrotic tissue increases, which may contribute to sustained CCN1 expression overall. In this study, we did not analyze CCN1 secretion from other cell populations, including fibroblasts; thus, further investigations are warranted.
After acute injury, surviving and injured tubules dedifferentiate and migrate towards the denuded basement membrane where tubular epithelia are lost, eventually proliferate, and redifferentiate to healthy tubular epithelia after complete tissue repair55,56. The present results on the effects of CCN1 on tubular epithelial cells showed the phosphorylation of ERK and AKT, which stimulated slight cell proliferation, but did not enhance migration. While previous lineage tracing analyses revealed that epithelial cells never transdifferentiate to myofibroblasts through EMT24,27,47,57CCN1 did not up-regulate genes related to partial or intratubular EMT. These findings suggest that during injury and repair, the effects of CCN1 on tubular epithelia were limited to mild proliferation.
Regarding the effect of CCN1 on macrophages, this study showed that CCN1 did not affect macrophage polarization but did enhance migration in a concentration-dependent manner. Previous reports have shown that CCN1 promotes macrophage migration in various models of organ injury58,59and that neutralizing antibodies against CCN1 inhibited macrophage infiltration in renal injury models60which is consistent with our CCN1-KO results. However, another report indicated that CCN1 itself induces polarization toward the M1 phenotype61which contrasts with our in vitro findings. A possible explanation for this discrepancy is that CCN1 has been reported to upregulate inflammatory cytokines shortly after stimulation and at high concentrations61suggesting that our experimental conditions may not have been sufficient to elicit this effect. Nevertheless, since migration was accelerated at the same concentrations, it is likely that CCN1 primarily promotes macrophage migratory function rather than sustained inflammatory activation. Combined with our finding that Tnfa mRNA expression in IRI kidneys was exacerbated by congestion, it is possible that migrated macrophages changes its cellular phenotypes in response to other proinflammatory cytokines and promoting inflammation and subsequent fibrosis.
Our study has several limitations. First, although we have shown that CCN1 is secreted from injured tubular epithelia during the acute phase after injury, its secretion persisted in the Cong-IRI kidney. This prolonged secretion may be due not only to sustained tubular damage, but also to continued production by other cell types, such as fibroblasts and vascular endothelial cells. Second, in this study, we focused on fibroblasts and macrophages as cells that migrate in response to CCN1. However, we were unable to determine which specific cell types primarily migrate in response to CCN1, nor could we assess its effects on the migration of other inflammatory or endothelial cells. Lastly, while immunofluorescence analysis showed that fibroblasts and macrophages migrated towards the injury site in IRI-Cong kidney, which was ameliorated in CCN1-KO mice, however, quantitative analysis was not performed due to technical limitations. Intravital imaging can provide the direct evidences the accumulation of these cells in the injury sites16but future investigations are needed.
In conclusion, we herein demonstrated that the paracrine network centered on injured tubule-derived CCN1 plays an important role in the acceleration of AKI to CKD transition by renal congestion. Approaches to hemodynamic abnormalities using diuretics and hypertensive drugs are important for attenuating the pathogenesis of renal congestion, but are sometimes challenging to perform. Therefore, if molecular biological approaches, such as CCN1, effectively reduce renal damage, they represent a new treatment option for CRS and will reduce the number of HF patients who progress from AKI to CKD.
Data availability
RNA-seq data for all samples were deposited in the Gene Expression Omnibus under the accession number GSE283859. Secure token for reviewer access: ulkpcgisjpyrxud.The data supporting the findings of this study are available in the main manuscript. In addition, by contacting the corresponding author (Kusaba T), we will share models, protocols, methods, and other useful materials and resources related to the article as far as possible. There are no large data files to be shared via online resources.
References
Khan, M. S. et al. Global epidemiology of heart failure. Nat. Rev. Cardiol. 21, 717–734. https://doi.org/10.1038/s41569-024-01046-6 (2024).
Chesnaye, N. C., Ortiz, A., Zoccali, C., Stel, V. S. & Jager, K. J. The impact of population ageing on the burden of chronic kidney disease. Nat. Rev. Nephrol. 20, 569–585. https://doi.org/10.1038/s41581-024-00863-9 (2024).
Zannad, F. & Rossignol, P. Cardiorenal syndrome revisited. Circulation 138, 929–944. https://doi.org/10.1161/CIRCULATIONAHA.117.028814 (2018).
Damman, K. et al. Increased central venous pressure is associated with impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J. Am. Coll. Cardiol. 53, 582–588. https://doi.org/10.1016/j.jacc.2008.08.080 (2009).
Mullens, W. et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J. Am. Coll. Cardiol. 53, 589–596. https://doi.org/10.1016/j.jacc.2008.05.068 (2009).
