
Abstract
Renal congestion is a key factor in renal dysfunction associated with heart failure. We previously reported that renal congestion worsened renal ischemia-reperfusion in a murine model. However, its impact on sepsis-associated acute kidney injury (SA-AKI), the leading cause of AKI, remains unclear. Therefore, we herein investigated the mechanisms by which renal congestion exacerbates SA-AKI, with a focus on Toll-like receptor (TLR) 2. After inducing sepsis with cecal ligation and puncture (CLP) in a unilateral renal congestion model, transient blood pressure reductions and persistent renal vein dilation were observed. A histological analysis showed increased fibrosis and its markers in congested kidneys post-CLP. Acute phase results revealed extensive tubular damage, macrophage infiltration, TLR2 up-regulation, and elevated high mobility group box 1 (HMGB1) levels. In TLR2-knockout mice, exacerbation of tissue fibrosis by renal congestion was attenuated after CLP. In vitro, oxidative stress and hypoxia up-regulated TLR2 expression. Collectively, these results suggest that renal congestion and sepsis synergistically worsened renal damage, likely through hypoxia and the oxidative stress-induced activation of the TLR2 pathway.
Similar content being viewed by others


Introduction
Renal dysfunction frequently occurs in patients with heart failure (HF), which is one of the categories of cardio-renal syndrome1,2. Decreased renal perfusion due to low cardiac output is a primary cause of renal dysfunction associated with acute decompensated HF. However, recent epidemiological studies showed that renal dysfunction associated with HF correlated more strongly with increased central venous pressure (CVP) than with decreased cardiac output, drawing attention to the importance of renal congestion3,4,5.
Renal dysfunction associated with renal congestion has been investigated in many clinical studies and basic experiments using large animals, including dogs6,7. Multiple pathophysiologies for the worsening of renal function due to increased venous pressure have been proposed and include the activation of sympathetic nerves and the renin-angiotensin system, reduced transglomerular pressure, elevated renal interstitial pressure, and enhanced proinflammatory pathways7,8,9,10. However, due to the lack of an appropriate murine congestion model, analyses of the underlying molecular mechanisms using gene-edited animals have not yet been performed. We recently developed a novel mouse model of unilateral renal congestion and demonstrated that renal blood flow velocity was reduced and the renal function reserve was decreased by renal congestion11. Furthermore, the induction of bilateral mild ischemia-reperfusion injury (IRI) in the same model further reduced blood flow velocity and exacerbated local inflammation by increasing the frequency of neutrophil adhesion11. However, other models of AKI have not been examined, and it is unclear whether the impact of renal congestion on AKI may be generalized to renal injury other than IRI.
Sepsis is characterized by host responses to various infections that lead to life-threatening organ failure, including the kidneys12. Sepsis-associated acute kidney injury (SA-AKI) is a common complication in patients with severe infection and accounts for 45–70% of AKI cases in critically ill patients13. Various mechanisms contribute to the development and progression of SA-AKI, including systemic and local inflammation, renin-angiotensin system dysregulation, metabolic reprogramming, mitochondrial dysfunction, microcirculatory dysfunction, and macrocirculatory abnormalities14,15,16. Regarding HF patients, the decompensation of chronic HF may be triggered by various causes, including sepsis17. Cardiovascular organ failure is the most common complication in septic patients, and was reported in 87% of patients in a cohort of intensive care unit (ICU) patients in the USA and Europe18. In consideration of the frequent association of HF with AKI, the impact of macrocirculatory abnormalities on SA-AKI is considered to be more prominent in HF patients. Right ventricular dysfunction is one of the entities of sepsis-induced cardiomyopathy and occurs in approximately one-third to one-half of patients with sepsis and septic shock19,20. Although increased CVP worsens renal function in HF patients, the clinical relevance of renal congestion in the development of SA-AKI has yet to be clarified.
In the present study, we induced sepsis by cecal ligation and puncture (CLP) in a mouse model of unilateral renal congestion and examined the effects of renal congestion on SA-AKI and subsequent renal tissue fibrosis. We also investigated the relationship between innate immune mechanisms and the renal congestion-mediated worsening of SA-AKI, with a focus on the involvement of Toll-like receptor (TLR)-2, a major component of the innate immune response to bacterial infection21 that is constitutively expressed in various kidney cells, including epithelia22. TLR2 recognizes not only pathogen-associated molecular patterns, but also damage-associated molecular patterns (DAMPs)21. Since these TLR2-mediated responses consequently trigger innate immune defenses as well as renal inflammation23,24 we examined the involvement of TLR2 in the congestion-mediated worsening of SA-AKI using TLR2-knockout (KO) mice.
Results
Hemodynamic changes in the presence of both renal congestion and sepsis
To examine the impact of renal congestion on SA-AKI, we induced unilateral renal congestion by surgical coarctation of the inferior vena cava (IVCC), followed by CLP the next day (Fig. 1a−d, Supplementary Figure S1a,b). Blood pressure (BP) measurements over time showed that IVCC alone did not affect BP, whereas it decreased to the lowest value 3 h after CLP and recovered to the previous value 24 h later (Fig. 1e). Ultrasonography of the kidneys revealed that the left renal vein was dilated and renal venous blood flow velocity decreased after IVCC, while no significant differences were observed in hemodynamics after CLP, in renal vein dilatation in the congestive kidney, or in renal venous blood flow velocity with or without CLP (Fig. 1f,g, Supplementary Figure S1c). The dilatation of renal veins in congested kidneys and decreased renal venous blood flow persisted 7 days after CLP (Fig. 1f,g).

Physiological analyses of sepsis-associated acute kidney injury (SA-AKI) in the congestive kidney (Cong). (a) Scheme of unilateral inferior vena cava (IVC) constriction. (b) Scheme of cecal ligation and puncture (CLP). (c) Macroscopic pictures of the IVC and left renal vein before and after surgery. The arrowhead indicates the site of constriction. Asterisks indicate the left renal vein. (d) Experimental scheme and groups for analyses. Surgical coarctation of the inferior vena cava (IVCC) followed by the induction of sepsis by CLP were performed on 10-week-old male mice, which were then sacrificed 1 or 7 days later. Echography of the kidney and blood pressure (BP) measurements were performed at the indicated time points. (e) Serial BP measurements in CLP and sham-operated mice. (f) Ultrasonography (upper panels) and Doppler echography (lower panels) of the left renal vein before, just after IVCC, and 7 days after CLP. (g) Serial measurements of the diameters of (left panel) and flow velocity (right panel) in the right and left renal veins (n = 6 each). In all groups, data are shown as means ± standard deviation (SD). * p < 0.05 vs. contralateral kidneys (CLK) at the same time points. # p < 0.05 vs. the baseline of Cong.
