
Abstract
Currently, effective prediction models for patients with advanced and postoperative gastric cancer (GC) are lacking. Programmed cell death (PCD) plays a crucial role in the development and metastasis of malignant tumors. This study aimed to investigate the underlying PCD-related molecular mechanisms and develop predictive models for GC. GC profiles were collected from TCGA-STAD, GSE84433, GSE62254, and GSE183904 databases. Differential expression analysis was conducted to identify PCD-related genes (differentially expressed genes (DEGs)), which were then subjected to functional analyses. Cox proportional hazards analyses were used to select PCD-related prognostic DEGs, and a cell death index (CDI) model was proposed. The performance of this model, tumor molecular subtypes, and the tumor microenvironment were assessed. Additionally, drug sensitivity and immune checkpoint expression were examined based on the CDI model. A total of 345 PCD-related DEGs were identified, enriched in processes such as autophagy, apoptosis, necroptosis, ferroptosis, and signaling pathways including p53, NOD-like receptor, IL-17, NF-kappa B, and PI3K-Akt. Subsequently, a CDI model comprising 17 PCD-related prognostic DEGs was constructed, demonstrating superior predictive capability. GC samples were classified into three distinct clustering subtypes, with cluster 1 exhibiting the best overall survival, followed by cluster 3 and cluster 2. Eight immune cell types were significantly associated with the CDI risk score. Furthermore, the CDI risk score exhibited positive correlations with most drugs (except for BMS.754807). Additionally, the expression of immune checkpoint genes PDCD1, CD274, and IDO1 was notably upregulated in the low-risk CDI group. Our developed CDI model, based on 17 PCD-associated prognostic genes, can be employed for risk assessment and prognosis prediction in patients with GC.
Similar content being viewed by others


Background
Gastric cancer (GC) ranks as the fifth most prevalent cancer worldwide and stands as the fourth leading cause of cancer-related fatalities1. In 2020 alone, over one million individuals received a GC diagnosis, with more than 750,000 succumbing to this malignancy2. Risk factors for GC include age, high salt consumption, Helicobacter pylori infection, and diets lacking in fruits and vegetables. Gastric adenocarcinoma (STAD) represents nearly 95% of all GC cases3. GC can be classified into various subtypes based on pathology, each with distinct clinical outcomes and therapeutic responses4. Advanced GC is characterized by high heterogeneity, invasiveness, and metastatic potential. While anti-angiogenic treatments, such as bevacizumab and apatinib, have shown promise in various tumors, including advanced GC, managing this stage of the disease remains a formidable challenge due to chemotherapy resistance and poor prognosis5,6. Currently, only 30% of patients with advanced GC survive beyond 5 years7. Surgery is the primary treatment for early-stage GC, but robust prediction models for patients with advanced and postoperative GC are lacking8,9. Therefore, identifying new prognostic markers for GC and developing effective models to enable targeted therapy are urgently required.
Programmed cell death (PCD) and accidental cell death are the two primary mechanisms of cell death. PCD encompasses a complex array of processes, including autophagy-dependent cell death, oxeiptosis, pyroptosis, alkaliptosis, apoptosis, lysosome-dependent cell death, necroptosis, ferroptosis, parthanatos, entotic cell death, cuproptosis, and NETotic cell death10. Apoptosis, a highly regulated form of cell death, is crucial for removing damaged or superfluous cells from the body, with apoptotic vesicles efficiently cleared by neighboring macrophages, thus avoiding inflammatory responses11. Pyroptosis, characterized by cell lysis, swelling, and the release of pro-inflammatory cytokines, represents a regulated inflammatory cell death type12. While necrosis was initially thought to be uncontrolled cell death, mounting evidence indicates it can be induced and occurs in a regulated manner13. Recently discovered cell death modes, such as ferroptosis and cuproptosis, are closely linked to life-threatening diseases14. Cancer cell PCD, including pyroptosis, apoptosis, and necrotic apoptosis, has emerged as a critical player in cancer immunity. A previous study identified three distinct PANoptosis modes in GC, each associated with unique immunological features, molecular profiles, and clinical outcomes, closely intertwined with GC progression15. Yu et al.16 identified three molecular subtypes related to the tumor microenvironment (TME) and demonstrated that PCD modes were more likely to occur in TME-related subtypes C2 or C3. Another study devised a cell death score model based on PCD-related genes, effectively predicting prognosis and treatment responses in patients with lung adenocarcinoma17.
Furthermore, as PCD research advances, an increasing number of related drugs have been developed and incorporated into clinical practice. BCL-2 inhibitors, by modulating the apoptosis process, have gained approval from the U.S. Food and Drug Administration for lymphoma treatment18. GSDME-mediated pyroptosis has emerged as a novel antitumor immunotherapy19, while suppressing ferroptosis can induce cellular resistance to anti-PD-1/PD-L1 treatment20. These findings underscore the fundamental role of PCD in the development and metastasis of malignant tumors. However, the intricate interplay between PCD and GC has not been comprehensively elucidated, and the specific functions of PCD in GC progression remain relatively unexplored.
In light of these considerations, this study leveraged GC-associated expression profiles from public databases to identify genes associated with 12 distinct PCD patterns in GC. By characterizing these PCD-related gene signatures, we aimed to elucidate how different modes of programmed cell death influence tumor biology and clinical outcomes. Furthermore, we analyzed the GC tumor microenvironment (TME) and developed a novel cell death index (CDI) model using multiple machine learning algorithms to predict both prognosis and therapeutic response21. These efforts align closely with our overarching objective: to integrate PCD mechanisms into a clinically relevant framework that enhances prognostic accuracy and guides personalized treatment strategies for GC patients.
Methods
Data acquisition and preprocessing
Expression profile data and phenotypic data for GC were obtained from the GDC TCGA Stomach Cancer (STAD) Xena database (https://xenabrowser.net/), which includes 350 samples with survival data and 32 control samples. Additionally, GC expression data from GSE84433 and GSE62254 were acquired from the NCBI GEO database(https://www.ncbi.nlm.nih.gov/gds/?term=). GSE62254 and GSE84433 contain 300 and 357 cancer samples, respectively. These two GEO datasets were selected based on the following criteria: (1) comprehensive clinical annotations, particularly survival data; (2) sufficient sample sizes to support statistically robust analysis; and (3) standardized microarray platforms to ensure consistency with the primary dataset. Their relevance to GC prognosis makes them suitable for validating both the prognostic value of PCD-related genes and the performance of the CDI model.
Single-cell sequencing data (GSE183904) of GC were also acquired from the GEO database, and 26 cancer tissue samples were selected for further analysis based on screening criteria (min. cells = 200, min. features = 200). Among these datasets, STAD, GSE62254, and GSE183904 were used as training datasets, while GSE62254 and GSE84433 served as validation datasets.
Following a previous report22, a list of 1,255 genes associated with 12 distinct PCD patterns was compiled for subsequent analysis.