Husain-Syed, F. et al. Congestive nephropathy: a neglected entity? Proposal for diagnostic criteria and future perspectives. ESC Heart Fail. 8, 183–203. https://doi.org/10.1002/ehf2.13118 (2021).
Bansal, S., Prasad, A. & Linas, S. Right heart Failure-Unrecognized cause of cardiorenal syndrome. J. Am. Soc. Nephrology: JASN. 29, 1795–1798. https://doi.org/10.1681/ASN.2018020224 (2018).
J, F. G., von Haehling, S., Anker, S. D., Raj, D. S. & Radhakrishnan, J. The relevance of congestion in the cardio-renal syndrome. Kidney Int. 83, 384–391. https://doi.org/10.1038/ki.2012.406 (2013).
Ross, E. A. Congestive renal failure: the pathophysiology and treatment of renal venous hypertension. J. Card. Fail. 18, 930–938. https://doi.org/10.1016/j.cardfail.2012.10.010 (2012).
Afsar, B. et al. Focus on renal congestion in heart failure. Clin. Kidney J. 9, 39–47. https://doi.org/10.1093/ckj/sfv124 (2016).
Herrler, T. et al. The intrinsic renal compartment syndrome: new perspectives in kidney transplantation. Transplantation 89, 40–46. https://doi.org/10.1097/TP.0b013e3181c40aba (2010).
Cops, J., Haesen, S., De Moor, B., Mullens, W. & Hansen, D. Current animal models for the study of congestion in heart failure: an overview. Heart Fail. Rev. 24, 387–397. https://doi.org/10.1007/s10741-018-9762-4 (2019).
He, L. et al. AKI on CKD: heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 92, 1071–1083. https://doi.org/10.1016/j.kint.2017.06.030 (2017).
Venkatachalam, M. A., Weinberg, J. M., Kriz, W. & Bidani, A. K. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J. Am. Soc. Nephrology: JASN. 26, 1765–1776. https://doi.org/10.1681/ASN.2015010006 (2015).
Goldstein, S. L., Jaber, B. L., Faubel, S. & Chawla, L. S. Acute kidney injury advisory group of American society of, N. AKI transition of care: a potential opportunity to detect and prevent CKD. Clin. J. Am. Soc. Nephrol. 8, 476–483. https://doi.org/10.2215/CJN.12101112 (2013).
Schiessl, I. M. et al. Renal interstitial Platelet-Derived growth factor Receptor-beta cells support proximal tubular regeneration. J. Am. Soc. Nephrology: JASN. 29, 1383–1396. https://doi.org/10.1681/ASN.2017101069 (2018).
Yang, L., Besschetnova, T. Y., Brooks, C. R., Shah, J. V. & Bonventre, J. V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nature medicine 16, 535–543, 531p following 143, (2010). https://doi.org/10.1038/nm.2144
Kishi, S. et al. Proximal tubule ATR regulates DNA repair to prevent maladaptive renal injury responses. J. Clin. Investig. 129, 4797–4816. https://doi.org/10.1172/JCI122313 (2019).
Qi, R. & Yang, C. Renal tubular epithelial cells: the neglected mediator of tubulointerstitial fibrosis after injury. Cell. Death Dis. 9, 1126. https://doi.org/10.1038/s41419-018-1157-x (2018).
Liu, B. C., Tang, T. T., Lv, L. L. & Lan, H. Y. Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int. 93, 568–579. https://doi.org/10.1016/j.kint.2017.09.033 (2018).
Gewin, L., Zent, R. & Pozzi, A. Progression of chronic kidney disease: too much cellular talk causes damage. Kidney Int. 91, 552–560. https://doi.org/10.1016/j.kint.2016.08.025 (2017).
Tan, R. J., Zhou, D. & Liu, Y. Signaling crosstalk between tubular epithelial cells and interstitial fibroblasts after kidney injury. Kidney Dis. (Basel). 2, 136–144. https://doi.org/10.1159/000446336 (2016).
Kitani, T. et al. Kidney vascular congestion exacerbates acute kidney injury in mice. Kidney Int. 101, 551–562. https://doi.org/10.1016/j.kint.2021.11.015 (2022).
Kusaba, T., Lalli, M., Kramann, R., Kobayashi, A. & Humphreys, B. D. Differentiated kidney epithelial cells repair injured proximal tubule. Proc. Natl. Acad. Sci. U.S.A. 111, 1527–1532. https://doi.org/10.1073/pnas.1310653110 (2014).
Jun, J. I. & Lau, L. F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell. Biol. 12, 676–685. https://doi.org/10.1038/ncb2070 (2010).
