
Abstract
In the modern age of drug discovery sulfanilamide derivatives are known to have great anti-cancerous potential, the current study aimed to synthesize these derivatives in order to evaluate their biological properties against carbonic anhydrase II (CA-II) and Dickkopf − 1(Dkk1) protein which are highly expressed in many cancers including lung cancer. A series of 10 sulfanilamide derivatives was synthesized under controlled conditions using reflux condensation method. Among all the synthesized derivatives (5a-5j), the compound 5d was found to possess highest antioxidant activity (90.7397 ± 0.0732 µg/mL) comparable to vitamin C (95.1571 ± 0.057 µg/mL) and also exhibited maximum inhibition against CA-II with an IC50 value of 0.00690 ± 0.1119 µM, indicating that 5d is significantly more potent as compared to standard i.e., acetazolamide IC50 = 0.9979 ± 0.0024 µM. Keeping in view the importance of Dkk1 protein in cancer progression, the molecular docking investigations were performed, where compound 5d was proved to be the potential dual inhibitor of CA-II as well as Dkk1 with the binding energy of 8.9 and 9.7 kcal/mol, respectively. In addition to this DNA binding studies also confirmed the significance of compound 5d where it had maximum binding constant value of 6.7 × 104 mol− 1, supporting the other biological investigations and was in agreement with the reported values. All the experimental and computational results reveals the excellent potential of 5d as a candidate medicine in future. Conclusively, the current study may lead to the new therapeutic strategies for the treatment of cancer associated with the aberrant expression of CA-II and less explored DDK1 target.
Similar content being viewed by others
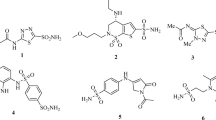
Introduction
Cancer is a major global health problem and is the second leading cause of human mortality after cardiovascular diseases1,2. In the present time the development of more safer and efficacious chemotherapeutic agents to treat such a dreadful disease is extremely needed3. The inhibition of carbonic anhydrase activity is one of the highly explored area in anticancer drug discovery. The human carbonic anhydrase (CA) family involves 15 members which can be divided according to their cellular location (intracellular or extracellular) or enzymatic activity4. Five active family members are located in the cytosol (CA I–III, VII, and XIII), four are membrane-associated (CA IV, IX, XII, and XIV), two are mitochondrial (CA VA and VB), and one is a secretory form (CA VI). In addition, there are three acatalytic forms, which are called CA-related proteins (CARPs)5,6,7. As the members of the CA isozyme family the CAPRs contribute to a number of important physiological functions, it is not surprising that their insufficient or excessive expression has been implicated in several diseases8. The functional involvement of CAs in cancer has become evident after the identification of two membrane-bound isoforms, CA IX and XII, which are overexpressed in several types of cancer9. These two CAs have become attractive research topics, given the hope that an understanding of their roles in tumor physiology could lead to the development of novel strategies for cancer detection, prognosis, and treatment10. Only few studies have been conducted on the CA II enzyme in tumors, Carbonic anhydrase isozyme II (CA II) is a cytosolic enzyme that is highly expressed in most organs, including the brain, where it is mainly located in the oligodendrocytes. Recent studies have shown that its expression is induced in the endothelium of neo-vessels in melanoma and esophageal, renal, and lung cancer11. Immunological studies further indicate that CA II represents a major target antigen stimulating an autoantibody response in melanoma patients. Thus CA II emerged as potential anticancer drug target.
Another Family, the dickkopf family encodes four main secreted glycoproteins of 255–350 amino acids: DKK1, DKK2, DKK3, and DKK412. DKK1, DKK2, and DKK4 can regulate canonical Wnt signaling pathway by binding to the same receptors due to their structural similarity13. Dkk1 is known to be involved and over-expressed in a variety of cancer through Wnt pathway. High serum level of Dkk1 has been found as biomarker in laryngeal squamous cell carcinoma, prostate cancer, cholangiocarcinoma, lung cancer, pancreatic cancer and gastric cancer14. DKK1 is a protein of 266 amino acids composed of two conservative cysteine-rich domains that are separated by a sequence possessing unknown functions and variable length. The C-terminal domain of DKK1 (DKK1c) has been identified as a necessary and sufficient domain for Wnt pathway inhibition, as well as being responsible for binding to LRP6. The function of the N-terminal domain may be associated with enhanced invasive activity and an anti-apoptotic signaling pathway that could be neutralized by the anti-DKK1 antibody. Owing to the over-expression of DKK1 and for the treatment of DKK1 related cancers at the protein and gene levels many studies to develop novel DKK1 inhibitors have been carried out in recent years, and several anti-DKK1 antibodies have been developed and tested in clinical trials15. However, there is still scarcity of potent anti-DKK1 drugs. The role of CA-II enzyme in tumor angiogenesis, endothelial tumors and particularly in lung cancer is coherent with DKK1. Therefore in an attempt to develop potent anti-cancer drugs, targeting lung cancer effectively and keeping in view the importance of both targets in cancer, it was speculated to synthesize the heterocyclic compound and to screen them against both CA-II and Dkk1 via in-vitro and in-silico investigations to assess the anticancer profile of the synthesized compounds.
Among various heterocyclic compounds, the sulfonamide class has remained one of the first available antibiotics for human use, but due to widespread resistance in common bacterial pathogens after decades of extensive use, applications for this class of agents are now focused in a few areas of remaining effectiveness16,17,18. However, for pathogens that remain susceptible to sulfonamides, either used alone or in combination with trimethoprim, these agents have a long history of efficacy with reasonable safety. Currently available sulfonamide agents are synthetic derivatives of the firstly described compound i.e., sulfanilamide. Sulfanilamide based derivatives are considered very important in medicinal chemistry because of their diverse applications. These derivatives are actually pharmacological agents, which are well known for their antitumor, antibacterial, anticarbonic anhydrase, diuretic, hypoglycemic, proteaseinhibitory activities and antithyroid activities19,20,21,22. Some of important antimicrobial drugs include sulfanilamide itself, sulfamethoxazole, sulfisoxazole, sulfametrole, sulfathiazole, sulfapyridine, sulfadiazine, sulfamerazine, slfamethazine, sulfadimethoxine, sulfamethoxypyridazine, sulfachloropyridazine, sulfalene, sulfaquinoxaline, sulfacetamide sodium, sulfaguanidine and sulfasalazine etc19,23,24,25. Moreover, currently many lead compounds with sulfonamide functionality also are in clinical test for the treatment of varied medical conditions.
Pyrimidine scaffold are widely used in drug devolpment due to their diverse biological activities26. For these reasons, the improvement of an efficient process for the synthesis of sulfonamides has continually been inside the recognition of studies in organic synthesis. Various sulfonamide drugs are being used for the treatment of cancer upon FDA approval e.g. Belinostat, Dabrafenib and Pazopanib etc18,27,28. Their chemical structures are given in (Fig. 1).