Variant landscape of PCD-related genes in GC
Differential gene expression analysis between GC and control samples was performed using the limma package in R, with criteria of adj. P value < 0.05 and |log2 fold change (FC)| > 0.523. The adjusted P-value cutoff was chosen to control the false discovery rate (FDR) and ensure statistical robustness in the context of multiple testing. A |log2FC| > 0.5 was applied to identify genes with biologically meaningful expression changes while maintaining sensitivity to moderate yet potentially significant transcriptional alterations commonly observed in cancer-related pathways such as PCD. The identified differentially expressed genes (DEGs) underwent functional enrichment analysis using the clusterProfiler package in R. The DEGs that overlapped with the PCD-related genes were selected for mutation analysis utilizing the maftools package in R.
Construction and validation of a CDI model
Univariate Cox analysis with P < 0.224, and subsequent multivariate Cox analysis with P < 0.125, were conducted on data from the STAD and GSE62254 datasets using the survival package in R. The glmnet package in R was employed to construct a CDI model with lambda.Min26. The model’s performance was assessed using Kaplan–Meier (KM) survival curves, receiver operating characteristic (ROC) curves with the survival and survminer packages in R, followed by univariate and multivariate Cox validation analyses. The CDI was calculated using the formula: CDI risk score = Gene1 × (coef1) + Gene2 × (coef2) + Gene3 × (coef3) + …. To stratify patients into low- risk and high-risk groups, we used the median CDI score within the training dataset as the cutoff. Additionally, the gene set variation analysis (GSVA) package in R was employed for differential biological function analysis between the high and low-risk CDI groups27. The model was further validated using data from the validation datasets (GSE62254 and GSE84432).
Molecular subtyping of GC based on CDI-containing genes
Utilizing the genes included in the CDI model and data from the STAD dataset (350 samples with survival data), consistent clustering analysis was performed using the Consensus Cluster Plus package in R. Parameters included distance = “euclidean,” reps = 100, pFeature = 1, clusterAlg = “km,” and pItem = 0.7. Subsequently, the expression of CDI-containing genes in the resulting three clustering subtypes was examined and differences in clinical characteristics were analyzed. Additionally, immune cell infiltration was assessed using the CIBERSORT package, calculating immune score, tumor purity, and matrix score in the three clustering subtypes. An alluvial diagram was employed to visualize relationships among molecular subtypes, survival status, and different CDI groups.
Analysis of TME based on the CDI model
ImmuneScore, StromalScore, and EstimateScore were calculated using the estimate package in R for the STAD dataset. Correlations between the model score and these three scores were analyzed, as well as differences between high and low-risk CDI groups. Additionally, proportions of immune cell infiltration in the STAD dataset were estimated using the CIBERSORT package, and comparisons were made between high- and low-risk CDI groups.
Prediction of drug sensitivity and immunotherapy
Drug sensitivity data for the STAD dataset were obtained using Genomics of Drug Sensitivity in Cancer (GDSC2) as a reference, utilizing the oncoPredict package in R28. The expression of immune checkpoints in the high- and low-risk CDI groups was assessed and immunotherapy was analyzed using the IM vigor 210 Core Biologies dataset29.
Establishment of the nomogram survival model
Data from the STAD dataset were used to create a nomogram survival model with the rms package in R. The established model underwent calibration and decision curve analysis (DCA) using the survival and ggDCA packages in R. Subsequently, the model was divided into high- and low-score groups, and survival analysis was conducted on these groups using ROC curves with the survivalROC package in R. Validation of the nomogram survival model was performed using the GSE62254 dataset.
Characterization of the TME landscape based on single-cell RNA-seq data
Data from GSE183904 were filtered using the Seurat package in R, resulting in a total of 119,878 cells. Clustering was performed using the tSNE method with 20 principal components. The distribution of cells, distribution of model scores, proportion of cells in different samples, differences in model scores among different cells, and average expression and percentage of genes were analyzed in different cell subtypes.
Results
Identification of PCD-related DEGs in GC and functional analysis
We identified 5,846 DEGs between cancer and control samples in GC based on the criteria of adj. P value < 0.05 and |log2FC| > 0.5. Among these DEGs, 4,263 were upregulated, and 1,583 were downregulated in GC samples (Table S1). When compared with the list of 1,255 PCD-related genes, we found 345 overlapping genes, which were designated as PCD-related DEGs. The Figure shows a volcano plot illustrating these PCD-related DEGs, highlighting genes significantly upregulated or downregulated based on adjusted P-value and log2 fold change thresholds. (Fig. 1A). Bidirectional hierarchical clustering of these 345 PCD-related DEGs demonstrated their ability to differentiate between cancer and control samples (Fig. 1B). Further analysis revealed the expression patterns, chromosomal locations, and correlations among these DEGs (Fig. 1C).

Identification of programmed cell death (PCD)-related differential expressed genes (DEGs) in gastric cancer (GC) and functional analysis. (A) Volcano plot displaying PCD-related DEGs between cancer and control samples. (B) Bidirectional hierarchical clustering heatmap of PCD-related DEGs. (C) Chromosome location, expression, and correlation of PCD-related DEGs. (D) Gene Ontology (GO) term analysis of identified PCD-related DEGs. (E) Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of identified PCD-related DEGs. (F) Mutation analysis of identified PCD-related DEGs. (G) Copy number variation (CNV) analysis of identified PCD-related DEGs.
Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed on these DEGs. Notably, the DEGs were significantly enriched in GO terms related to processes such as “secretory granule lumen,” “regulation of autophagy,” “signaling pathway in response to DNA damage,” “lysosomal membrane,” and “p53 binding” (Fig. 1D), indicating active involvement in cell stress responses, survival mechanisms, and tumor suppression. Additionally, KEGG pathway analysis further highlighted enrichment in key cancer-related and immune-regulatory pathways including “NOD-like receptor signaling pathway,” “lysosome,” “apoptosis,” “IL-17 signaling pathway,” “necroptosis,” “p53 signaling pathway,” “ferroptosis,” “NF-kappa B signaling pathway,” and “PI3K-Akt signaling pathway” (Fig. 1E). These pathways are not only central to distinct PCD modalities but are also closely linked to tumor immune evasion, inflammation, and therapeutic resistance in GC. Functional enrichment was performed using the KEGG database30.
Mutation analysis of PCD-related DEGs revealed that missense mutations were the most common, followed by frame shift deletions and nonsense mutations. Single nucleotide polymorphisms were the predominant variant type (Fig. 1F). The mutation status of 20 randomly selected genes, including PRKDC, LARP1, and FANCD2, is shown in (Fig. 1F). Copy number variation (CNV) analysis indicated frequent alterations in the PCD-related DEGs, with extensive copy number amplification and deletion observed in genes such as NUPR1 and PYCARD, which are known to regulate autophagy and inflammasome activation, respectively (Fig. 1G). Collectively, these findings suggest that PCD-related DEGs are not only statistically enriched but also biologically relevant to GC pathogenesis, with potential implications in tumor cell survival, genomic instability, immune interaction, and treatment response.