Exacerbation of tubular injury by renal congestion in SA-AKI
We initially investigated whether renal congestion alone induced changes in normal kidney tissue in our model. qPCR revealed no significant differences in the mRNA expression of the kidney injury markers hepatitis A virus cellular receptor 1 (Havcr1, encoding kidney injury molecule (KIM)-1) and lipocalin 2 (Lcn2, encoding neutrophil gelatinase-associated lipocalin), the tubule integrity marker low density lipoprotein receptor-related protein 2 (Lrp2, encoding megalin), the neutrophil marker lymphocyte antigen 6 complex locus G6D (Ly6g, encoding lymphocyte antigen 6 family member G), the macrophage marker cluster of differentiation 68 (Cd68, encoding lysosome-associated membrane glycoprotein-4), or the pro-inflammatory markers tumor necrosis factor-α (Tnfα) and C-C motif chemokine ligand 2 (Ccl2, encoding monocyte chemoattractant protein-1) between congestive kidneys (Cong) and contralateral kidneys (CLK) (Supplementary Figure S2a). Furthermore, Periodic Acid-Schiff (PAS) staining showed no significant differences in renal histology score between Cong and CLK (Supplementary Figure S2b,c). These results suggest that our mouse model is a latent renal congestion model that does not solely induce tubular injury or tissue inflammation.
We then examined the impact of renal congestion on SA-AKI (Fig. 2a). One day after CLP surgery, serum TNFα levels were higher in CLP mice than in uninjured mice (Fig. 2b). PAS staining revealed extensive tubular injury in Cong-CLP kidneys and no significant histological differences between CLK-CLP and uninjured kidneys (Fig. 2c,d). An immunofluorescence analysis showed a decrease in Lotus Tetragonolobus Lectin (LTL) + tubules, indicating healthy tubules, and an increase in KIM-1 + tubules, indicating injured tubules, in Cong-CLP kidneys (Fig. 2c). Immunostaining for a macrophage marker (F4/80) revealed an increase in macrophage infiltration in Cong-CLP kidneys (Fig. 2c). qPCR showed the up-regulated expression of Havcr1, Lcn2, Ccl2, Tnfα, and Cd68 and the down-regulated expression of Lrp2 in Cong-CLP kidneys (Fig. 2e).

Renal congestion exacerbated SA-AKI. (a) Experimental scheme and groups for analyses. (b) Serum TNFα measurements by ELISA in mice 1 day after CLP. (c) Representative pictures of Periodic Acid-Schiff (PAS), immunohistochemistry for F4/80, and dual immunofluorescence for KIM1 and LTL. (d) Semi-quantitative analysis of tubular injury by PAS staining. (e) qPCR of RNA from the whole kidney for representative markers of tubular injury (Havcr1 and Lcn2), tubular integrity (Lrp2), proinflammatory cytokines (Tnfα and Ccl2), macrophages (Cd68), and leukocytes (Ly6g). Bars indicate 50 μm in PAS and F4/80 staining and 100 μm in immunofluorescence staining in (c). In all groups, data are shown as means ± SD. N = 8 for uninjured mice and n = 12 for CLP mice. *P < 0.05. The paired t-test was used for comparisons of variables between CLK-CLP and Cong-CLP. ANOVA and Tukey’s post hoc test were used for comparisons of multiple variables.
Seven days after CLP, Sirius red staining revealed extensive tissue fibrosis in Cong-CLP kidneys, but no significant difference between CLK-CLP and uninjured kidneys (Fig. 3a,b). Immunostaining for F4/80 showed the infiltration of macrophages in Cong-CLP kidneys (Fig. 3a). qPCR revealed increases in the gene expression of tissue fibrosis-related factors (transforming growth factor β1 (Tgfb1), actin α2 smooth muscle (Acta2), collagen type I α1 (Col1a1), and fibronectin 1 (Fn1)) in Cong-CLP kidneys (Fig. 3c). The up-regulated expression of the Havcr1, Lcn2, Ccl2, Cd68, and Tnfα genes was still observed in Cong-CLP kidneys at this time point. Collectively, these results suggest that renal congestion exacerbated sepsis-induced tubular injury at earlier time points and subsequent tissue fibrosis at a later time point, even though sepsis was mild because it did not solely induce AKI.

Renal congestion exacerbated SA-AKI-mediated kidney fibrosis. (a) Representative images of Sirius red staining and immunohistochemistry for F4/80 7 days after CLP (b) Semi-quantitative analysis of tissue fibrosis by Sirius red staining. (c) qPCR of RNA from the whole kidney for representative markers of tubular injury (Havcr1 and Lcn2), tubular integrity (Lrp2), proinflammatory and profibrotic cytokines (Ccl2, Tnfα, and Tgfb1), macrophages (Cd68), and fibrosis (Acta2, Col1a1, and Fn1). Bars indicate 100 μm in (b). In all groups, data are shown as means ± SD. N = 5 for uninjured mice and n = 14 for CLP mice. The paired t-test was used for comparisons of variables between CLK-CLP and Cong-CLP. ANOVA and Tukey’s post hoc test were used for comparisons of multiple variables.
Up-regulated expression of TLR-2 by renal congestion in SA-AKI
Innate immune responses play an essential role in the progression of SA-AKI and TLRs are essential components of this system. A previous study reported the expression of TLR2 and TLR4 in renal tissue and their involvement in SA-AKI25. In addition, TLR9 is primarily expressed in immune cells, and renal damage associated with CLP was shown to be ameliorated in TLR9-KO mice26. Based on these findings, we investigated the expression of TLR2, TLR4, and TLR9 in kidney tissue and found that the up-regulated expression of TLR2 was the most pronounced in Cong-CLP kidneys (Fig. 4a). We also examined the expression of TLRs in congestive kidneys without CLP and found no changes (Supplementary Figure S3). The immunostaining of kidney tissue showed a marked increase in the expression of TLR2 in Cong-CLP mice (Fig. 4b). Co-staining for TLR2 and various markers revealed the expression of TLR2 in KIM1 + injured tubular epithelia, platelet-derived growth factor receptor β (PDGFRβ) + interstitial fibroblasts, and F4/80 + macrophages in Cong-CLP mice (Fig. 4c). Extracellular high mobility group box 1 (HMGB1) functions as a ligand of TLR2 and is one of the strong mediators of experimental sepsis27. Serum HMGB1 levels were not elevated in CLP mice and Hmgb1 gene expression was not up-regulated in Cong-CLP kidneys (Fig. 4d,e). The immunostaining of kidney tissue revealed the cytoplasmic expression of HMGB1 at low levels in uninjured or CLK-CLP kidneys and high levels in Cong-CLP kidneys (Fig. 4f,g,h).