The development of dual inhibitors targeting DKK1 and CA II represents a promising strategy for cancer therapy. This study is going to be the first study to explore the dual inhibition of Dickkopf-1 (DKK1) and Carbonic Anhydrase II (CA II) enzymes. Based upon the previous studies, here we are interested in integrating benzenesulfonamide and the versatile pharmacological profile of thiazolo[4,5-d]pyrimidines which offered a promising lead structure for dual inhibition of CA-II and Dkk1, with potential implications in treating cancer, osteoporosis, and inflammation. Conclusively, the study is focused by combining synthetic chemistry, computational modeling, and biological evaluation. All the compounds exhibited promising results suggesting that these compounds can potentially offer an enhanced efficacy and reduced side effects compared to single-target drugs.
Experimental
Methods and materials
All the solvents used in this study were dried and then distilled according to standard procedures prior to use. All reagents were purchased from Sigma Aldrich and used without further purification. Melting point of all the compounds were determined on a Stuart SMP3 melting point apparatus. The NMR spectra were recorded on a Bruker 300 (1H-NMR at 300 MHz and for 13CNMR at 75.5 MHz) in DMSO-d6 solvent and chemical shifts are reported in ppm. Tetramethylsilane (TMS) or the residual solvent resonance was used as an internal reference standard. The electronic absorption spectra of synthesized compounds were recorded using a Shimadzu 1800 UV–Vis spectrophotometer with a pair of quartz cells of 1 cm path length. The solution of all the compound (8 × 10− 6 M) were prepared in distilled ethanol, and same solvent was used as a reference. The spectrum of all the compounds were at 200–800 nm. The progress of all the reactions was monitored by Thin layer chromatography technique using thin aluminum sheets coated with silica gel F254 (Merck), detection by means of UV light at 254 and 360 nm.
Chemistry
1.72 g of sulfanilamide (10 mmol) was treated in THF solvent with 0.92 ml of chloroacetyl chloride (10 mmol) in ice bath to obtain a product 2-chloro-N-(4-sulfamoylphenyl) acetamide. In the 2nd step this freshly prepared dry intermediate (2.48 g, 10 mmol) was treated with potassium thiocyanate (0.97 g, 10 mmol) in dry acetone (30 mL) under reflux conditions for 8 h resulting in the formation of 4-(4-oxo-4,5-dihydrothiazol-2-ylamino)benzene sulfonamide. This intermediate (2.71 g, 10 mmol) was further treated with different aromatic aldehydes and sodium acetate (0.82 g, 10 mmol), (dissolved in glacial acetic acid solvent) in equimolar ratio (10 mmol) to obtain the condensed product (Z)-4-(5-benzylidene-4-oxo-4,5-dihydrothiazol-2-ylamino)benzenesulfonamide. About 10 mmol solution of this condensed product was treated with equimolar amount of thiourea (0.76 g, 10 mmol) and potassium hydroxide (0.56 g, 10 mmol) in absolute ethanol to obtain the final product.
4-((5-thioxo-7-(p-tolyl)-5,6-dihydrothiazolo[4,5-d]pyrimidin-2-yl)amino) benzenesulfonamide (5a)
Yellow solid, Yield; 71%, m.p: 300 °C, Rf =00.54 (chloroform: methanol, 9:1), FTIR υ (cm-1) 3325, 3255, 3125 (NH2, NH), 3081 (C-H Ar), (sp2 C-H), 2981 (CH3), 1673 (C = N), 1479 (Ar-C), 1152(C = S), 1H-NMR (300 MHz, DMSO-d6); δ (ppm) 12.20 (s, 1H, NH), 7.96 (d, 2H, J = 8.1, Hz Ar-H), 7.87 (d, 2H, J = 8.2, Hz Ar-H), 7.66 (d, 2H, J = 8.9, Hz Ar-H), 7.46 (d, 2H, J = 8.8, Hz Ar-H), 7.27 (s, 1H, NH-C = S), 7.23 (s, 2H, NH2), 1.65 (s, 3 H, CH3); 13C-NMR (75 MHz,0DMSOd6); δ (ppm) 181.0 (C = S), 172.3 (C-S), 166.1 (C = N), 152.1 (C-N), 142.0, 134.5, 133.2, 131.9, 128.9, 128.0, 127.6, 124.2 (ArC), 121.5 (C = C), 18.4 (CH3) Anal.Calcd. for C18H15N5O2S3: C, 50.33; H, 3.52; N, 16.30; S, 22.39; found: C, 50.34; H, 3.63; N, 16.43. S, 22.36; HRMS (ESI): m/z Calcd for [C18H15N5O2S3 + H]+ 429.0388, Found 429.0390.
4-((7-(2,4-dichlorophenyl)-5-thioxo-5,6-dihydrothiazolo[4,5-d]pyrimidin-2-yl)amino) benzenesulfonamide (5b)
Yellow solid, Yield; 66%, m.p: >300 °C, Rf = 0.55 (chloroform: methanol, 9:1), FTIR υ (cm-1) 3251, 3216 (NH2, NH), 3055 (Ar-Hs), 1696 (C = N), 1546 (Ar-C), 1241 (C = S), 1162 (C-N), 1102 (C-S), 756 (C-Cl); 1H-NMR (300 MHz, DMSO-d6); δ0 (ppm) 12.40 (s, 1H), 7.70–6.84 (ArH), 7.29 (s, 2 H, NH2), 7.24 (s, 1H, NH-C = S); 13C-NMR (75 MHz, DMSO-d6); δ (ppm) 184.69 (C = S), 182.1, 172.8 (C-S), 168.9 (C = N), 153.1 (C-N), 144.6, 136.4, 133.7, 131.9, 129.4, 128.2, 127.5, 125.0 (ArC), 123.9(C = C), Anal.Calcd. for C17H11Cl2N5O2S3: C, 42.15; H, 2.29; N, 14.46; S, 19.86; found: C, 42.34; H, 3.63; N, 14.56. S, 19.84 HRMS (ESI): m/z Calcd for [C17H11Cl2N5O2S3 + H] + 482.9452, Found 482.9454.