Identification of PCD-related prognostic DEGs and assessment of the CDI model
We conducted univariate Cox analysis on the 345 PCD-related DEGs, resulting in 124 and 206 genes with significant associations (P < 0.2) in the STAD and GSE62254 datasets, respectively. An overlapping set of 89 genes was subjected to multivariate Cox analysis, which identified 18 genes with significant associations (P < 0.1). Subsequently, a CDI model was constructed using least absolute shrinkage and selection operator (LASSO) Cox regression analysis, which included 17 PCD-related prognostic DEGs: FANCD2, DDIAS, CRYAB, PLA2G15, PDK4, SERPINE1, SRPX, APAF1, TRIM65, TPP1, CXCL12, DNASE2, ABCA2, CD24, PYCARD, ATF3, and CYP1B1 (Fig. 2A).

Identification of PCD-related prognostic DEGs and evaluation of the constructed cell death index (CDI) model in GC. (A) Construction of the CDI model and coefficients of the 17 PCD-related prognostic DEGs. (B) Kaplan–Meier (KM) survival curves in samples with high-risk and low-risk CDI scores. (C) Receiver operating characteristic (ROC) curves for 1, 3, and 5 years. (D) Expression of the 17 PCD-related prognostic DEGs in high- and low-risk CDI groups. (E) Risk score of the CDI model in different clinical characteristics.
The CDI model was defined by the following formula:
Risk score of CDI = 0.32288 × FANCD2 + (–0.43662) × DDIAS + (–0.04725) × CRYAB + 0.61807 × PLA2G15 + 0.09553 × PDK4 + 0.17529 × SERPINE1 + (–0.03216) × SRPX + (–0.43708) × APAF1 + 0.13882 × TRIM65 + 0.25287 × TPP1 + (–0.11492) × CXCL12 + (–0.537) × DNASE2 + (–0.40868) × ABCA2 + 0.12131 × CD24 + (–0.14339) × PYCARD + 0.09819 × ATF3 + 0.13558 × CYP1B1.
Using the median CDI risk score as a cutoff, patients in the STAD dataset were stratified into high- and low-risk CDI groups (n = 175 each)(Figure S2). Kaplan–Meier (KM) survival curves in samples with high-risk and low-risk CDI scores, the high-risk CDI group exhibited significantly lower overall survival (OS) compared to the low-risk CDI group (P < 0.0001, Fig. 2B). ROC curve analysis demonstrated good predictive performance of the CDI model, with AUC values of 0.723 for 1-year, 0.762 for 3-year, and 0.784 for 5-year survival (Fig. 2C).
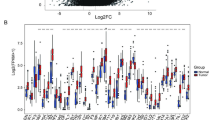
The expression levels of the 17 PCD-related prognostic DEGs in different risk CDI groups are depicted in Fig. 2D. PDK4, CRYAB, SRPX, CXCL12, and CYP1B1 were upregulated in the high-risk CDI group. Correlation analysis between the CDI risk score and clinical characteristics revealed significant differences in risk scores between various clinical features, including tumor grade (G2 vs. G3, P = 0.006), age (> 65 years old vs. ≤ 65 years old, P = 0.042), lymph node status (N0 vs. N3, P = 0.046; N0 vs. N2, P = 0.043), tumor size (T1 vs. T3, P = 0.023), gender (female vs. male, P = 0.043), and cancer stage (stage I vs. stage III, P = 0.037, Fig. 2E and Table S2). The expression patterns of the 17 PCD-related prognostic DEGs across different clinical features are shown in Fig. 3A. Differential biological function analysis using GSVA revealed significant processes associated with high- and low-risk CDI groups, including “O glycosylation of TSR domain containing proteins,” “neurofascin interactions,” “molecules associated with elastic fibers,” and “formation of fibrin clot clotting cascade” (Fig. 3B). Cox regression analysis confirmed that the CDI risk score was an independent prognostic factor in patients with GC (Fig. 3C).

Correlation between the 17 PCD-related prognostic DEGs and clinical characteristics and functional analysis. (A) Heatmap illustrating the relationship between the 17 PCD-related prognostic DEGs and clinical characteristics. (B) Top 10 biological processes with the most significant differences between high- and low-risk CDI groups. (C) Univariate and multivariate Cox regression analysis of the risk score of CDI and other clinical characteristics.
Verification of the CDI model
Validation datasets GSE84433 and GSE62254 were utilized to validate the performance of the CDI model (Figure S3). In both datasets, patients in the low-risk CDI group had significantly better prognosis than those in the high-risk CDI group (Fig. 4A). The AUC values for 1-year, 3-year, and 5-year survival in the GSE84433 dataset were 0.606, 0.581, and 0.587, respectively, while in the GSE62254 dataset, the AUC values were 0.604, 0.604, and 0.624, respectively (Fig. 4A), confirming the reliability of the CDI model. Cox analyses identified age and risk score as independent prognostic factors in GC in the GSE84433 dataset (P < 0.05, Fig. 4B) and age, pathologic-N, pathologic-M, and risk score as independent prognostic factors in the GSE62254 dataset (P < 0.05, Fig. 4C).

Verification of the constructed CDI model. (A) KM survival curves for high- and low-risk CDI groups and ROC curves for 1, 3, and 5 years in the validation datasets (GSE84433 and GSE62254). Univariate and multivariate Cox regression analysis of the risk score of CDI and other clinical characteristics in GSE84433 (B) and GSE62254 (C).
Different molecular subtypes of GC based on CDI-containing genes
The 17 PCD-related prognostic DEGs included in the CDI model were used for consistent clustering analysis of the STAD dataset, resulting in the identification of three clustering subtypes: cluster 1, 2, and 3 (Fig. 5A, S4). Examination of the expression of these DEGs in the three clustering subtypes revealed no significant differences in ABCA2, DNASE2, PLA2G15, PYCARD, and TRIM65 expression among the clusters (P > 0.05, Fig. 5B, Table S3).

Different molecular subtypes of GC based on the 17 PCD-related prognostic DEGs. (A) Identification of three clustering subtypes in the STAD dataset. (B) Expression patterns of the 17 PCD-related prognostic DEGs in the three clustering subtypes. (C) Kaplan–Meier (KM) survival curves for samples in different clustering subtypes. (D) Variation in clinical characteristics across the three clustering subtypes. (E) ImmuneScore, StromalScore, and EstimateScore in the three clustering subtypes. (F) Heatmap depicting immune cell infiltration in different subtypes. (G) Alluvial diagram illustrating relationships among different clustering subtypes, overall survival, and risk. 0: live; 1: dead. * P < 0.05, ** P < 0.01, *** P < 0.001, ns: not significant.
Comparison of clinical characteristics showed that cluster 1 had the best OS, followed by cluster 3 and 2 (Fig. 5C). Additionally, significant differences were observed in age, gender, and pathologic-M among the three clusters (P < 0.05, Fig. 5D). Evaluation of immune, stromal, and estimate scores revealed the highest scores in cluster 2, followed by cluster 3 and 1 (Fig. 5E, Table S3). Hierarchical clustering of immune cell proportions showed marked differences in the infiltration ratios of 11 immune cell types (such as B cells naïve, T cells follicular helper, monocytes, macrophages M0, macrophages M2, and neutrophils) among the three clustering subtypes (P < 0.05, Fig. 5F, Table S3). An alluvial diagram effectively demonstrated the relationships among molecular subtypes, survival outcomes, and CDI risk stratification, further highlighting the potential clinical utility of CDI-based molecular subtyping in personalized therapeutic decision-making (Fig. 5G).