Renal congestion up-regulated the expression of toll-like receptors (TLR) and High Mobility Group Box 1 (HMGB1) in congestive kidneys with SA-AKI. (a) qPCR of RNA from the whole kidney for toll-like receptors (TLR)-2, -4, and -9 1 day after CLP (b) Representative immunofluorescence images of the kidney for TLR2 1 day after CLP. (c) Representative co-immunofluorescence images of the kidney for TLR2 and KIM1, PDGFRβ, and F4/80 1 day after CLP (d) Serum HMGB1 measurements by ELISA in mice 1 day after CLP (e) qPCR of RNA from the whole kidney for Hmgb1 1 day after CLP (f) Representative immunostaining images for HMGB1 1 day after CLP. The dotted square indicates high (*) and no (§) cytoplasmic HMGB1 expression in tubules. (g) Average HMGB1 scores in tubules. (h) Frequency of tubules in each score. Bar = 50 μm in (b) and (c) and = 30 μm in (f). In all groups, data are shown as means ± SD. The paired t-test was used for comparisons of variables between CLK-CLP and Cong-CLP. ANOVA and Tukey’s post hoc test were used for comparisons of multiple variables. * p < 0.05.
Hypoxia and oxidative stress up-regulate TLR2 expression in tubular epithelia
Since the expression of TLR2 was lower in CLK-CLP than in Cong-CLP kidneys, we hypothesized that hemodynamic changes, e.g., a transient decrease in BP and subsequent reperfusion, rather than circulating factors were contributing to the up-regulated expression of TLR2 in Cong-CLP kidneys. To examine the causal effects of these hemodynamic changes and humoral mediators on TLR2 expression in tubular epithelia, we performed an in vitro analysis using normal rat kidney epithelial cells (NRK52E), a rat tubular epithelial cell line. Due to the slight increase observed in serum TNFα levels in our CLP model (Fig. 2b), we examined the effects of TNFα in vitro and found that it slightly increased the expression of Tlr2 (Fig. 5a). After a 6-h exposure to hypoxia, glyceraldehyde-3-phosphate dehydrogenase (Gapdh), one of the hypoxia-inducible factor (HIF) target genes, Havcr1, Tlr2, and Tlr4, but not Tlr9, were up-regulated in NRK52E (Fig. 5b). To clarify whether the up-regulated expression of these genes was mediated by the activation of HIF, we performed qPCR after a treatment with Cobalt chloride (CoCl2), a potent activator of HIF through the inhibition of its degradation in the cytoplasm. The CoCl2 treatment up-regulated the expression of Gapdh, whereas that of Havcr1, Tlr2, Tlr4, and Tlr9 remained unchanged (Fig. 5c), indicating that the expression of Tlr2 was not regulated by HIF. Regarding the reperfusion period, oxidative stress by hydrogen peroxide (H2O2) significantly up-regulated the expression of oxidative stress markers (heme oxygenase 1 (Hmox1) and catalase (Cat)), Havcr1, Tlr2, Tlr4, and Tlr9 (Fig. 5d, Supplementary figure S4).

Oxidative stress increased TLRs in tubular epithelia. (a–d) qPCR of RNA from NRK-52E exposed to TNFα (a), hypoxia (b), CoCl2 (c), and H2O2 (d). (e) qPCR of RNA from the whole kidney for oxidative stress markers. (f) Representative immunohistochemical images of 3-nitrotyrosine 1 day after CLP. Bar = 100 μm in (f). In all groups, data are shown as means ± SD. The unpaired t-test was used for comparisons of variables between the 2 groups in vitro. The paired t-test was used for comparisons of variables between CLK-CLP and Cong-CLP. ANOVA and Tukey’s post hoc test were used for comparisons of multiple variables. * p < 0.05.
Since H2O2 markedly increased the expression levels of Tlr2, we investigated the effects of oxidative stress on Cong-CLP kidneys in more detail. Hmox1 and superoxide dismutase 1 (Sod1) gene expression was up-regulated in Cong-CLP kidneys (Fig. 5e). Immunostaining for 3-nitrotyrosine, a marker of oxidative stress, revealed a marked increase in its expression in Cong-CLP kidneys (Fig. 5f).
To generalize the up-regulation of Tlr2 after injury, we further analyzed its expression in various kidney injury models (Supplemental Figure S5). In the IRI model, the expression of Tlr2 was up-regulated 3 days after injury (Supplementary Figure S5). In the cisplatin model, its expression was up-regulated 7 days after the injection (Supplementary Figure S5). In the repeated cisplatin injection model as a chronic kidney injury model, the expression of Tlr2 was not up-regulated. Overall, the up-regulation of Tlr2 after injury was noted in multiple independent AKI models, but not chronic kidney disease (CKD) models.
Systemic TLR2-KO ameliorates the renal congestion-mediated exacerbation of SA-AKI
To investigate whether the loss of systemic TLR2 expression ameliorated the congestion-mediated worsening of SA-AKI, we performed in vivo experiments using TLR2-KO mice (Fig. 6a). We confirmed the loss of TLR2 expression in IRI kidneys by PCR (Supplementary Figure S6). After IVCC and CLP, BP transiently decreased in both TLR2-KO and wild-type (WT) mice, with no significant difference (Fig. 6b). Serum TNFα levels 1 day after CLP slightly decreased in TLR2-KO mice (Fig. 6c). Seven days after CLP, Sirius red staining showed extensive tissue fibrosis in Cong-CLP WT mice, whereas no significant difference was observed between the CLK-CLP and Cong-CLP kidneys of TLR2-KO mice (Fig. 6d,e). Immunostaining for F4/80 revealed a decrease in the infiltration of macrophages in the Cong-CLP kidneys of TLR2-KO mice (Fig. 6d). qPCR showed the significant congestion-mediated up-regulation of tubular injury markers (Havcr1 and Lcn2), macrophage markers (Cd68), a pro-inflammatory cytokine (Tnfα), and extracellular matrix markers (Col1a1 and Fn1) in WT mice, while these differences was canceled in TLR2-KO mice (Fig. 6f). The congestion-mediated down-regulation of a tubular integrity marker (Lrp2) was not observed in TLR2-KO mice (Fig. 6f).