4-((7-(4-(dimethylamino)phenyl)-5-thioxo-5,6–dihydrothiazolo[4,5-d]pyrimidin-2-yl)amino) benzenesulfonamide (5c)
Yellow solid, Yield; 71%, m.p: >300 °C, Rf =00.42 (chloroform: methanol, 9:1), FTIR υ (cm-1)) 3251, 3208, (NH2, NH), 3031(Ar-Hs), 1694 (C = N), 1545 (Ar-C), 1376 (CH3), 1230 (C = S), 1162 (C-N), 1064 (C-S); 1H-NMR (300 MHz, DMSO-d6); δ (ppm) 12.39 (s, 1H), 7.71–6.63 (ArH), 7.29 (s, 2 H, NH2), 7.24 (s, 1H, NH-C = S), 2.94 (s, 6 H, N-CH3); 13C-NMR (75 MHz, DMSO-d6); δ (ppm) 181.18(C = S), 172.90 (C-S), 163.94 (C = N), 152.90 (C-N), 145.82, 140.77, 137.57, 128.27, 128.16, 125.01, 122.24, 119.27 (ArC), 115.4 (C = C), Anal.Calcd. for C19H18N6O2S3: C, 49.76; H, 3.96; N, 18.33; S, 20.98 found: C, 49.34; H, 3.68; N, 18.86. S, 20.96 HRMS (ESI): m/z Calcd for [C19H18N6O2S3 + H] + 458.0653, Found 458.0655.
4-((7-(9-ethyl-9 H-carbazol-3-yl)-5-thioxo-5,6-dihydrothiazolo[4,5-d]pyrimidin-2-yl)amino) benzenesulfonamide (5d)
Yellowsolid, Yield; 66%, m.p: >300 °C, Rf =00.50 (chloroform: methanol, 9:1), FTIR υ (cm-1) 3449, 3256, 3171 (NH2, NH), 3049 (sp2 C-H Ar-H) 2956 (sp3 C-H ), 1627 (C = N), 1583 (Ar-C), 1226 (C = S), ,1H-NMR (300 MHz, DMSO-d6); δ(ppm) 12.46 (s, 1H, NH), 10.47 (s, 1H, NH-C = S), 8.45–7.24 (m 11 H, ArH), 7.38 (s, 2H, NH2), 4.36 (q, 2H, N-CH2), 1.46 (t, J = 6.3 Hz, 3H, CH3); 13C-NMR (75 MHz, DMSO-d6); δ (ppm) 183.6 (C = S), 175.8, 171.8 (C-S), 151.4 (C = N), 140.6 (C-N), 140.4, 142.72, 132.2, 127.7, 127.0 ,124.4, 124.1, 123.2, 122.3, 121.1, 120.6, 120.1, 118.9, 110.5, 110.1, (Ar-C), 120.1(C = C), 37.1 (CH2), 14.1(CH3), Anal.Calcd. for C25H20N6O2S3: C, 56.37; H, 3.78; N, 15.78; S, 18.06 found: C, 56.74; H, 3.98; N, 15.46. S, 18.08 HRMS (ESI): m/z Calcd for [C25H20N6O2S3 + H] + 532.0810, Found 532.0812.
4-((7-(4-chlorophenyl)-5-thioxo-5,6-dihydrothiazolo[4,5-d]pyrimidin-2-yl)amino) benzenesulfonamide (5e)
Yellow solid, Yield; 67%, m.p: >300 °C, Rf =0.59 (chloroform: methanol, 9:1), FTIR υ (cm-1) 3188, 3051 (NH2, NH), 1691 (C = N), 1508 (Ar-C), 1292 (C = S), 1177 (C-N), 1050 (C-S), 777 (C-Cl); 1H-NMR(300 MHz, 0DMSO-d6); δ (ppm) 12.6 (s, 1H), 7.85–7.20 (ArH), 7.54 (s, 2 H, NH2), 7.39 (s, 1H, NH-C = S); 13C-NMR (75 MHz, 0DMSO-d6); δ (ppm)180.6 (C = S), 171.7 (C-S), 167.7(C = N), 151.1 (C-N), 141.4, 135.0, 132.54, 130.4, 129.8, 129.1, 127.7, 123.8 (ArC), 120.9(C = C), Anal.Calcd. for C17H12ClN5O2S3: C, 45.38; H, 2.69; N, 15.56; S, 21.38 found: C, 45.84; H, 2.58; N, 15.78. S, 21.35 HRMS (ESI): m/z Calcd for [C17H12ClN5O2S3 + H] + 448.9842, Found 448.9845.
4-((7-(4-hydroxy-3-methoxyphenyl)-5-thioxo-5,6-dihydrothiazolo[4,5-d]pyrimidin-2-yl)amino)benzenesulfonamide (5f)
Yellow solid, Yield; 73%, m.p: >300 °C, Rf =0.45 (chloroform: methanol, 9:1), FTIR υ (cm-1) 3401, 3333, 3286 (NH2, NH), 3044 (sp2 C-H Ar-H) 2944 (sp3 C-H ), 1673 (C = N), 1597 (Ar-C), 1151 (C = S), 1H-NMR (300 MHz, DMSO-d6); δ (ppm) 12.20 (s, 1H, NH), 9.59 (s, 1H, OH), 8.33 (d, 2H, J = 7.8, Hz Ar-H), 7.84 (d, 2H, J = 7.8, Hz Ar-H), 8.49 (s, 2H, NH2), 7.35–7.17 (m, 3H, ArH), 7.15 (s, 1H, NH-C = S), 6.85 (s, 1H, NH), 3.82 (s, 3H, OCH3); 13C-NMR (75 MHz, DMSO-d6); δ (ppm) 168.6 (C = S), 158.7(C = N), 148.4 (C-S), 146.4 (C-N), 141.6, 140.3, 137.6, 127.6, 125.5, 123.9, 122.0, 121.0, 120.2, 119.6 (Ar-C), 114.3 (C = C), 65.5 (OCH3) Anal.Calcd. for C19H19N5O4S3: C, 47.78; H, 4.01; N, 14.66; S, 20.14 found: C, 47.75; H, 4.04; N, 14.68; S, 20.12 HRMS (ESI): m/z Calcd for [C19H19N5O4S3 + H] + 477.0599, Found 477.0601.
4-((7-(3-hydroxyphenyl)-5-thioxo-5,6-dihydrothiazolo[4,5-d]pyrimidin-2-yl)amino)benzenesulfonamide (5 g)
Yellow solid, Yield; 80.22%, m.p: >300 °C, Rf =0.62 (chloroform: methanol, 9:1), FTIR υ (cm-1) 3326, 3248, 3128 (NH2, NH, OH), 1671 (C = N), 1588 (Ar-C), 1404 (C = S), 1154 (C-S), 1H-NMR (300 MHz, DMSO-d6), δ (ppm)12.57 (s, 1H, NH) 7.94 (d, 2 H, J = 8.1, Hz Ar-H), 7.92 (s, 1H, OH), 7.86 (m 4 H, ArH), ), 7.53 (s, 2 H, NH2), 7.38 (s, 1H, NH-C = S), 7.21 (d, 2 H, J = 8.1, Hz Ar-H), 13C-NMR (75 MHz, DMSO-d6), δ (ppm) 181.0 (C = S), 172.3 (C-S), 166.1 (C = N), 152.1 (C-N),142.0, 134.5, 132.2, 131.9, 128.9, 128.0, 127.6, 124.2, (Ar-C), 121.5, (C = C), Anal.Calcd. for C17H13N5O3S3: C, 47.32; H, 3.04; N, 16.23; S, 22.29 found: C, 47.54; H, 3.24; N, 16.45. S, 22.26 HRMS (ESI): m/z Calcd for [C17H13N5O3S3 + H] + 431.0181, Found 431.0883.