Immune cell infiltration based on CDI model score
Based on the CDI risk score, the STAD dataset samples were divided into high- and low-risk CDI groups. The StromalScore and EstimateScore, derived from the ESTIMATE algorithm, were significantly lower in the low-risk group compared to the high-risk group (P < 0.05), indicating a reduced presence of stromal and non-tumor components in tumors with a more favorable prognosis (Fig. 6A). Although the ImmuneScore showed no significant difference between groups (P = 0.1), correlation analysis revealed that all three scores—ImmuneScore, StromalScore, and EstimateScore—were positively associated with the CDI risk score (P < 0.05, Fig. 6B), suggesting that higher CDI scores are linked to a more complex and potentially immunosuppressive tumor microenvironment. Furthermore, CIBERSORT analysis revealed significant differences in the infiltration proportions of eight immune cell types between the high- and low-risk CDI groups (P < 0.05, Fig. 6C). These findings indicate that the CDI model not only reflects cell death-related molecular alterations but also captures variations in the tumor immune landscape, thereby enhancing its clinical applicability in stratifying patients and potentially guiding immunotherapy decisions.

Analysis of immune cell infiltration based on the CDI model score. (A) ImmuneScore, StromalScore, and EstimateScore in the high- and low-risk CDI groups. (B) Correlation between ImmuneScore, StromalScore, EstimateScore, and risk score. (C) Proportions of different immune cell infiltrations in the high-risk and low-risk CDI groups. * P < 0.05, ** P < 0.01, *** P < 0.001, ns: not significant.
Prediction of drug sensitivity based on the CDI model and CDI-associated immunotherapy
The relationships among drug sensitivity, the risk score of the CDI model, and the 17 PCD-related prognostic DEGs are illustrated in Fig. 7A and Table S4. It was observed that the risk score of the CDI model, similar to SRPX, PDK4, CYP1B1, CXCL12, and CRYAB, exhibited a positive correlation with most drugs, with the exception of BMS.754,807 (Figure S5). Subsequently, the risk score of the CDI model was evaluated in samples with partial response (PR), progressive disease (PD), stable disease (SD), and complete response (CR). Notably, the risk score of the CDI model was significantly different only between the PD and CR groups (P < 0.05, Fig. 7B). Subsequently, the proportions of CR/PR and PD/SD were determined in the high-risk and low-risk CDI groups. It is evident that in both the high- and low-risk CDI groups, the proportion of CR/PR was higher than that of PD/SD, with the lowest proportion of PD/SD observed in the low-risk CDI group (Fig. 7C). Additionally, the expression of the 17 PCD-related prognostic DEGs was analyzed between the CR/PR and PD/SD groups. Compared to the CR/PR group, the expression of ABCA2, APAF1, and DDIAS was significantly downregulated in the PD/SD group, while the expression of PLA2G15 and CYP1B1 was markedly upregulated in the PD/SD group (Fig. 7D).

Prediction of drug sensitivity based on the CDI model and CDI-associated immunotherapy. (A) Relationships among drug sensitivity, CDI model risk score, and 17 PCD-related prognostic DEGs. (B) CDI model risk score in samples with progressive disease (PD), stable disease (SD), complete response (CR), and partial response (PR). (C) Percentage of CR/PR and PD/SD in high-risk and low-risk CDI groups. (D) Expression of the 17 PCD-related prognostic DEGs between CR/PR and PD/SD groups. (E) Top 10 immune checkpoints in high-risk and low-risk CDI groups. (F) KM survival curves in high- and low-risk CDI groups. (G) CDI model risk score in samples with PD, SD, CR, and PR. * P < 0.05, ** P < 0.01, *** P < 0.001, ns: not significant.
Furthermore, we examined immune checkpoints in the high- and low-risk CDI groups. In comparison to the high-risk CDI group, PDCD1, CD274, and IDO1 were significantly upregulated in the low-risk CDI group (P < 0.05, Fig. 7E). Additionally, it was observed that samples with a high-risk CDI score had lower OS than those with a low-risk CDI score (Fig. 7F). In the CDI-associated immunotherapy analysis, the risk score in the SD group was markedly higher than in the PR group (P = 0.049, Fig. 7G).
Establishment and evaluation of the nomogram survival model
To better elucidate the relationship between independent clinical factors and survival, we established a nomogram survival model. After Cox regression analyses, a nomogram survival model predicting survival at 1, 3, and 5 years was constructed (Fig. 8A). Calibration curves demonstrated the accuracy of the model in predicting 1-, 3-, and 5-year survival rates (Fig. 8B). Decision curve analysis (DCA) indicated that the performance of the nomogram survival model was superior to other factors in the first part of the threshold probability, while the model constructed by LASSO regression performed better in the latter part of the threshold probability (Fig. 8C). The DCA demonstrates that the nomogram model incorporating the CDI risk score provides a higher net clinical benefit across a wide range of threshold probabilities, compared to models based on individual clinical variables. KM analysis revealed that samples with a high nomogram score had lower OS than those with a low nomogram score (Fig. 8D). The 1-, 3-, and 5-year AUC values in the TCGA-STAD dataset were 0.7264, 0.79, and 0.78, respectively (Fig. 8E), and in the validation dataset (GSE62254), the corresponding AUC values were 0.81, 0.786, and 0.78 (Fig. 8F). These results suggest that the established nomogram survival model is reliable for predicting GC outcomes, as it demonstrates consistency between the training and validation datasets.

Establishment and evaluation of the nomogram survival model. (A) Nomogram survival model incorporating CDI risk score and other clinical characteristics. (B) Calibration plots of the nomogram survival model for predicting survival probability at 1, 3, and 5 years. (C) Decision curve analysis of the nomogram survival model and other clinical factors. (D) KM survival curves in high- and low-risk nomogram score groups. ROC curves for 1, 3, and 5 years in the TCGA-STAD database (E) and GSE62254 database (F).
Characterization of immune cells according to the single-cell RNA-seq data
The single-cell RNA-seq data from GSE183904 were utilized to investigate the distribution of model scores using LASSO regression in patients with GC. After annotation, the primary cell types were identified as NK/T cells (Fig. 9A and B). The distribution of different cell types across different samples is depicted in Fig. 9C. The model score was higher in fibroblasts and epithelial cells compared to other cell types (Fig. 9D). Additionally, the correlation between the 17 PCD-related prognostic DEGs and cell types revealed that SERPINE1 and CXCL12 were closely related to fibroblasts, while ATF3 and CD24 were correlated with epithelial cells (Fig. 9E).

Characterization of immune cells based on single-cell RNA-seq data. (A) Distribution of immune cells in the single-cell RNA-seq dataset GSE183904. (B) Score distribution of immune cells in GSE183904. (C) Proportion of different cell types in various samples in GSE183904. (D) Scores of different cell types. (E) Correlation between the 17 PCD-related prognostic DEGs and different cell types.
Discussion
GC is a prevalent malignant tumor that poses a significant threat to people’s lives and health. PCD plays a pivotal role in the regulation of the TME and the determination of clinical outcomes in cancer treatments31. In this study, we identified 345 DEGs closely associated with 12 distinct PCD patterns. These genes are involved in essential cellular processes, such as autophagy, apoptosis, necroptosis, ferroptosis, and signaling pathways, including p53, NOD-like receptor (NLR), IL-17, NF-kappa B, and PI3K-Akt.