SA-AKI-mediated kidney fibrosis was ameliorated in TLR2-KO mice. (a) Experimental scheme and groups for analyses. (b) Serial BP measurements in WT and TLR2-KO mice that underwent IVCC and CLP. (c) Serum TNFα measurements by ELISA in mice 1 day after CLP. (d) Representative images of Sirius red staining and immunohistochemistry for F4/80 7 days after CLP (e) Semi-quantitative analysis of tissue fibrosis by Sirius red staining. (f) qPCR of RNA from the whole kidney for representative markers of tubular injury (Havcr1 and Lcn2), tubular integrity (Lrp2), a profibrotic cytokine (Tgfb1), proinflammatory cytokine (Tnfa), macrophages (Cd68), and fibrosis (Acta2, Col1a1, and Fn1). Bar = 100 μm in (d). In all groups, data are shown as means ± SD. N = 9 for WT mice and n = 8 for TLR2-KO mice. The paired t-test was used for comparisons of variables between CLK-CLP and Cong-CLP. * p < 0.05.
Discussion
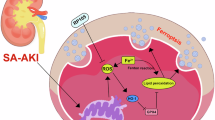
The common and major cause of the acute exacerbation of cardiac and renal failure is infection17,28 and sepsis is the leading cause of AKI entering the ICU29 and requiring renal replacement therapy30. Conversely, 80% of patients with sepsis are complicated by cardiovascular events and 40% have renal dysfunction18. Infections may induce cardiac and renal dysfunctions due to an increased systemic oxygen demand, circulatory changes associated with elevated levels of inflammatory cytokines, and acute myocardial damage associated with sepsis15,16,20. Since elevated CVP and subsequent renal congestion have been shown to play an important role in the deterioration of renal function associated with HF31 we herein focused on the relationship between the following three disease conditions: congestion, sepsis, and renal fibrosis. We found that the concomitant occurrence of congestion and CLP, neither of which induces renal injury by itself, may exacerbate SA-AKI and induce tissue fibrosis at the chronic phase. In vivo and in vitro experiments showed that transient hypotension and elevated levels of inflammatory cytokines due to sepsis up-regulated the expression of TLR2 and increased the cytoplasmic translocation of HMGB1 in renal tissues, resulting in tubular injury and inflammation in renal tissues, which, in turn, led to fibrosis (Fig. 7). Recent clinical trials showing that fluid restriction improved the survival of septic patients with hypotension provide support for volume overload worsening organ congestion and causing mortality in these patients32. TLR2 deficiency prevented fibrosis at the chronic phase, indicating the potential of TLR2-targeted therapy for the prevention of fibrosis at the chronic phase of SA-AKI complicated by congestion.

Proposed mechanisms underlying the exacerbation of SA-AKI by renal congestion.
CLP is the most widely used rodent polymicrobial sepsis model and is also considered to be more similar to the human model of sepsis than the sepsis model induced by the administration of lipopolysaccharide (LPS)33,34,35. CLP allows disease severity to be adjusted by the length of the ligated cecum and the size/number of punctures34. In the present study, transient hypotension similar to human sepsis was observed after CLP, while tissue injury only occurred in congestive, not non-congestive, kidneys, suggesting that the strength of CLP was not sufficient to solely induce SA-AKI. Previous studies demonstrated that, in the case of renal congestion, higher than normal BP was required to maintain renal blood flow, the glomerular filtration rate, and urine output36. In addition, diffuse microcirculatory flow abnormalities, namely, a decrease in the percentage of capillaries with continuous blood flow and an increase in the percentage of capillaries with intermittent or no flow, have been reported in SA-AKI14,15,16. In congestive kidneys, blood flow velocity decreases due to a smaller arterio-venous pressure gradient. SA microcirculatory abnormalities and congestion-mediated reductions in blood flow velocity may synergistically increase susceptibility to SA-AKI. These findings provide support for patients with HF and congestion being susceptible to renal failure following infection17.
TLRs play important roles not only in host defenses against pathogenic infection, but also in the progression of inflammatory responses through their recognition of endogenous molecules, including DAMPs37. TLRs trigger an inflammatory response through nuclear factor kappa-B and mitogen-activated protein kinases, and inflammatory cytokines are eventually released37. Regarding renal involvement, TLRs, particularly TLR2 and TLR4, are involved in the progression of both infectious and non-infectious kidney diseases, including ischemic AKI, autoimmune diseases, metabolic diseases, and chronic kidney injury24. Previous studies showed that TLR2 deficiency attenuated AKI due to IRI22,38. In terms of the pathophysiology regulating TLR2 expression, since transient hypotension was observed after CLP in in vivo experiments, we examined the expression of TLRs induced by various sepsis-mediated humoral factors and hemodynamic changes, including hypoxia and reperfusion. Our in vitro experiments using tubular epithelial cells revealed that hypoxia, TNFα, and oxidative stress up-regulated the expression of TLR2. The hypoxic stimulation up-regulated the expression of TLR2, whereas CoCl2, an activator of HIF, did not, indicating that increases in the expression levels of TLR2 by the hypoxic stimulation were due to a HIF-independent pathway.
Furthermore, oxidative stress by H2O2 strongly up-regulated the expression of TLR2 in tubular epithelia. In vivo, we found the marked up-regulation of oxidative stress-related genes and positive staining for nitrotyrosine in Cong-CLP kidneys, not in CLK-CLP kidneys. Based on previous findings showing the up-regulated expression of TLR2 in IRI kidneys38 the present results indicate that hemodynamic changes, rather than the SA up-regulation of humoral mediators, promoted oxidative stress and the subsequent expression of TLR2 in Cong-CLP kidneys. In addition, the expression of HMGB1 was not up-regulated in blood after CLP, while its translocation from the nucleus to the cytoplasm was significant in Cong-CLP kidneys, suggesting that a vicious cycle between HMGB1 and TLR2 formed locally and contributed to tissue fibrosis at the chronic phase after injury.