4-((7-(2-bromophenyl)-5-thioxo-5,6-dihydrothiazolo[4,5-d]pyrimidin-2-yl)amino) benzenesulfonamide (5 h)
Yellow solid, Yield; 74%, m.p: >300 °C, Rf =0.49 (chloroform: methanol, 9:1), FTIR υ (cm-1) 3553, 3253, 3211 (OH NH2, NH), 1698 (C = N), 1453 (Ar-C), 1244 (C = S), 1153 (C-N), 1022 (C-S), 756 (C-Br); 1H-NMR (300 MHz, DMSO-d6); δ (ppm) 12.42 (s, 1H, NH), 7.29 (s, 2 H, NH2), 7.31–6.85 (ArH), 7.09 (s, 1H, NH-C = S); 13C-NMR (75 MHz, DMSO-d6); δ (ppm) 184.7 (C = S), 172.6 (C-S), 169.9(C = N), 153.1(C-N), 148.2, 140.8, 137.9, 137.6, 132.3, 131.4, 129.5, 128.2, 128.0, 125.0 (ArC), 122.9(C = C), Anal.Calcd. for C17H12BrN5O2S3; C, 41.30; H, 2.45; N, 14.17; S, 19.46 found: C, 42.34; H, 2.42; N, 14.65. S, 19.43 HRMS (ESI): m/z Calcd for [C17H12BrN5O2S3 + H] + 492.9336, Found 492.9338.
4-((7-(4-hydroxyphenyl)-5-thioxo-5,6-dihydrothiazolo[4,5-d]pyrimidin-2-yl)amino) benzenesulfonamide (5i)
Yellow solid, Yield; 65%, m.p: >300 °C, Rf =00.54 (chloroform: methanol, 9:1), FTIR υ (cm-1) 3450, 3150, 3050 (NH2, NH, OH), 1685 (C = N), 1445 (Ar-C), 1276 (C = S), 1129 (C-N), 1016 (C-S); 1H-NMR (300 MHz, 0DMSO-d6); δ (ppm) 12.51 (s, 1H, NH), 7.71 (d, J = 7.5 Hz, 2ArH), 7.29 (s, 2 H, NH2), 7.27 (d, J = 7.5 Hz, 2ArH), 7.19 (s, 1H, NH-C = S), 6.85 (d, J = 7.5 Hz, 2ArH), 6.80 (s, 1H, OH), 6.77 (d, J = 7.5 Hz, 2ArH); 13C-NMR (75 MHz, DMSO-d6); δ (ppm) 184.7 (C = S), 163.9 (C-S), 159.6 (C = N), 154.1 (C-N), 159.82, 145.82, 140.77, 137.57, 129.85, 128.27, 126.15, 125.01 (ArC), 119.2 (C = C), Anal.Calcd. for C17H13N5O3S3; C, 47.32; H, 3.04; N, 16.23; S, 22.29 found: C, 47.36; H, 3.52; N, 16.75. S, 22.26. HRMS (ESI): m/z Calcd for [C17H13N5O3S3 + H] + 431.0181, Found 431.0184.
4-((7-(2-fluorophenyl)-5-thioxo-5,6-dihydrothiazolo[4,5-d]pyrimidin-2-yl)amino) benzenesulfonamide (5j)
Yellow solid, Yield; 69%, m.p: >300 °C, Rf =0.44 (chloroform: methanol, 9:1), FTIR υ (cm-1) 3250, 3210, (NH2, NH), 1597 (C = N), 1453 (Ar-C), 1243 (C = S), 1153 (C-N), 1022 (C-S), 866 (C-F); 1H-NMR (300 MHz, DMSO-d6); δ (ppm) 12.61 (s, 1H, NH), 7.71 (d, J = 7.5 Hz, 2ArH), 7.54 (s, 2 H, NH2), 7.39–6.93 (ArH), 7.12 (s, 1H, NH-C = S); 13C-NMR (75 MHz, DMSO-d6); δ (ppm) 184.69 (C = S), 173.9 (C-S), 169.9 (C = N), 153.8 (C-N), 161.87 (d, J = 246 Hz, 163.51, 160.23 CF aromatic), 149.8, 140.7, 137.6, 134.2, 130.0, 129.9, 128.3, 126.2, 125.01 (Ar-C), 124.1 (C = C), Anal.Calcd. for C17H12FN5O2S3; C, 47.10; H, 2.79; N, 16.16; S, 22.19 found: C, 47.45; H, 2.83; N, 16.41 S, 22.16. HRMS (ESI): m/z Calcd for [C17H12FN5O2S3; +H] + 433.0137, Found 433.01399.
Biological evaluation
Anti-oxidant activity: free radical scavenging activity
The Radical scavenging activity of all the derivatives was determined by an already reported method after modification29,30 by using 2, 2-diphenyl-1 picrylhydrazyl (DPPH) assay. The total assay volume was 200 µL which consisted of 100 µL of DPPH (150 µM) and 20 µL of test compounds (100 µM), rest of the volume was adjusted to 200 µL in each well with Methanol. The reaction mixture was then incubated for 30 min at room temperature. Ascorbic acid was used as a reference inhibitor. The assay measurements were carried out by using standard microplate reader (OPTI Max, Tunable) at 517 nm. The reaction rates were compared and the percent inhibition of tested inhibitors was calculated. Each concentration test was analyzed in three independent experiments run in triplicate.
Carbonic anhydrase II Inhibition assay
Carbonic anhydrase II inhibition was measured as described previously with some modifications31,32.The method was based on the principle that p-nitrophenyl acetate is hydrolyzed by Carbonic anhydrase to form yellow colored p-nitrophenol which was measured spectrophotometrically. Briefly, Reaction mixture contained 120 µL of 50 mM Tris-sulfate buffer (pH 7.6 containing 0.1 mM ZnCl2), 20 µL of inhibitor (100 µM) and 20 µL (50 U) bovine enzyme per well. Contents were well mixed and pre-incubated at 25 °C for 10 min. Then substrate p-nitrophenyl acetate was prepared (6 mM stock using < 5% acetonitrile in buffer and used fresh every time) and 40 µL was added per well to achieve 0.6 mM concentration per well. Total reaction volume was made to 200 µL. After 30 min incubation at 25 °C all the content was mixed and absorbance was measured at 348 nm using a microplate reader. Acetazolamide was used as a reference inhibitor. Each concentration was analyzed in three independent experiments. IC50 values were calculated by nonlinear regression using GraphPad Prism 5.0.