Autophagy is a critical cellular homeostasis mechanism that involves the degradation and recycling of defective proteins and organelles, particularly under conditions of stress, such as hypoxia and nutrient deprivation32. Autophagy can both promote cell survival and accelerate cell death, making it a complex process with implications for normal and tumor cells alike. Signaling pathways related to autophagy encompass mTOR-dependent pathways, such as PI3K/Akt/mTOR, as well as non-mTOR-dependent pathways, such as p5333. Qiu et al.34 have shown that HBXIP can modulate glucose metabolism through PI3K/AKT and p53 signaling, contributing to GC development.
Apoptosis, necroptosis, and ferroptosis are distinct PCD patterns that have been implicated in GC progression35. In our study, enrichment analyses revealed that several key signaling pathways—such as NF-κB, PI3K-Akt, IL-17, and NOD-like receptor (NLR) pathways—were significantly associated with the identified PCD-related DEGs, suggesting potential mechanistic links. Notably, NLRs are critical mediators of inflammation-associated chemoresistance, tumorigenesis, cancer cell death, and angiogenesis. Aberrant activation of NLRs occurs in various cancers, shaping the TME and increasing the risk of tumorigenesis36. Similarly, IL-17 is closely linked to immunopathology, and elevated serum IL-17 levels have been observed in patients with GC, suggesting its potential as a diagnostic marker for GC37. Dysregulation of NF-κB signaling is known to modulate the immune response to infection and has been widely associated with cancer development, particularly metastasis38. For instance, Deng et al.39 elucidated that the long non-coding RNA ANRIL enhances GC progression via the NF-κB signaling pathway. In our analysis, several PCD-related DEGs (e.g., SERPINE1, CXCL12, PYCARD) are known to interact with or regulate these pathways, potentially contributing to tumor proliferation, apoptosis resistance, and immune modulation. In summary, our findings suggest that signaling pathways involving autophagy, apoptosis, necroptosis, ferroptosis, p53, NLR, IL-17, NF-kappa B, and PI3K-Akt may play significant roles in the initiation and metastasis of GC. Nevertheless, further experimental validation is required to elucidate the precise molecular interactions and biological consequences of these pathway–gene networks in GC.
Subsequently, we constructed a CDI model consisting of 17 PCD-related prognostic DEGs: FANCD2, DDIAS, CRYAB, PLA2G15, PDK4, SERPINE1, SRPX, APAF1, TRIM65, TPP1, CXCL12, DNASE2, ABCA2, CD24, PYCARD, ATF3, and CYP1B1. These genes are implicated in various cancer-associated biological processes, highlighting their potential utility as prognostic biomarkers in gastric cancer (GC). For example, FANCD2 for instance, plays a crucial role in regulating tumorigenesis and apoptosis in cancers. Elevated FANCD2 levels are strongly correlated with poor prognosis in both primary and recurrent glioblastoma, and its silencing has been shown to inhibit cell survival40. DDIAS, a DNA damage-induced apoptosis inhibitor, exhibits a positive correlation with tumor size and regional lymph node metastasis and is known to enhance proliferation and invasion of non-small cell lung cancer (NSCLC) cells when overexpressed41. CRYAB, a protein involved in multiple signaling pathways including cell apoptosis, inflammation, and oxidative stress, is upregulated in NSCLC42. FATC3-PLA2G15 overexpression has been associated with aggressive T-acute lymphoblastic leukemia (T-ALL) biology and poor prognosis in human T-ALL43. PDK4, highly expressed in GC cell, regulates various cellular processes, including metal ion transport, growth, migration, and cancer cell metabolism, making it a potential diagnostic biomarker and therapeutic target for patients with GC44. SERPINE1, upregulated in GC tissues, promotes GC occurrence and development through the VEGF and JAK-STAT3 signaling pathways45. SRPX expression is enhanced in glioblastoma cells exposed to temozolomide, and its knockdown inhibits cell viability, suggesting it may serve as a novel tumor marker for glioblastomas46. APAF1, TRIM65, and CXCL12 are upregulated in GC, and their downregulation induces GC cell apoptosis and inhibits invasion47,48,49. TPP1, significantly upregulated in esophageal cancer, can inhibit DNA damage response and chemical sensitivity, potentially influencing the progression of esophageal cancer50. ATF3 and CD24 have been identified as hub genes in diabetic nephropathy, possibly promoting its progression51. DNASE2 plays a key role in hepatocellular carcinoma (HCC) development, with its silencing repressing HCC cell growth and promoting apoptosis52. ABCA2 has been linked to cisplatin resistance in different cancer cells53, and lncRNA UCA1 can enhance cisplatin resistance by regulating CYP1B1-mediated apoptosis in human GC through miR-513a-3p54. High PYCARD abundance has been identified as an independent predictor of poor prognosis and chemotherapy resistance in glioma55.
The mutation and CNV analyses of the 345 PCD-related DEGs provided important insights into their genomic instability in gastric cancer (GC). Mutation profiling revealed that missense mutations were the most frequent type, followed by frameshift deletions and nonsense mutations, with single nucleotide polymorphisms being the dominant variant class. While informative, these mutation patterns also have functional implications. For example, mutations in FANCD2 and PYCARD, which are involved in DNA damage repair and pyroptosis regulation respectively, could impair their normal tumor-suppressive functions, leading to enhanced genomic instability, immune evasion, or resistance to programmed cell death—key features of aggressive tumor behavior and treatment resistance.
Furthermore, the CNV analysis showed widespread amplifications and deletions among the PCD-related DEGs. Notably, genes such as NUPR1 and PYCARD exhibited recurrent copy number alterations, which may contribute to dysregulated expression and downstream pathway disruption. Integrating CNV data with expression profiles revealed that many genes with copy number gains, such as SERPINE1 and PDK4, were also overexpressed in tumor samples, suggesting a possible dosage effect that could drive tumor progression. Correlation of these alterations with clinical data indicated that CNV-driven upregulation of certain genes was associated with poorer prognosis and higher CDI risk scores, further supporting their role in GC pathogenesis. These findings underscore the functional relevance of genetic alterations in PCD-related DEGs and highlight their potential as biomarkers for prognosis or therapeutic targets.
These findings not only underscore the functional relevance of genomic alterations in PCD-related DEGs but also support the robustness of our predictive models. Specifically, the 1-, 3-, and 5-year AUC values in both the CDI model and the nomogram survival model exceeded 0.7, indicating their potential for robust predictive ability in assessing GC risk and prognosis22. Building upon these results and prior evidence, we propose that the 17 PCD-related prognostic DEGs included in the CDI model may play pivotal roles in gastric cancer development and progression. Accordingly, the CDI and nomogram models based on these genes hold promise for improving individualized risk stratification and outcome prediction in clinical settings. Nonetheless, further experimental and clinical validation is required to fully elucidate the functional roles of these genes and their potential as therapeutic targets in GC.One of the primary factors allowing tumor cells to evade destruction and thrive is the TME, which provides them with opportunities to escape immune surveillance and resist drug treatments56. The TME is a complex composition of inflammatory mediators, mesenchymal cells, tumor-infiltrating immune cells, and extracellular matrix molecules. The immune composition within the TME has been closely linked to the prognosis of patients with cancer57. In theory, a high-risk CDI group might exhibit reduced levels of checkpoints and antitumor infiltrating immune cells, indicating an overall impairment of immune function58. Increased immune cell infiltration in patients with cancer is associated with better clinical outcomes, whereas lower immune cell infiltration is linked to worse prognosis59. Consequently, identifying immune signatures in human cancers may reveal valuable biomarkers for evaluating clinical outcomes.