After mice are subjected to CLP, the levels of early inflammatory mediators, including TNFα and interleukin-1β, immediately increase, while HMGB1 as a late mediator begins to be detected 8 h after a septic insult and reaches a plateau after 16 h27. Previous studies showed that HMGB1 was also locally up-regulated in renal resident cells after various injuries, including IRI, and passively leaked from apoptotic/necrotic cells39,40. In our model, HMGB1 was not detected in blood 24 h after CLP, while the immunostaining of tissues showed a high expression level of cytoplasmic HMGB1 in Cong-CLP kidneys only. These results support our CLP model not being a severe sepsis model with the systemic up-regulation of HMGB1 and suggest that the local expression of HMGB1 was induced in the congestive kidney by an abnormality in the macrocirculation. HMGB1 is a potent ligand for TLR2, and the tissue co-localization of HMGB1 and TLR2 in congested kidneys may have triggered further inflammation in Cong-CLP kidneys. In addition, it is important to note that HMGB1-mediated inflammation is an important factor driving renal fibrosis39,41 and the activation of TLR2/HMGB1 signaling positively correlated with the severity of renal fibrosis42. The present results showing less severe fibrosis in the Cong-CLP kidneys of TLR2-KO mice are consistent with these findings, and TLR2/HMGB1 is an attractive target for preventing SA-AKI and its subsequent transition to CKD.
There are several limitations that need to be addressed. Since congestion was only introduced in the left kidney in our model, it was not possible to evaluate the impact of the congestive kidney on renal function, which is a potential disadvantage of our unilateral congestion model. In addition, renal congestion is commonly associated with HF in clinical fields; however, we did not investigate whether renal congestion was truly exacerbated in the HF mouse model, and, thus, future studies are needed. While TLR4 has also been shown to play important roles in the septic and non-septic progression of AKI24,43,44,45 we only focused on TLR2 in our experiments. Therefore, the involvement of other TLR family members, such as TLR4 and TLR9, in the congestion-mediated exacerbation of SA-AKI remains unknown. Furthermore, since we only examined the CLP model as SA-AKI, it remains unclear whether our results may be generalized to other sepsis models, such as the LPS injection model.
We presumed that IRI associated with hypotension after CLP was important for the expression of TLRs; however, it has yet to be confirmed whether SA-AKI is ameliorated when BP is maintained with inotropic agents, as is performed in clinical practice. Moreover, the congestive status of IVCC surgery may be affected by local peritonitis caused by CLP, resulting in tissue injury in the chronic phase.
Conclusion
The concomitant occurrence of renal congestion and sepsis synergistically exacerbated renal damage and subsequent tissue fibrosis, even if each was not severe enough to induce AKI by itself. TLR2-mediated inflammation plays a critical role in this process. With the aging of society, the number of patients with heart and renal failure will increase worldwide. Infections are a major life-threatening disease among the elderly and elderly patients with sepsis have more comorbid illnesses, including renal dysfunction and congestive HF46. Therefore, it is important to manage patients with HF who develop infections with attention to congestion-mediated AKI.
Methods
Animal experiments
In mouse experiments, we used 9- to 12-week-old male BL6 mice or TLR2-KO mice47 (purchased from Shimizu, Inc. and Oriental Bio Service, Inc. Kyoto, Japan, respectively). IVCC was performed as previously described11. At the age of 10 weeks, male mice were anesthetized with isoflurane. After a midline abdominal incision, the inferior vena cava (IVC) between the right and left renal veins was carefully exposed. The isolated IVC and a 22-gauge (22G) needle were tied by a surgical suture (7-0 nylon). Silk surgical sutures and smaller gauge needles need to be avoided in order to prevent the thrombotic occlusion of IVC or the left renal vein. The needle was then removed and the wound was closed. The same surgical procedure was performed for sham-operated mice, except for the application of coarctation, and was assigned as the uninjured mouse kidney. CLP surgery was performed to induce sepsis, as previously described48. After a midline incision, the cecum was located and ligated with 5-0 silk suture 10 mm from the tip, and a 22G needle was passed through the ligated cecum. The cecum was returned to the peritoneal cavity and the incision was closed. One or 7 days after surgery, mice were euthanized after isoflurane anesthesia by cervical dislocation.
In the cisplatin injury model, 10-week-old male C57/BL6 mice received a single intraperitoneal injection of cisplatin (15 mg/kg body weight, Nippon Kayaku, Tokyo, Japan) in saline49. Three days after the cisplatin injection, mice were euthanized after isoflurane anesthesia by cervical dislocation.
In the repeated cisplatin injection model, 10-week-old male C57/BL6 mice were injected repeatedly with cisplatin (10 mg/kg body weight) 4 times at 1-week intervals50. Cisplatin was administered in the morning. Seven days after the last cisplatin injection, mice were euthanized after isoflurane anesthesia by cervical dislocation.
In the unilateral IRI model, 10-week-old male C57/BL6 mice were anesthetized with isoflurane, and ischemia was induced through the retroperitoneal approach on the left kidney at 37 °C for 35 min by clamping the left kidney pedicle51. Regarding volume supplementation, 0.5 ml of saline was injected intraperitoneally after surgery. Three days after IRI surgery, mice were euthanized after isoflurane anesthesia by cervical dislocation.
Although mice were randomly allocated to control groups or models, investigators were not blinded to surgical or treatment groups. Mice were housed under pathogen‑free conditions (temperature, 25 °C; 12-h light/dark cycle) and had free access to normal chow and water. All experiments were conducted following approval from the Kyoto Prefectural University of Medicine Animal Experiment Committee and in accordance with facility guidelines, the Guidelines for Proper Conduct of Animal Experiments of the Japanese Ministry of Education, Culture, Sports, Science and Technology, and ARRIVE guidelines.
Doppler echography and BP measurements
Doppler echography of the renal veins was performed using the 2100 Imaging System (VisualSonics Inc., Toronto, Canada) as previously described11. During echography, the heart rate of mice was maintained at between 400 and 500 /min by adjusting the depth of anesthesia with isoflurane (3.0–5.0% for induction; 1.0–2.0% for maintenance). Heart rate and BP were measured using the non-invasive tail cuff method (BP-98 A, Softron Co., Ltd., Tokyo, Japan) at the indicated time points.
Tissue preparation and histological analysis
To obtain paraffin sections, the kidneys were fixed in 4% paraformaldehyde and embedded in paraffin by Applied Medical Research Laboratory (Osaka, Japan). Paraffin-embedded mouse and human tissues were cut into 4-µm-thick sections. PAS and Sirius Red staining were performed according to standard procedures. The histology of the kidney was examined using formalin sections stained with PAS and Sirius red.