Kim, K. H., Chen, C. C., Monzon, R. I. & Lau, L. F. Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol. Cell. Biol. 33, 2078–2090. https://doi.org/10.1128/MCB.00049-13 (2013).
Yamashita, N. et al. Intratubular epithelial-mesenchymal transition and tubular atrophy after kidney injury in mice. Am. J. Physiol. Renal. Physiol. 319, F579–F591. https://doi.org/10.1152/ajprenal.00108.2020 (2020).
Kamezaki, M. et al. Comprehensive renoprotective effects of Ipragliflozin on early diabetic nephropathy in mice. Sci. Rep. 8, 4029. https://doi.org/10.1038/s41598-018-22229-5 (2018).
Kramann, R., Wongboonsin, J., Chang-Panesso, M., Machado, F. G. & Humphreys, B. D. Gli1(+) pericyte loss induces capillary rarefaction and proximal tubular injury. J. Am. Soc. Nephrology: JASN. 28, 776–784. https://doi.org/10.1681/ASN.2016030297 (2017).
Uehara-Watanabe, N. et al. Direct evidence of proximal tubular proliferation in early diabetic nephropathy. Sci. Rep. 12, 778. https://doi.org/10.1038/s41598-022-04880-1 (2022).
Uehara-Watanabe, N. et al. Proximal tubular epithelia-specific transcriptomics of diabetic mice treated with Dapagliflozin. Heliyon 8, e10615. https://doi.org/10.1016/j.heliyon.2022.e10615 (2022).
Uehara, M. et al. Pharmacological Inhibition of ataxia-telangiectasia mutated exacerbates acute kidney injury by activating p53 signaling in mice. Sci. Rep. 10, 4441. https://doi.org/10.1038/s41598-020-61456-7 (2020).
Wada, N. et al. Maternal high-fat diet exaggerates diet-induced insulin resistance in adult offspring by enhancing inflammasome activation through noncanonical pathway of caspase-11. Mol. Metab. 37, 100988. https://doi.org/10.1016/j.molmet.2020.100988 (2020).
Weischenfeldt, J. & Porse, B. Bone Marrow-Derived macrophages (BMM): isolation and applications. CSH Protoc. 2008 (pdb prot5080). https://doi.org/10.1101/pdb.prot5080 (2008).
Tomita-Yagi, A. et al. The importance of Proinflammatory failed-repair tubular epithelia as a predictor of diabetic kidney disease progression. iScience 27, 109020. https://doi.org/10.1016/j.isci.2024.109020 (2024).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. EdgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. https://doi.org/10.1093/bioinformatics/btp616 (2010).
Kirita, Y., Wu, H., Uchimura, K., Wilson, P. C. & Humphreys, B. D. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc. Natl. Acad. Sci. U.S.A. https://doi.org/10.1073/pnas.2005477117 (2020).
Malek, M. & Nematbakhsh, M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J. Ren. Inj Prev. 4, 20–27. https://doi.org/10.12861/jrip.2015.06 (2015).
Shimada, S. et al. Pathophysiological and molecular mechanisms involved in renal congestion in a novel rat model. Sci. Rep. 8, 16808. https://doi.org/10.1038/s41598-018-35162-4 (2018).
Ito, H. et al. Pericyte detachment and renal congestion involve interstitial injury and fibrosis in Dahl salt-sensitive rats and humans with heart failure. Hypertens. Res. 46, 2705–2717. https://doi.org/10.1038/s41440-023-01451-3 (2023).
Husain-Syed, F. et al. Doppler-Derived renal venous stasis index in the prognosis of right heart failure. J. Am. Heart Association. 8, e013584. https://doi.org/10.1161/JAHA.119.013584 (2019).
Seo, Y. et al. Doppler-Derived intrarenal venous flow mirrors Right-Sided heart hemodynamics in patients with cardiovascular disease. Circulation Journal: Official J. Japanese Circulation Soc. 84, 1552–1559. https://doi.org/10.1253/circj.CJ-20-0332 (2020).
Winton, F. R. The influence of venous pressure on the isolated mammalian kidney. J. Physiol. 72, 49–61. https://doi.org/10.1113/jphysiol.1931.sp002761 (1931).
Hinshaw, L. B., Brake, C. M., Iampietro, P. F. & Emerson, T. E. Jr. Effect of increased venous pressure on renal hemodynamics. Am. J. Physiol. 204, 119–123. https://doi.org/10.1152/ajplegacy.1963.204.1.119 (1963).
Kim, K. H., Won, J. H., Cheng, N. & Lau, L. F. The matricellular protein CCN1 in tissue injury repair. J. Cell. Commun. Signal. 12, 273–279. https://doi.org/10.1007/s12079-018-0450-x (2018).