In our study, we identified eight immune cell types that exhibited a close correlation with the risk score of the CDI model. These included activated NK cells, macrophages M1, monocytes, activated memory CD4 T cells, macrophages M2, and follicular helper T cells. Analysis of drug sensitivity predictions revealed that the risk score of the established CDI model had a negative correlation with sensitivity to BMS.754,807 but a positive correlation with 23 other drugs, including cytarabine, gefitinib, afatinib, and lapatinib. These findings imply that patients in the high-risk CDI group may be more responsive to certain targeted therapies or chemotherapeutic agents. However, further experimental validation and clinical data are needed to confirm the practical utility of these predictions and to evaluate their potential for guiding individualized treatment strategies.
Furthermore, we analyzed the expression of 10 immune checkpoint genes and observed that PDCD1, CD274 and IDO1 were significantly upregulated in the low-risk CDI group compared to the high-risk CDI group. PDCD1, an immunomodulator expressed on activated T cells, is upregulated by chemotherapy drugs in GC cells, and anti-PDCD1 treatment can promote tumor infiltration by CD8 T cells60. CD274, which encodes programmed death ligand 1, participates in major co-inhibitory checkpoint signaling that controls T cell behavior61. IDO1, abundant in many cancers, suppresses the antitumor immune response within the host by consuming tryptophan in the TME62. Moreover, IDO1 and COL12A1 collaboratively promote GC metastasis, suggesting their potential as promising targets for GC anticancer treatment63.
Therefore, our hypothesis is that these eight immune cell types, such as macrophages M1, monocytes, follicular helper T cells, macrophages M2, activated NK cells, and memory CD4 T cells, could serve as valuable predictors of clinical outcomes in GC. Additionally, PDCD1, CD274, and IDO1 may represent crucial immune checkpoints with promising implications for GC therapy.
The identification of three distinct molecular subtypes based on CDI-related genes provides valuable insight into the heterogeneity of gastric cancer (GC). While significant differences in overall survival were observed among the subtypes, their relevance extends beyond prognosis. Our analysis revealed that these subtypes also differ in immune and stromal scores, as well as in the composition of tumor-infiltrating immune cells, suggesting that they may represent distinct tumor microenvironment (TME) phenotypes. For instance, Cluster 2 exhibited elevated stromal and immune infiltration, potentially indicating a more immunosuppressive TME, which may influence responsiveness to immunotherapies or targeted treatments.
These findings highlight the potential clinical utility of molecular subtyping for guiding individualized therapeutic strategies. However, we acknowledge that further investigation into the biological functions and treatment sensitivities of each subtype is warranted to enhance their translational relevance. Additionally, although our immune infiltration analysis provides preliminary insights into TME characteristics, its robustness would be further improved by functional validation using methods such as immunohistochemistry or high-resolution single-cell transcriptomics. Future studies incorporating these approaches are needed to confirm the immune landscape and clarify its implications for GC therapy.
Conclusions
In summary, we have systematically developed and assessed a CDI risk score based on 17 PCD-associated prognostic genes, and subsequently correlated this CDI model with the TME. Our findings suggest that the CDI model, relying on the identified 17 PCD-related prognostic DEGs (FANCD2, DDIAS, CRYAB, PLA2G15, PDK4, SERPINE1, SRPX, APAF1, TRIM65, TPP1, CXCL12, DNASE2, ABCA2, CD24, PYCARD, ATF3, and CYP1B1) holds promise for risk assessment, prognosis prediction, and guiding immunotherapy decisions in GC. These results not only expand our comprehension of the role of PCD in GC but also offer potential avenues for personalized GC treatment, ultimately enhancing clinical outcomes.
Data availability
All datasets involved in this study can be viewed in Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/gds/?term=), The Cancer Genome Atlas (TCGA) (https://tega-data.nci.nih.gov/tega), and the Xena (https://xenabrowser), or data availability part of the corresponding articles. All the other data supporting the findings of this study are available within the article and its Supplementary Information Files, or from the corresponding authors upon reasonable request.
Abbreviations
- CDI:
-
Cell death index
- DCA:
-
Decision curve analysis
- DEG:
-
Differentially expressed gene
- FC:
-
Fold change
- GC:
-
Gastric cancer
- GO:
-
Gene Ontology
- GSVA:
-
Gene set variation analysis
- HCC:
-
Hepatocellular carcinoma
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- KM:
-
Kaplan–Meier
- LASSO:
-
Least absolute shrinkage and selection operator
- NLR:
-
NOD-like receptor
- NSCLC:
-
Non-small cell lung cancer
- OS:
-
Overall survival
- PCD:
-
Programmed cell death
- ROC:
-
Receiver operating characteristic
- STAD:
-
GDC TCGA Stomach Cancer
- T-ALL:
-
T-acute lymphoblastic leukemia
- TME:
-
Tumor microenvironment
References
Smyth, E. C., Nilsson, M., Grabsch, H. I., van Grieken, N. C. & Lordick, F. Gastric cancer. Lancet 396(10251), 635–648. https://doi.org/10.1016/s0140-6736(20)31288-5 (2020).
Sung, H. et al. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71(3), 209–249. https://doi.org/10.3322/caac.21660 (2021).
Karimi, P., Islami, F., Anandasabapathy, S., Freedman, N. D. & Kamangar, F. Gastric cancer: Descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol. Biomarkers Prev. 23(5), 700–713. https://doi.org/10.1158/1055-9965.Epi-13-1057 (2014).
Onoyama, T., Ishikawa, S. & Isomoto, H. Gastric cancer and genomics: Review of literature. J. Gastroenterol. 57(8), 505–516. https://doi.org/10.1007/s00535-022-01879-3 (2022).
Hironaka, S. Anti-angiogenic therapies for gastric cancer. Asia Pac. J. Clin. Oncol. 15(4), 208–217. https://doi.org/10.1111/ajco.13174 (2019).
Zeng, Y. & Jin, R. U. Molecular pathogenesis, targeted therapies, and future perspectives for gastric cancer. Semin Cancer Biol. 86(Pt 3), 566–582. https://doi.org/10.1016/j.semcancer.2021.12.004 (2022).
De Re, V. Molecular features distinguish gastric Cancer subtypes. Int. J. Mol. Sci. 19(10). https://doi.org/10.3390/ijms19103121 (2018).
Guerrini, G. P. et al. Robotic versus laparoscopic gastrectomy for gastric cancer: The largest meta-analysis. Int. J. Surg. 82, 210–228. https://doi.org/10.1016/j.ijsu.2020.07.053 (2020).