The tubular injury score was evaluated using PAS-stained specimens. According to previously described methods52,53,54 the degree of tubular injury was scored semi-quantitatively in 5 randomly selected images of 25 consecutive non-overlapping fields in each kidney section stained with PAS under high magnification (n = 5) using the following scoring system: 0, 0%; 1, 1–10%; 2, 11–25%; 3, 26–50%; 4, 51–75%; and 5, 76–100%. Tubular injury was judged by tubular atrophy, tubular dilation, protein casts, necrotic cells, and brush border loss. The degrees of interstitial fibrosis were scored semi-quantitatively in 5 randomly selected images of 25 consecutive non-overlapping fields in each kidney section stained with Sirius red under high magnification (n = 5) using the following scores: 0, 0%; 1, 1–10%; 2, 11–25%; 3, 26–50%; 4, 51–75%; and 5, 76–100%.
Cell culture
NRK52E obtained from the JCRB Cell Bank were cultured in Dulbecco’s modified Eagle’s medium (Wako, Osaka, Japan) containing 1% penicillin and streptomycin (Invitrogen, Carlsbad, CA) and 5% fetal bovine serum (Invitrogen) at 37 °C in a humidified 5% CO2 and 95% air atmosphere. At the indicated time points, rat recombinant TNFα (PeproTech, Rocky Hill, NJ, USA) or vehicle was added to the medium. To induce hypoxia, cells were cultured in a gas barrier pouch bag for 6 h using the nBIONIX Hypoxic Cell Culture Kit (AR Brown Co., Ltd., Tokyo, Japan) as previously described [11]. Regarding the CoCl2 or H2O2 treatment, 100 µM of CoCl2 or H2O2 was added to the culture medium and incubated for 6 h.
RNA extraction and real-time quantitative PCR
Total RNA was extracted from the kidneys and NRK-52E using TRIzol (Life Technologies, Carlsbad, CA) and DirectzolTM RNA MiniPrep (Zymo Research, Irvine, CA) according to the manufacturer’s protocol. Complementary DNA was synthesized using a Prime Script reverse transcription reagent kit (RR0471A Takara Bio Inc., Shiga, Japan). The real-time detection of PCR products was performed using KAPA SYBR FAST qPCR Master Mix (2×) (KK4602: Kapa Biosystems, Wilmington, MA) and a Thermal Cycler Dice Real Time System (Takara Bio Inc., Shiga, Japan). An initial denaturation step was performed at 95 °C for 3 min and was followed by 40 cycles of amplification at 95 °C for 10 s, 62 °C for 10 s, and 55 °C for 30s. The specific primers pairs used for PCR are provided in Supplemental Table S2.
Immunofluorescence analysis
In immunofluorescence, sections were rehydrated and permeabilized with 0.5% Triton X-100 in phosphate-buffered saline (PBS) for 5 min. Samples were blocked with 10% normal goat serum in PBS and sequentially incubated with the primary antibodies shown in Supplementary Table S1 at 4 °C overnight, followed by an incubation with dye-conjugate secondary antibodies (Supplementary Table S1) at room temperature for 1 h. Nuclear counterstaining was performed using 4’,6-diamidino-2-phenylindole or DRAQ5 (DR50050; BioStatus, Leicestershire, UK; 1:2000), followed by mounting in Prolong-Gold (Thermo Fisher Scientific, Waltham, MA). Images were obtained by confocal microscopy (FV1000, Olympus).
Immunohistochemistry analysis
After deparaffinization, sections were placed in citrate-buffered solution (pH 6.0) and boiled for 5 min to retrieve antigens. Endogenous peroxidase was quenched with 3.0% H2O2 in methanol for 20 min. Blocking was performed using 3.0% bovine serum albumin (BSA) (Nacalai Tesque, Kyoto, Japan) in PBS for 30 min. Regarding nitrotyrosine staining, blocking was conducted using 1.0% fish gelatin in PBS instead of 3.0% BSA in PBS55. Sections were then incubated with primary antibodies, as provided in Supplemental Table S1, followed by an incubation with horseradish peroxidase-conjugated secondary antibodies (Supplemental Table S1). Diaminobenzidine chromogenic substrate (K3468, Agilent Technologies, Santa Clara, CA) was used for color visualization followed by counterstaining with hematoxylin. All sections were observed using an Eclipse E600 microscope (Nikon, Tokyo, Japan).
The degree of the cytoplasmic translocation of HMGB1 in tubular epithelia was scored semi-quantitatively in 5 randomly selected images of 25 consecutive non-overlapping fields in each kidney section immunostained with HMGB1 under a high magnification (n = 5) using the following arbitrarily defined values: 0, none (no positive cytoplasmic staining); 1, mild (faint positive cytoplasmic staining); 2, moderate (definitely positive cytoplasmic staining); and 3, strong (positive cytoplasmic staining with an unclear border of the nuclear membrane) (Supplementary Figure S7). The average tubular score and frequency of tubules with each score were calculated.
Statistical analysis
Results are expressed as means ± standard deviation. Each experiment was performed using at least 6 mice/group. At least 5 high power field images for each kidney were used for quantification. Mouse kidney sections were scored semi-quantitatively by two nephrologists in a blinded manner, and average scores were calculated for all parameters. Based on histological scores, the statistical power analysis was performed using G*Power software56 and the sample size was selected to achieve power of at least 0.8. The paired t-test was used for comparisons of variables between congestive and contralateral kidneys. Where data were normally distributed, an analysis of variance (ANOVA) and Tukey’s post hoc test were used for comparisons of multiple variables. Where data were not normally distributed, non-parametric tests (Kruskal-Wallis test) were used for comparisons of multiple variables. P values < 0.05 were considered to be significant.
Data availability
The data supporting the findings of this study are available in the main manuscript or the Supplementary Material. In addition, by contacting the corresponding author (Kusaba T), we will share models, protocols, methods, and other useful materials and resources related to the article as far as possible. There are no large data files to be shared via online resources.
References
Savira, F. et al. Cardiorenal syndrome: Multi-organ dysfunction involving the heart, kidney and vasculature. Br. J. Pharmacol. 177, 2906–2922. https://doi.org/10.1111/bph.15065 (2020).
Junho, C. V. C. et al. Cardiorenal syndrome: Long road between kidney and heart. Heart Fail. Rev. 27, 2137–2153. https://doi.org/10.1007/s10741-022-10218-w (2022).