Lau, L. F. CCN1/CYR61: the very model of a modern matricellular protein. Cell. Mol. Life Sci. 68, 3149–3163. https://doi.org/10.1007/s00018-011-0778-3 (2011).
Humphreys, B. D. et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 176, 85–97. https://doi.org/10.2353/ajpath.2010.090517 (2010).
Lin, S. L., Kisseleva, T., Brenner, D. A. & Duffield, J. S. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am. J. Pathol. 173, 1617–1627. https://doi.org/10.2353/ajpath.2008.080433 (2008).
Schrimpf, C. & Duffield, J. S. Mechanisms of fibrosis: the role of the pericyte. Curr. Opin. Nephrol. Hypertens. 20, 297–305. https://doi.org/10.1097/MNH.0b013e328344c3d4 (2011).
Nakata, T. et al. Injured tubular epithelia-derived CCN1 promotes the mobilization of fibroblasts toward injury sites after kidney injury. iScience 28, 112176. https://doi.org/10.1016/j.isci.2025.112176 (2025).
Bonventre, J. V., Vaidya, V. S., Schmouder, R., Feig, P. & Dieterle, F. Next-generation biomarkers for detecting kidney toxicity. Nat. Biotechnol. 28, 436–440. https://doi.org/10.1038/nbt0510-436 (2010).
Vaidya, V. S., Ferguson, M. A. & Bonventre, J. V. Biomarkers of acute kidney injury. Annu. Rev. Pharmacol. Toxicol. 48, 463–493. https://doi.org/10.1146/annurev.pharmtox.48.113006.094615 (2008).
Ferguson, M. A., Vaidya, V. S. & Bonventre, J. V. Biomarkers of nephrotoxic acute kidney injury. Toxicology 245, 182–193. https://doi.org/10.1016/j.tox.2007.12.024 (2008).
Li, C. et al. Cysteine-rich protein 61, a specific ultra-early biomarker in kidney ischemia/reperfusion injury. Nephrol. (Carlton). 24, 798–805. https://doi.org/10.1111/nep.13513 (2019).
Bonventre, J. V. & Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 121, 4210–4221. https://doi.org/10.1172/JCI45161 (2011).
Bonventre, J. V. Dedifferentiation and proliferation of surviving epithelial cells in acute renal failure. J. Am. Soc. Nephrol. 14 (Suppl 1), S55–61 (2003).
Kramann, R. et al. Parabiosis and single-cell RNA sequencing reveal a limited contribution of monocytes to myofibroblasts in kidney fibrosis. JCI Insight. 3 https://doi.org/10.1172/jci.insight.99561 (2018).
Shigeoka, M. et al. Cyr61 promotes CD204 expression and the migration of macrophages via MEK/ERK pathway in esophageal squamous cell carcinoma. Cancer Med. 4, 437–446. https://doi.org/10.1002/cam4.401 (2015).
Singh, B., Bhatowa, R., Tripathi, C. B. & Kapil, R. Developing micro-/nanoparticulate drug delivery systems using design of experiments. Int. J. Pharm. Investig. 1, 75–87. https://doi.org/10.4103/2230-973X.82395 (2011).
Lai, C. F. et al. Blockade of cysteine-rich protein 61 attenuates renal inflammation and fibrosis after ischemic kidney injury. Am. J. Physiol. Renal. Physiol. 307, F581–592. https://doi.org/10.1152/ajprenal.00670.2013 (2014).
Bai, T., Chen, C. C. & Lau, L. F. Matricellular protein CCN1 activates a Proinflammatory genetic program in murine macrophages. J. Immunol. 184, 3223–3232. https://doi.org/10.4049/jimmunol.0902792 (2010).
Acknowledgements
The authors would like to thank Prof. Lester F. Lau for his kind gift of CCN1-floxed mice.
Author information
Authors and Affiliations
Contributions
A.M., T.N., and T. K. designed the study, performed the experiments, analyzed the data, and wrote the manuscript. S.S., M.N., and M.U. performed the experiments, Y.K. analyzed the data. R.K., H.Y-S., Y.S., Y.M., N.O-O, I.N., K.N., N.Y., K.T., and S.M. contributed to discussions.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Disclosure
The authors declare that there is no conflict of interest associated with this study.
Data sharing statement
Data supporting the present results are available in the main manuscript or Supplementary Material. In addition, we will share models, protocols, methods, and other useful materials and resources related to the article as much as possible. There are no large data files to be shared via online resources.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 2
Supplementary Material 3
Supplementary Material 4
Supplementary Material 5
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Minamida, A., Nakata, T., Kurose, R. et al. Injured tubule derived CCN1 exacerbates renal congestion-mediated acute kidney injury and fibrosis. Sci Rep 15, 20840 (2025). https://doi.org/10.1038/s41598-025-05723-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-05723-5