Here, the B and S are the absorbance values for the blank and sample.
DNA binding studies
For DNA binding study; Commercial Solman DNA was taken and dissolved in double distilled H2O to make a stock solution of 2.6 × 10− 4 M. Working solutions of different DNA concentration were prepared from this stock solution, as discussed in previous work33,34. Concentration of the stock solution was determined by UV absorbance at 260 nm having Ɛ value 6600 M-1cm-1. DNA was free of protein because A260/A280 was ~ 1. For DNA binding study working solutions were prepared by reported method35. UV-visible spectroscopic data was obtained on Shimadzu 1800 spectrophotometer. Quartz cells with 1 cm path length were used for running samples. First, in the absence of DNA spectra, pure compound solution was recorded at 200–800 nm and then equal amount of different DNA concentrations were added to both sample and reference cells in order to avoid its interference.
Computational studies
All the synthesized sulfonamide derivatives were analyzed using well-designed computational study. The DFT calculations were performed with Gaussian and gauss-view software36,37, molecular docking studies were performed using autodock Vina38 with carbonic anhydrase II, Dickkopf 1(Dkk1) and Low-density lipoprotein receptor-related protein 6 (LRP6). In addition to this, protein-protein docking studies39,40 of Dkk1 and LRP6 proteins and MD simulation studies41 were also performed.
Density functional theory
DFT is the well-known mechanical modelling technique that offers a unique way to study the complex electronic structure e.g. protein-ligand complexes, solids, and nano-materials, with a good balance between accuracy and computational cost. DFT can be used to understand how atoms are bonded with each other and predict how molecules or materials will react making it valuable tool in modern drug discovery42.
All the DFT calculations were performed employing B3LYP/6-31G and 3-21G method in the Gaussian 9 software and analyzed with gauss-view 6 software35,36.
Computational methodology for molecular Docking
Retrieval of carbonic anhydrase II and Dkk1 protein from RCSB data bank
The three dimensional (3D) crystal structure of both proteins i.e. carbonic anhydrase II and DKK1-LRP6 protein complex was accessed form the Protein Data Bank (PDB) with PDB ID 1V9E and 3S2K, respectively43,44. Energy minimization of both targeted structures was carried out by using conjugate gradient algorithm and Amber force field in UCSF Chimera 1.10.145. The stereo-chemical properties, Ramachandran graph and values of Carbonic anhydrase II structure were assessed by Molprobity server46,47, while the hydrophobicity graph was generated by Discovery Studio 4.1 Client48.
Molecular Docking
In order to execute molecular docking all the synthesized ligands (5a-5j) were sketched in Chemdraw 12.0.2 and their energy was minimized by using Open babel tools49,50. Then all the proteins were prepared for docking by adding Gasteiger charges and necessary kollman charges using AutoDock software. Molecular docking experiment was employed on all the synthesized ligands with AutoDock Vina software. The grid box center values were adjusted as to include all the residues of active pocket. The size of grid box was adjusted big enough to include all the amino acid residues of active pocket al.lowing ligand to move freely in the search space. The graphical depictions of all the docking complexes were carried out using Pymol Software51.
Protein-protein Docking
The crystal structure of 3s2k protein downloaded from RCSB data bank is a protein-protein complex of DKK1 with LRP6 protein attached through CDR1 domain of Dkk1. The binding residues of both protein were analyzed using online server i.e. PDBsum. In order to confirm the binding site of Dkk1 with receptor LRP6 both proteins were separated and then redocked using online CLUSPRO server with the default parameters and 70,000 rotations. The results were analyzed using PDBsum platform52,53.
Molecular dynamic simulations
Molecular dynamic simulation is a modern age computer technique used to determine the stability of the protein-ligand complex under simulated conditions41. All the MD calculations were carried out using Nano Scale Molecular Dynamics (NAMD) software on a CUDA-accelerated GPU system. In this study MD simulation was performed with the best docked structures of LRP6-5d and dkk1-5d complex. The CHARMM36 forcefield with TIP3P water model was used for the simulating system. In order to neutralize the system 0.15 M NaCl ion were added for the 100 ns simulation. The system was brought to equilibrium at 300 K and 1.01 bar pressure, under isothermal and isobaric (NPT) environment. The other criteria were established as previously mentioned. After 500,000 steps in an NVT ensemble, the system was equilibrated in an NPT ensemble for an additional 500,000 steps54,55. All the simulation results were analyzed using Visual Molecular Dynamics (VMD) software.
Results and discussion
Synthesis of 7-Aryl-5-thioxo-5,6-dihydrothiazolo[4,5-d]pyrimidin-2-yl)amino) benzenesulfonamides
Sulfanilamide derivatives possess many important biological and non-biological applications. In this study a series of ten novel sulfanilamide derivatives was synthesized (5a-5j) under short reaction time and mild conditions that resulted in fine yield.
Initially, sulfanilamide was converted into corresponding acetylated product by treating it with chloroacetylchloride. The freshly prepared acetylated product was then reacted with potassium thiocyanate in dry acetone to obtain a key thiazol-4(5 H)-one based intermediate, which played a pivotal role in the synthesis of desired product. The key intermediate was then condensed with desired substituted aromatic aldehyde. In the last step the condensed product was treated with hydroxylamine hydrochloride (anhydrous) to obtain the thiazole(4,5 d)pyrimidin-2-thione based fused ring heterocyclic system. The crude product obtained was then recrystallized, using ethanol as solvent to get the purified product with excellent yield, as shown in Fig. 2.

Synthesis of 7-Aryl-5-thioxo-5,6-dihydrothiazolo[4,5-d]pyrimidin-2-yl)amino)benzenesulfonamides.