Mullens, W. et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J. Am. Coll. Cardiol. 53, 589–596. https://doi.org/10.1016/j.jacc.2008.05.068 (2009).
Damman, K. et al. Increased central venous pressure is associated with impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J. Am. Coll. Cardiol. 53, 582–588. https://doi.org/10.1016/j.jacc.2008.08.080 (2009).
Guglin, M., Rivero, A., Matar, F. & Garcia, M. Renal dysfunction in heart failure is due to congestion but not low output. Clin. Cardiol. 34, 113–116. https://doi.org/10.1002/clc.20831 (2011).
Cops, J., Heesen, S., De Moor, B., Mullens, W. & Hansen, D. Current animal models for the study of congestion in heart failure: An overview. Heart Fail. Rev. 24, 387–397. https://doi.org/10.1007/s10741-018-9762-4 (2019).
Afsar, B. et al. Focus on renal congestion in heart failure. Clin. Kidney J. 9, 39–47. https://doi.org/10.1093/ckj/sfv124 (2016).
Ross, E. A. Congestive renal failure: The pathophysiology and treatment of renal venous hypertension. J. Card. Fail. 18, 930–938. https://doi.org/10.1016/j.cardfail.2012.10.010 (2012).
Bansal, S., Prasad, A. & Linas, S. Right heart failure-unrecognized cause of cardiorenal syndrome. J. Am. Soc. Nephrol. JASN. 29, 1795–1798. https://doi.org/10.1681/ASN.2018020224 (2018).
Gnanaraj, J. F., von Haehling, S., Anker, S. D., Raj, D. S. & Radhakrishnan, J. The relevance of congestion in the cardio-renal syndrome. Kidney Int. 83, 384–391. https://doi.org/10.1038/ki.2012.406 (2013).
Kitani, T. et al. Kidney vascular congestion exacerbates acute kidney injury in mice. Kidney Int. 101, 551–562. https://doi.org/10.1016/j.kint.2021.11.015 (2022).
Singer, M. et al. The third international consensus definitions for Sepsis and septic shock (Sepsis-3). Jama 315, 801–810. https://doi.org/10.1001/jama.2016.0287 (2016).
Ronco, C., Bellomo, R. & Kellum, J. A. Acute kidney injury. Lancet 394, 1949–1964. https://doi.org/10.1016/S0140-6736(19)32563-2 (2019).
Zarbock, A. et al. Sepsis-associated acute kidney injury: Consensus report of the 28th acute disease quality initiative workgroup. Nat. Rev. Nephrol. 19, 401–417. https://doi.org/10.1038/s41581-023-00683-3 (2023).
Peerapornratana, S., Manrique-Caballero, C. L., Gomez, H. & Kellum, J. A. Acute kidney injury from sepsis: Current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 96, 1083–1099. https://doi.org/10.1016/j.kint.2019.05.026 (2019).
Gomez, H. et al. A unified theory of sepsis-induced acute kidney injury: Inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock 41, 3–11. https://doi.org/10.1097/SHK.0000000000000052 (2014).
Arfaras-Melainis, A. et al. Heart failure and sepsis: Practical recommendations for the optimal management. Heart Fail. Rev. 25, 183–194. https://doi.org/10.1007/s10741-019-09816-y (2020).
Levy, M. M. et al. Outcomes of the surviving Sepsis campaign in intensive care units in the USA and Europe: A prospective cohort study. Lancet Infect. Dis. 12, 919–924. https://doi.org/10.1016/S1473-3099(12)70239-6 (2012).
Lanspa, M. J. et al. Right ventricular dysfunction in early Sepsis and septic shock. Chest 159, 1055–1063. https://doi.org/10.1016/j.chest.2020.09.274 (2021).
Shvilkina, T. & Shapiro, N. Sepsis-Induced myocardial dysfunction: Heterogeneity of functional effects and clinical significance. Front. Cardiovasc. Med. 10, 1200441. https://doi.org/10.3389/fcvm.2023.1200441 (2023).
Kawai, T. & Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 11, 373–384. https://doi.org/10.1038/ni.1863 (2010).
Shigeoka, A. A. et al. TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and -independent pathways. J. Immunol. 178, 6252–6258. https://doi.org/10.4049/jimmunol.178.10.6252 (2007).
Gluba, A. et al. The role of Toll-like receptors in renal diseases. Nat. Rev. Nephrol. 6, 224–235. https://doi.org/10.1038/nrneph.2010.16 (2010).
Liu, M. & Zen, K. Toll-Like receptors regulate the development and progression of renal diseases. Kidney Dis. (Basel). 7, 14–23. https://doi.org/10.1159/000511947 (2021).
Tammaro, A., Kers, J., Scantlebery, A. M. L. & Florquin, S. Metabolic flexibility and innate immunity in renal ischemia reperfusion injury: The fine balance between adaptive repair and tissue degeneration. Front. Immunol. 11, 1346. https://doi.org/10.3389/fimmu.2020.01346 (2020).
Tsuji, N. et al. Role of mitochondrial DNA in septic AKI via toll-like receptor 9. J. Am. Soc. Nephrol. JASN. 27, 2009–2020. https://doi.org/10.1681/ASN.2015040376 (2016).
Wang, H., Ward, M. F. & Sama, A. E. Novel HMGB1-inhibiting therapeutic agents for experimental sepsis. Shock 32, 348–357. https://doi.org/10.1097/SHK.0b013e3181a551bd (2009).
Ponikowski, P. et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 37, 2129–2200 https://doi.org/10.1093/eurheartj/ehw128 (2016).
Hoste, E. A. et al. Epidemiology of acute kidney injury in critically ill patients: The multinational AKI-EPI study. Intens. Care Med. 41, 1411–1423. https://doi.org/10.1007/s00134-015-3934-7 (2015).
Yasuda, H., Kato, A., Fujigaki, Y. & Hishida, A. Shizuoka Kidney Disease Study Group. Incidence and clinical outcomes of acute kidney injury requiring renal replacement therapy in Japan. Ther. Apher Dial. 14, 541–546. https://doi.org/10.1111/j.1744-9987.2010.00826.x (2010).
Husain-Syed, F. et al. Congestive nephropathy: A neglected entity? Proposal for diagnostic criteria and future perspectives. ESC Heart Fail. 8, 183–203. https://doi.org/10.1002/ehf2.13118 (2021).