Li, X. et al. An artificial intelligence model to predict survival and chemotherapy benefits for gastric cancer patients after gastrectomy development and validation in international multicenter cohorts. Int. J. Surg. 105, 106889. https://doi.org/10.1016/j.ijsu.2022.106889 (2022).
Tang, D., Kang, R., Berghe, T. V., Vandenabeele, P. & Kroemer, G. The molecular machinery of regulated cell death. Cell. Res. 29(5), 347–364. https://doi.org/10.1038/s41422-019-0164-5 (2019).
Zhao, R. et al. Novel roles of apoptotic caspases in tumor repopulation, epigenetic reprogramming, carcinogenesis, and beyond. Cancer Metastasis Rev. 37(2–3), 227–236. https://doi.org/10.1007/s10555-018-9736-y (2018).
Tang, R. et al. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 13(1), 110. https://doi.org/10.1186/s13045-020-00946-7 (2020).
Xia, Y. et al. Development and validation of a necroptosis-related gene prognostic score to predict prognosis and efficiency of immunotherapy in gastric cancer. Front. Immunol. 13, 977338. https://doi.org/10.3389/fimmu.2022.977338 (2022).
Tong, X. et al. Targeting cell death pathways for cancer therapy: Recent developments in necroptosis, pyroptosis, ferroptosis, and Cuproptosis research. J. Hematol. Oncol. 15(1), 174. https://doi.org/10.1186/s13045-022-01392-3 (2022).
Pan, H., Pan, J., Li, P. & Gao, J. Characterization of PANoptosis patterns predicts survival and immunotherapy response in gastric cancer. Clin. Immunol. 238, 109019. https://doi.org/10.1016/j.clim.2022.109019 (2022).
Yu, L., He, R. & Cui, Y. Characterization of tumor microenvironment and programmed death-related genes to identify molecular subtypes and drug resistance in pancreatic cancer. Front. Pharmacol. 14, 1146280. https://doi.org/10.3389/fphar.2023.1146280 (2023).
Zhang, Y., Wang, Y., Chen, J., Xia, Y. & Huang, Y. A programmed cell death-related model based on machine learning for predicting prognosis and immunotherapy responses in patients with lung adenocarcinoma. Front. Immunol. 14, 1183230. https://doi.org/10.3389/fimmu.2023.1183230 (2023).
Carneiro, B. A. & El-Deiry, W. S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 17(7), 395–417. https://doi.org/10.1038/s41571-020-0341-y (2020).
Zhang, Z. et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579(7799), 415–420. https://doi.org/10.1038/s41586-020-2071-9 (2020).
Jiang, Z. et al. TYRO3 induces anti-PD-1/PD-L1 therapy resistance by limiting innate immunity and tumoral ferroptosis. J. Clin. Invest. 131(8). https://doi.org/10.1172/jci139434 (2021).
Djilali, S., Sarmad, G. & Tridane, A. Dynamics and asymptotic profiles of a local-nonlocal dispersal SIR epidemic model with Spatial heterogeneity. Infect. Dis. Model. 10(2), 387–409. https://doi.org/10.1016/j.idm.2024.12.003 (2025).
Zou, Y. et al. Leveraging diverse cell-death patterns to predict the prognosis and drug sensitivity of triple-negative breast cancer patients after surgery. Int. J. Surg. 107, 106936. https://doi.org/10.1016/j.ijsu.2022.106936 (2022).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. EdgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1), 139–140. https://doi.org/10.1093/bioinformatics/btp616 (2010).
Gomes, E. C., Falci, D. R., Bergo, P., Zavascki, A. P. & Rigatto, M. H. Impact of polymyxin-B-associated acute kidney injury in 1-year mortality and renal function recovery. Int. J. Antimicrob. Agents. 52(1), 86–89. https://doi.org/10.1016/j.ijantimicag.2018.02.016 (2018).
Li, R., Zhang, S. & Liu, G. Identification and validation of a pyroptosis-related prognostic model for colorectal cancer. Funct. Integr. Genomics. 23(1), 21. https://doi.org/10.1007/s10142-022-00935-8 (2022).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33(1), 1–22 (2010).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7. https://doi.org/10.1186/1471-2105-14-7 (2013).
Maeser, D., Gruener, R. F. & Huang, R. S. OncoPredict: an R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief. Bioinform. 22(6). https://doi.org/10.1093/bib/bbab260 (2021).
Fu, J. et al. Large-scale public data reuse to model immunotherapy response and resistance. Genome Med. 12(1), 21. https://doi.org/10.1186/s13073-020-0721-z (2020).
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M. & Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 49(D1), D545–D51. https://doi.org/10.1093/nar/gkaa970 (2021).
Liu, J. et al. Programmed cell death Tunes tumor immunity. Front. Immunol. 13, 847345. https://doi.org/10.3389/fimmu.2022.847345 (2022).
Onorati, A. V., Dyczynski, M., Ojha, R. & Amaravadi, R. K. Targeting autophagy in cancer. Cancer 124(16), 3307–3318. https://doi.org/10.1002/cncr.31335 (2018).
Cao, Y. et al. Autophagy and its role in gastric cancer. Clin. Chim. Acta. 489, 10–20. https://doi.org/10.1016/j.cca.2018.11.028 (2019).
Qiu, L. et al. HBXIP regulates gastric Cancer glucose metabolism and malignancy through PI3K/AKT and p53 signaling. Onco Targets Ther. 13, 3359–3374. https://doi.org/10.2147/ott.S243250 (2020).
Wang, H. et al. Cell death affecting the progression of gastric cancer. Cell. Death Discov. 8(1), 377. https://doi.org/10.1038/s41420-022-01161-8 (2022).
Liu, P. et al. NOD-like receptor signaling in inflammation-associated cancers: From functions to targeted therapies. Phytomedicine 64, 152925. https://doi.org/10.1016/j.phymed.2019.152925 (2019).
Karabulut, M., Usul Afsar, C., Serimez, M. & Karabulut, S. Serum IL-17 levels can be diagnostic for gastric cancer. J. Buon. 24(4), 1601–1609 (2019).
Rinkenbaugh, A. L. & Baldwin, A. S. The NF-κB pathway and Cancer stem cells. Cells 5(2). https://doi.org/10.3390/cells5020016 (2016).
Deng, W. et al. LncRNA-ANRIL promotes gastric cancer progression by enhancing NF-kB signaling. Exp. Biol. Med. (Maywood). 244(12), 953–959. https://doi.org/10.1177/1535370219860207 (2019).
Song, L. et al. Abnormally expressed Ferroptosis-Associated FANCD2 in mediating the Temozolomide resistance and immune response in glioblastoma. Front. Pharmacol. 13, 921963. https://doi.org/10.3389/fphar.2022.921963 (2022).
Liu, N. et al. DDIAS promotes invasion and proliferation of non-small cell lung cancer and predicts poor survival of lung cancer patients. Int. J. Clin. Exp. Pathol. 10(12), 11506–11515 (2017).
Zhang, J. et al. Progression of the role of CRYAB in signaling pathways and cancers. Onco Targets Ther. 12, 4129–4139. https://doi.org/10.2147/ott.S201799 (2019).