Spectroscopic characterization
The synthesized series of aryl substituted fused ring heterocyclic system was characterized on the basis of spectroscopic data. 1H and 13C NMR were recorded in deuterated DMSO-d6 solvent. All the compounds were characterized by the appearance of their characteristic (C = N) and (C = S) absorption at 1631and 1231 cm− 1 respectively in the IR spectra. In the 1H-NMR spectra, signals for NH, NH2 and NH-C = O appeared at 12.57, 7.53 and 7.38 respectively. In 1H NMR spectra a singlet signal integrating 1H appeared at δ 11–12 ppm. The most de-shielded signal represented the N-H protons of the sulfanilamide ring as this NH group was under the influence of two electron-withdrawing groups i.e. one sulfanilamide moiety and the second was heterocyclic fused ring moiety. The withdrawing groups always shift signals to the higher ppm value which justified the bond between sulfanilamide and thiazole[4,5 c]pyrimidin-2-thione heterocyclic system. The NH2 group was de-shielded due to SO2 (withdrawing group) directly attached to it. Characteristic peak for NH-C = S was appeared as singlet in aromatic region as it was acidic proton, appeared anywhere in spectrum. Secondly it was slightly de-shielded due to heavy atom effect of sulfur atom. The protons of the aromatic rings were observed in the typical region around δ 7.5 ppm. The 13C NMR analysis further confirmed the structure of the title compounds by displaying the characteristic signals for the amide (C = S) carbon at 184 − 180 ppm, which was the most de-shielded signal due to heavy atom effect and the peak for (C-N) of pyrimidin-2-thione ring was observed in the range of 150–155 ppm for synthesized compounds. Almost all the signals for heterocyclic carbons appeared de-shielded due to heavy atom effect due to sulfur atom. The characterization data of allthe compound is given in supplementary file (Fig. S1-Fig.S21). The purity of synthesized compounds was determined through elemental analysis as shown in Table 1.
Biological evaluation
Carbonic anhydrase activity and free radical scavenging activity result
All the synthesized compounds (5a-5j) were evaluated for DPPH free radical scavenging activity and carbonic anhydrase activity using the reported methods53. The compound 5f showed significant activity in comparison to Standard (Vitamin C), while other compounds were not having significant radical scavenging activity even at the higher concentration (100 µg/mL) Table 2.
UV spectroscopy
According to the results of UV spectroscopic studies, all the newly synthesized derivatives had maximum absorption at wavelength ranging 210–422 nm. Two types of transitions were observes in synthesized derivatives (5a-5j) including π-π* and n-π* transitions due to the presence of conjugated double bonds and heteroatoms (with lone pairs) respectively. The absorption spectra toward the high wavelength originated from the n-π* transitions whereas the peaks toward the lower wavelength region was due to π-π* transition.

UV spectrums of DNA Binding for all the derivatives (5a-j).
The compound 5 h showed a higher λmax value as compared to 5e as down the group the size of atom increases, the donating tendency of atoms also increases. The 5 h derivative with 1-ethylindoline substituent absorbs at higher wavelength as compared to 5 h (bromo substitution) and 5e (4-Cl) due to large size of bromine (Table 3), and band Gape plots are given in supplementary file. In the same way 5i (4-OH) absorbs at higher λmax value as compared to 5f (4-OH, 3-OMe) as OH is strong electron donating group, present at para position, which results in its increased conjugation effect and decreased inductive effect, which is true in case of 5i, so it absorbs at higher λmax value. But 5f showed a decreased λmaxvalue as OH group may forms the hydrogen bond with methoxy group. All the results are given in (Fig. 3), and also given in supplementary file (Fig.S22-Fig. S31).
DNA binding results
UV-spectrum of best compound 5d, for DNA binding studies of the synthesized compounds showing absorption maximum at 240 nm, has been demonstrated. These peaks showed hyperchromism after the formation of drug-DNA adduct by injecting 5 different concentrations of the DNA keeping the dilution same. Binding constant was determined by Benesi-Hildebrand equation.
\(\frac{{{A_0}}}{{A - {A_0}}}=\frac{{{\varepsilon _G}}}{{{\varepsilon _{H - G}} - {\varepsilon _G}}}+\frac{{{\varepsilon _G}}}{{{\varepsilon _{H - G}} - {\varepsilon _G}}}\frac{1}{{K\left[ {DNA} \right]}}\)
Where K denotes the binding constant, A₀ is the absorbance of the free compound, A represents Compound-DNA adduct, ƐG and ƐG-H are the absorption co-efficients of the free compound and compound-DNA adduct. The Plot of (A₀/A-A₀) vs. DNA concentration shows a straight line from that slope to intercept ratio, giveing binding constant of 6.7 × 104 mol-1 for 5 h respectively that are in agreement with the reported values56,57,58. These values shows the importance of these derivatives to be evaluated as a candidate drugs in future. The negative value of gibbs free energy proved that the interaction of synthesized derivatives with DNA, is spontaneous (Fig. 4).

Plots of Absorbance vs. Wavelength for 5d. Respective Plots of 5d, Relative absorbance (A₀/A-A₀) vs. [1/DNA] for Calculation of DNA binding.
Density functional theory
DFT calculations helps to predict the reactivity of a molecule by analyzing its structure and energy levels. These energy levels are described by quantum chemical parameters, such as hardness, softness, electronegativity, and electrophilicity etc. Hardness of a chemical compound tells us how well a molecule resists losing electrons i.e. stability, while softness indicates its ability to gain electrons from its immediate vicinity. Electronegativity of a compound quantifies ability of a molecule to attract electrons. Finally, electrophilicity reflects a molecule’s tendency to accept electrons59.
The study found that molecule 5d is the most stable compound with all the parameters in optimum range. This conclusion is based on calculations of softness, hardness, and the energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). Molecules with a small HOMO-LUMO gap are generally more reactive because they are more easily polarized. The study again identified 5d as the most stable and reactive molecule based on these energy levels. The surfaces of these frontier molecular orbitals were also visualized to provide further insight into the molecule’s reactivity. The values of all the parameters are given in the Table 4.
Electrical parameters
Electrical parameters such as dipole moment, polarizability, and optimization energy, have been estimated at the same theoretical level for understanding nonlinear optical activities of all the sulfonamide derivatives using the B3LYP with two distinct basis sets, namely 3-21G and 6-31G. According to the results compound 5c has a highest dipole moment value of 17.36 debye calculated with a basis set 3–21 G and 16.86 computed with 6–31 G. One of the most important factors in predicting the conductivity of a compound is its electric polarizability. Furthermore, polarizability and ligand binding propensity are directly correlated; a highly polarizable ligand may attach to its target more strongly than a weakly polarizable ligand60. In the same way, polarizability of compound 5d has the highest mean value of all the derivatives, indicating a greater possibility for the development of molecular interactions because of a more widely dispersed electronic cloud. The higher estimated polarizability value of a compound could increase its bioactivity. It is observed that 5d compound has best polarizability value of 417.26 and optimum dipole moment values. Most of the values are almost same for both 3-21G and 6-31G basis sets (Table 5). Highest dipole moment and electron affinity was recorded for 5c due to the N,N-dimethyl groups at para position. As the lone pair of nitrogen is in conjugation with benzene ring as a result it occupies a positive charge which is stabilized by methyl group through hyperconjugation which actually creates dipoles in the molecule which results in highest electron affinity of molecule.