Jorda, A. et al. Fluid management for sepsis-induced hypotension in patients with advanced chronic kidney disease: A secondary analysis of the CLOVERS trial. Crit. Care. 28, 231. https://doi.org/10.1186/s13054-024-05019-6 (2024).
Cai, L., Rodgers, E., Schoenmann, N. & Raju, R. P. Advances in rodent experimental models of Sepsis. Int. J. Mol. Sci. https://doi.org/10.3390/ijms24119578 (2023).
Doi, K., Leelahavanichkul, A., Yuen, P. S. & Star, R. A. Animal models of sepsis and sepsis-induced kidney injury. J. Clin. Investig. 119, 2868–2878. https://doi.org/10.1172/JCI39421 (2009).
Poston, J. T. & Koyner, J. L. Sepsis associated acute kidney injury. BMJ 364, k4891. https://doi.org/10.1136/bmj.k4891 (2019).
Hinshaw, L. B., Brake, C. M., Iampietro, P. F. & Emerson, T. E. Jr. Effect of increased venous pressure on renal hemodynamics. Am. J. Physiol. 204, 119–123. https://doi.org/10.1152/ajplegacy.1963.204.1.119 (1963).
Luo, L., Lucas, R. M., Liu, L. & Stow, J. L. Signalling, sorting and scaffolding adaptors for Toll-like receptors. J. Cell. Sci. https://doi.org/10.1242/jcs.239194 (2019).
Leemans, J. C. et al. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J. Clin. Investig. 115, 2894–2903. https://doi.org/10.1172/JCI22832 (2005).
Liu, T. et al. Targeting HMGB1: A potential therapeutic strategy for chronic kidney disease. Int. J. Biol. Sci. 19, 5020–5035. https://doi.org/10.7150/ijbs.87964 (2023).
Wu, H. et al. HMGB1 contributes to kidney ischemia reperfusion injury. J. Am. Soc. Nephrol. JASN 21, 1878–1890. https://doi.org/10.1681/ASN.2009101048 (2010).
Li, L. C., Gao, J. & Li, J. Emerging role of HMGB1 in fibrotic diseases. J. Cell. Mol. Med. 18, 2331–2339. https://doi.org/10.1111/jcmm.12419 (2014).
Yuan, Y. et al. Aggravated renal fibrosis is positively associated with the activation of HMGB1-TLR2/4 signaling in STZ-induced diabetic mice. Open. Life Sci. 17, 1451–1461. https://doi.org/10.1515/biol-2022-0506 (2022).
Nakano, D. et al. Reduction of tubular flow rate as a mechanism of oliguria in the early phase of endotoxemia revealed by intravital imaging. J. Am. Soc. Nephrol. JASN. 26, 3035–3044. https://doi.org/10.1681/ASN.2014060577 (2015).
Pulskens, W. P. et al. Toll-like receptor-4 coordinates the innate immune response of the kidney to renal ischemia/reperfusion injury. PLoS ONE. 3, e3596. https://doi.org/10.1371/journal.pone.0003596 (2008).
Wu, H. et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J. Clin. Investig. 117, 2847–2859. https://doi.org/10.1172/JCI31008 (2007).
Wester, A. L., Dunlop, O., Melby, K. K., Dahle, U. R. & Wyller, T. B. Age-related differences in symptoms, diagnosis and prognosis of bacteremia. BMC Infect. Dis. 13, 346. https://doi.org/10.1186/1471-2334-13-346 (2013).
Takeuchi, O. et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11, 443–451. https://doi.org/10.1016/s1074-7613(00)80119-3 (1999).
Tsuji, N. et al. BAM15 treats mouse sepsis and kidney injury, linking mortality, mitochondrial DNA, tubule damage, and neutrophils. J. Clin. Investig. https://doi.org/10.1172/JCI152401 (2023).
Uehara, M. et al. Pharmacological Inhibition of ataxia-telangiectasia mutated exacerbates acute kidney injury by activating p53 signaling in mice. Sci. Rep. 10, 4441. https://doi.org/10.1038/s41598-020-61456-7 (2020).
Yamashita, N. et al. Cumulative DNA damage by repeated low-dose cisplatin injection promotes the transition of acute to chronic kidney injury in mice. Sci. Rep. 11, 20920. https://doi.org/10.1038/s41598-021-00392-6 (2021).
Kusaba, T., Lalli, M., Kramann, R., Kobayashi, A. & Humphreys, B. D. Differentiated kidney epithelial cells repair injured proximal tubule. Proc. Natl. Acad. Sci. U.S.A. 111, 1527–1532. https://doi.org/10.1073/pnas.1310653110 (2014).
Yamashita, N. et al. Intratubular epithelial-mesenchymal transition and tubular atrophy after kidney injury in mice. Am. J. Physiol. Renal. Physiol. 319, F579–F591. https://doi.org/10.1152/ajprenal.00108.2020 (2020).
Kramann, R., Wongboonsin, J., Chang-Panesso, M., Machado, F. G. & Humphreys, B. D. Gli1(+) pericyte loss induces capillary rarefaction and proximal tubular injury. J. Am. Soc. Nephrol. JASN. 28, 776–784. https://doi.org/10.1681/ASN.2016030297 (2017).
Yang, L., Besschetnova, T. Y., Brooks, C. R., Shah, J. V. & Bonventre, J. V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 16, 535–543 https://doi.org/10.1038/nm.2144 (2010).
Kamezaki, M. et al. Comprehensive renoprotective effects of Ipragliflozin on early diabetic nephropathy in mice. Sci. Rep. 8, 4029. https://doi.org/10.1038/s41598-018-22229-5 (2018).
Kang, H. Sample size determination and power analysis using the G*Power software. J. Educ. Eval. Health Prof. https://doi.org/10.3352/jeehp.2021.18.17 (2021).
Funding
The authors received no specific funding for this work.
Author information
Authors and Affiliations
Contributions
N.I. and T.K. designed the study, performed the experiments, analyzed the data, and wrote the manuscript. M.U., A.Y-T., and S.Y. performed experiments. S.S., M.S., A.M., H.Y-S., Y.S., Y.M., N.O-O, K.N., T.N., T.K., N.Y., K.K., Y.K., K.T., and S.M. contributed to the discussion.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nakamura, I., Umehara, M., Yagi-Tomita, A. et al. Toll like receptor 2 mediated exacerbation of sepsis associated acute kidney injury by renal congestion in mice. Sci Rep 15, 22081 (2025). https://doi.org/10.1038/s41598-025-05878-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-05878-1