Bond, J. et al. Novel intergenically spliced chimera, NFATC3-PLA2G15, is associated with aggressive T-ALL biology and outcome. Mol. Cancer Res. 16(3), 470–475. https://doi.org/10.1158/1541-7786.Mcr-17-0442 (2018).
Zhang, Z. et al. PDK4 constitutes a novel prognostic biomarker and therapeutic target in gastric Cancer. Diagnostics (Basel). 12(5). https://doi.org/10.3390/diagnostics12051101 (2022).
Chen, S. et al. SERPINE1 overexpression promotes malignant progression and poor prognosis of gastric Cancer. J. Oncol. 2022, 2647825. https://doi.org/10.1155/2022/2647825 (2022).
Ampudia-Mesias, E. et al. SRPX emerges as a potential tumor marker in the extracellular vesicles of glioblastoma. Cancers (Basel). 14(8). https://doi.org/10.3390/cancers14081984 (2022).
Wang, Y. C. et al. miR-1273 h-5p suppresses CXCL12 expression and inhibits gastric cancer cell invasion and metastasis. Open. Med. (Wars). 17(1), 930–946. https://doi.org/10.1515/med-2022-0486 (2022).
Wang, Q. et al. APAF1-Binding long noncoding RNA promotes tumor growth and multidrug resistance in gastric Cancer by blocking apoptosome assembly. Adv. Sci. (Weinh). 9(28), e2201889. https://doi.org/10.1002/advs.202201889 (2022).
Liu, C., Sun, W., Yang, K. & Xia, B. Knockdown of TRIM65 suppressed the proliferation and invasiveness of gastric cancer cells by restricting the ubiquitin degradation of PPM1A. Exp. Cell. Res. 416(2), 113154. https://doi.org/10.1016/j.yexcr.2022.113154 (2022).
Wen, J. et al. TPP1 inhibits DNA damage response and chemosensitivity in esophageal Cancer. Crit. Rev. Eukaryot. Gene Expr. 33(8), 77–91. https://doi.org/10.1615/CritRevEukaryotGeneExpr.2023048720 (2023).
Zhang, Y., Li, W. & Zhou, Y. Identification of hub genes in diabetic kidney disease via multiple-microarray analysis. Ann. Transl Med. 8(16), 997. https://doi.org/10.21037/atm-20-5171 (2020).
Bai, L. et al. Circ_0073228 serves as a competitive endogenous ribonucleic acid to facilitate proliferation and inhibit apoptosis of hepatocellular carcinoma cells by the miR-139-5p/deoxyribonuclease II axis. J. Gene. Med. 25(8), e3507. https://doi.org/10.1002/jgm.3507 (2023).
Gotovdorj, T. et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induced cell-specific drug transporters with acquired cisplatin resistance in cisplatin sensitive cancer cells. J. Korean Med. Sci. 29(9), 1188–1198. https://doi.org/10.3346/jkms.2014.29.9.1188 (2014).
Cheng, H., Sharen, G., Wang, Z. & Zhou, J. LncRNA UCA1 enhances cisplatin resistance by regulating CYP1B1-mediated apoptosis via miR-513a-3p in human gastric Cancer. Cancer Manag Res. 13, 367–377. https://doi.org/10.2147/cmar.S277399 (2021).
Liang, A. et al. High expression of PYCARD is an independent predictor of unfavorable prognosis and chemotherapy resistance in glioma. Ann. Transl Med. 9(12), 986. https://doi.org/10.21037/atm-21-2346 (2021).
Zou, Y. et al. The role of circular RNA CDR1as/ciRS-7 in regulating tumor microenvironment: A Pan-Cancer analysis. Biomolecules 9(9). https://doi.org/10.3390/biom9090429 (2019).
Liu, X. et al. The prognostic landscape of tumor-infiltrating immune cell and immunomodulators in lung cancer. Biomed. Pharmacother. 95, 55–61. https://doi.org/10.1016/j.biopha.2017.08.003 (2017).
Zou, Y. et al. Discordance of immunotherapy response predictive biomarkers between primary lesions and paired metastases in tumours: A systematic review and meta-analysis. EBioMedicine 63, 103137. https://doi.org/10.1016/j.ebiom.2020.103137 (2021).
Pan, H. et al. Immunological analyses reveal an immune subtype of uveal melanoma with a poor prognosis. Aging (Albany NY). 12(2), 1446–1464. https://doi.org/10.18632/aging.102693 (2020).
Kim, W. et al. PD-1 signaling promotes Tumor-Infiltrating Myeloid-Derived suppressor cells and gastric tumorigenesis in mice. Gastroenterology 160(3), 781–796. https://doi.org/10.1053/j.gastro.2020.10.036 (2021).
Cha, J. H., Chan, L. C., Li, C. W., Hsu, J. L. & Hung, M. C. Mechanisms controlling PD-L1 expression in Cancer. Mol. Cell. 76(3), 359–370. https://doi.org/10.1016/j.molcel.2019.09.030 (2019).
Zhai, L. et al. IDO1 in cancer: A gemini of immune checkpoints. Cell. Mol. Immunol. 15(5), 447–457. https://doi.org/10.1038/cmi.2017.143 (2018).
Xiang, Z. et al. A positive feedback between IDO1 metabolite and COL12A1 via MAPK pathway to promote gastric cancer metastasis. J. Exp. Clin. Cancer Res. 38(1), 314. https://doi.org/10.1186/s13046-019-1318-5 (2019).
Acknowledgements
This research was supported by Key Project of Scientifc Research from Jiangsu Commission of Health (Grant numbers: ZDB2020026); Wuxi Taihu Lake Talent Plan Team in Medical and Health Profession,2021; Wuxi Medical Key Discipline Construction Project, Medical Development Discipline; Wuxi Science and Technology Development Fund from Wuxi Science and Technology Bureau (Grant numbers: N20201002); Project of Wuxi science and Technology Development Fund Y20212004; Natural Science Foundation of Jiangsu Province Youth Fund (Grant numbers: BK20210065); Youth Fund Project from Wuxi Commission of Health (Grant numbers: Q2025).
Author information
Authors and Affiliations
Contributions
Jiazeng Xia, Huiheng Qu, and Peng Zhou conceived and designed the study, analyzed and interpreted the data and results, performed experiments, and wrote the manuscript. Huiheng Qu and Peng Zhou prepared Figs. 1, 2, 3, 4, 5, 6, 7, 8 and 9. Jiazeng Xia, Qian Yang and Yigang Chen supervised and guided the study design. Huiheng Qu, Zhihui Yang, Kaiyuan Deng, and Hao Wang performed most of the experiments and conducted the statistical analyses. Huiheng Qu, Peng Zhou, Zhihui Yang, and Hao Wang contributed equally to this work. Nan Wang and Yuyang Li collected the literature and revised the manuscript. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qu, H., Zhou, P., Yang, Z. et al. Comprehensive analysis of prognosis and drug sensitivity of programmed cell death pattern-related genes in gastric cancer patients. Sci Rep 15, 22535 (2025). https://doi.org/10.1038/s41598-025-06424-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-06424-9