A specific pattern was observed in ionization energy of the derivatives like the halogen substituents (at ortho or para) show a trend of decrease in ionization energy with increase in size including compounds 5 h and 5j. 5c has shown the highest ionization energy as in this compounds benzene ring is substituted with two chlorine atoms at ortho and para position which are electronegative in nature. The compounds, in which the benzene ring is substituted with electron donating group, have shown higher ionization energies.
FMOs analysis
Frontier Molecular Orbitals (FMOs) are a common tool used in DFT calculations to predict the various different chemical characteristics. Therefore FMOs are most important and are widely analysed in DFT results. In order to understand the reactivity of a compound basic understanding of its Frontier molecular orbitals especially the Lowest Unoccupied Molecular Orbital (LUMO) and the Highest Occupied Molecular Orbital (HOMO) is required. The orbital where incoming electrons can be accommodated is represented by LUMO, whereas the electrons most likely to engage in bonding are represented by HOMO. The HOMO-LUMO gap, or energy difference between HOMO and LUMO, is frequently associated with the reactivity of the sample. Higher reactivity is often indicated by a smaller HOMO-LUMO gap61. The DFT results indicate that all the derivatives low HOMO-LUMO energy gap ranging from 0.105 to 0.112 eV showing that all the derivatives are nearly equal reactive (Table 6).
The possible movement of electrons during a reaction can be predicted by looking at the shape and locations of HOMO and LUMO orbitals indicating the mechanism of a reaction and reactive sites in the large molecules. FMOs also contribute to the understanding of how light interacts with molecules. HOMO and LUMO are frequently involved in electronic transitions, which occur when electrons change orbitals as a result of absorbing light. We may be able to forecast the wavelengths of light that a molecule would either absorb or emit by examining the FMOs. The HOMO and LUMO orbitals of 5d compound along with ESP map and optimized structure is given in (Fig. 4), and the data of other compounds is given in supplementary file (Fig. S32-Fig.S34).
Energy optimization
Energy optimization is a critical step in DFT calculations since it enables you to determine most stable configuration of a system or molecule62. DFT calculations use the positional arrangement of atoms and electrons to predict the electronic structure and characteristics of a system. A molecule, however, can have several configurations, each with a unique energy state. Energy optimization helps in finding the ground state configuration with the lowest energy. In the energy optimized structure of a compounds all forces acting on the atoms are balanced and the molecule is at its most stable state which is the ground state (Fig. 5).

HOMO and LUMO diagrams of 5d derivative along with Optimized structures and ESP bar.
Electrostatic potential (ESP) bar
DFT analysis can be used to calculate the electrostatic potential surrounding a molecule. The colored surface map is generated, where different colors indicate regions with positive, negative, or neutral electrostatic potential, is a common way to visualize and interpret the data63. ESP defines the potential energy that an electron could encounter due to the distribution of nuclei and electrons at a certain location around a molecule. Electrons are attracted to areas with positive Electrostatic potential is present and are repelled by those with negative Electrostatic potential. The susceptibility of a particular molecular site to electrophilic or nucleophilic attack is indicated by a strong positive or negative ESP region providing an insight into the reactivity of a compound.
In the same way the possible hydrogen bonding sites may be indicated by regions that exhibit strong positive and negative ESP next to one another. Molecular interactions and mutual recognition may be affected by variations in ESP. It can be inferred from the ESP bar that in 5d compound the higher electron density is present around the sulfonamide moiety while the thizole ring is electrically neutral. In the same way blue shade indicates that the pyrimidine ring is the electron deficient area of the compound.
Structure-activity relationship
The structure-activity relationship (SAR) of a drug molecule is the connection that exists between a molecule’s biological property and its chemical structure. The SAR is very significant piece of study and is especially important in the search and development of new drugs. After the analysis on how structural variations impact activity of molecule with biological targets, scientists may create more precise and potent compounds.
In this study a series of ten sulfanilamide based derivatives are designed and their carbonic anhydrase inhibition activity was measured using acetazolamide as a reference drug. The IC50 value of all the derivatives was lower than acetazolamide showing that all the compounds are potent anti-cancer molecules. It can be inferred from the structure and activity results of all the compounds that the activity of this sulfonamide series is significantly varying according to the nature of substituent present at the pyrimidine neighboring ring at the far side of compound. The activity of compounds is increasing by the presence of electron withdrawing group and is decreasing by electron donating groups.
The maximum activity of compound 5d may be attributed to the presence of carbazol ring where all the electrons are resonance stabilized imparting significant stability to the compounds. The activity of compound 5a is lower than all the compounds due the presence of CH3 group which in an electron donating group. In the compound 5b the activity is significantly increased where methyl group is removed and two chlorine atoms are introduced at the ortho and meta position. In the same way activity of 5e, 5 h and 5j is higher due to the presence of electron withdrawing groups i.e. Cl, Br and F in their structures.
The amino group present in compound 5c slightly lowers its activity compared to similar compound with electronegative atoms. The hydroxyl group is an electropositive group present at meta and para position in compound 5 g and 5i affecting the activity in the similar fashion. In the chemical structure of compound 5f two electron donating groups are present i.e. OH and OCH3 and the activity of compound is significantly decreased.
Molecular Docking
Chemo-informatic properties and Lipinski rule (RO5) evaluation of ligands
The designed ligands were analyzed computationally to predict the best ligand on the basis of biological properties and Rule of five. The predicted chemo-informatic properties like Mol Wt (g/mol) HBD, HBA, LogP, polar surface area (PSA), molar volume and drug likeness score of ligand molecules are mentioned in Table 1. It has been confirmed from previous research data that the standard values for Mol Wt ( < = 500 g/mol)65. The ligands predicted results showed that compounds possessed good Mol Wt as compared to standard value. The compounds 5 g showed little higher Mol Wt value (532 g/mol) than the standard value. Research data revealed poor permeation is more likely to be observed when the HBA and HBD are exceeded then 10 and 5 respectively36, . The chemo-informatics analysis justified that all the designed compounds possess < 10 HBA and < 5 HBD. Moreover, their logP value were also comparable with standard value except the 5d which showed higher than 5. All the synthetic compounds fully obeyed the RO5 except the SA7S. However there are plenty of examples are available for RO5 violation amongst the existing drugs66,67.
Furthermore, the polar surface area (PSA) or total polar surface area is also known as important parameter for drug development67. The PSA parameter is commonly used for drug’s optimization ability to permeate cells. Prior research data showed the standard value of PSA (< 89 A2)68. Our predicted results showed that compounds possessed comparable results however, few were with exceeded values than standard values of PSA results. The drug score is a complex of basic molecular parameters are hydrophobicity, electronic distribution, hydrogen bonding characteristics, molecule size and flexibility. Our results showed that all the synthetic compounds (5a-5j) showed good drug score values which depicts its good drug like behavior and may be considered as drug candidate molecules. Whereas, couple of compounds showed negative valued results which may depicts its less drug likeness behavior. The overall predicted results values of all compounds are mentioned in Table 7.
Binding pocket analysis of carbonic anhydrase II
The binding pocket analysis showed that all ligands were confined in the active region of target protein nearby zinc metal. All the synthesized derivatives were separately docked into the active pocket of all the 3 protein including Carbonic Anhydrase, Dkk1 and LRP6 and the binding score of all the docking interactions along with amino acid residues forming hydrogen bonds are given in Table 8.
The docking analysis of compound 5d with CA-II indicate that there are various bonding and non-bonding interactions including 6 conventional hydrogen bonds and 3 pi-sigma bonds. In molecular docking studies the hydrogen bond is considered the strongest of all the bonds. The amino acid residues that are involved in the bonding interactions includes Thr198, Asn66, Val59, Thr197, Ser1 and His2. In addition to this various non-bonding weak interactions are also present including Van der Waals forces. 3D depiction of all the derivatives within the active pocket and binding interactions of 5d are given in (Fig. 6). While the structural assessment of carbonic anhydrase II and Dkk1 Complex is given in supplementary file (Fig.S35).

Binding interactions of Carbonic Anhydrase II with 5d. (A) Active pocket of CA-II with all derivatives (B) 3D interactions with bond length.
Active pocket analysis of Dkk1-LRP6 complex through protein-protein Docking
The crystal structure of protein 3s2k obtained from the RCSB data bank is a complex of two different proteins i.e. Dickkopf 1 and LRP 6. Both protein are attached with each other through the cysteine rich domain of Dkk1 protein. The binding interaction analysis of Dkk1 protein was carried using online serve PDBsum. The result of binding studies shows that chain B (LRP6) is attached with chain C (Dkk1) through 8 hydrogen bonds, 4 salt bridges and 101 non-bonded interactions. The amino acid residues of protein Dkk1 forming hydrogen bonds include Arg236, Phe234, Glu232, His204, Arg224 and Glu185.
In order to confirm the active pocket residues of Dkk1-LRP6 complex and validate the results both the protein were separated and docked with each other via protein-protein docking tool i.e. CLUSPRO. Among the various models generated by CLUSPRO after 70,000 rotations the best docked configuration with lowest energy (-1464.7) was selected for further analysis. The active pocket of docked complex indicates the 90% similarity of amino acid residues with the original RCSB complex (Fig. 7).

The detailed interpretations of Dkk1-LRP6 Complex along with active pocket amino acid residues before and after protein-protein docking.
Detailed binding interactions of 5d-Dkk1 complex
The binding analysis of 5d with Dkk1 reveals that the derivative Interacts within the Active Pocket through Various different bonding and non-bonding interactions. The following amino acid residues i.e. Arg259, Arg224, His261, Lys222, Phe232, Glu234, Arg236 from the active pocket form connections with 5d derivative. These connections are crucial for the stability of molecules. Among all the observed interactions hydrogen bonds are comparatively most stable and strong. The docking results indicate that 5d forms four hydrogen bonds with the 3 different amino acid residues. All the 4 hydrogen bonds are formed by the aminosulfonyl group with Arg259, Arg224 and His261. Two hydrogen bonds are formed by Arg224 with 2 hydrogen atoms, Nitrogen atom forms 1 hydrogen bond with Arg224 and the fourth hydrogen bond is formed by His261 and hydrogen atom. In addition to hydrogen bonds, other interactions were identified including Interactions involving pi-electrons, such as pi-sigma and pi-alkyl bonds, along with van der Waals forces within the active pocket provides a detailed view of these interactions using both 3D and 2D representations (Fig. 8). The binding interactions of all other derivatives with DKK1 has been given in the supplementary file (Fig S36-Fig.S45).

(A) 3D and (B) 2D representation of docking interaction of 5d with Dkk1 protein.
Binding analyses of synthesized compounds against carbonic anhydrase II
The ligands-protein binding analyses showed that 5d confined in the active binding pocket of target protein. The carbonic anhydrase II has an active site cleft (15 Å in diameter and 15 Å deep), and contains a zinc ion which is coordinated in a tetrahedral geometry with three Histidine residues (His94, His96 and His119) and a water molecule/hydroxide ion. The 5d -docked complex reveals the good conformational state with hydrogen bond interactions within the receptor binding pocket. The docking result of 5d-receptor docked complex showed that six hydrogen bonds were observed with Asn66, His93, Thr197, Thr198, and His95 residues, respectively. Moreover, various hydrophobic interactions were also observed between 5d and Gln91 and His93, respectively. The aminosulfonyl moiety of 5d interacts with Thr197 forming 2 hydrogen bonds with bond distance 2.81Å and 3.10Å. The thiazol ring and 2-amino group attached with thiazol ring form two hydrogen bonds against Asn66 having bond length of 3.14, respectively. Similarly, our substituted functional group (CH3) involves in two hydrophobic interactions. The binding interaction pattern within binding pocket and against residues is mentioned in (Fig. 9). Literature reports also favor our docking results which strengthen our ligand binding and its conformational position within active region of target protein69. The binding interactions of all other derivatives with CA-II has been given in the supplementary file (Fig S46-Fig.S55).

Docking interaction of 5d with receptor molecule. A) 3D and 2D interaction of 5d with Carbonic anhydrase II.
Detailed binding interactions of 5d-LRP6 complex
The docking results of 5d-LRP6 complex reveals that there are seven hydrogen bonds formed by the following amino acid residues i.e. Gln740, Ser954, His919, Thr933 and Thr934. In addition to these strong bonding interactions various other non-bonding interactions are also present which includes Pi-Pi interactions and van der Waals forces. The 1st hydrogen bond of 3.24 Å is formed by the sulfur atom of thiazole ring with amino acid Gln740. In the same way two hydrogen bonds of 3.12 Å and 3.18 Å are formed by the nitrogen atom of thiazole ring with two different amino acids i.e. Ser994 and His919. The nitrogen atom of aminosulfonyl group form 2 hydrogen bonds with Thr933 and 2 H-bonds with Thr934. The detailed depiction of 2D and 3D structures of 5d-LRP6 complex is given in the (Fig. 10). The binding interactions of all other derivatives with CA-II has been given in the supplementary file (Fig S56-Fig. 65).

(A) 3D and (B) 2D representation of docking interaction of 5d with LRP6 protein.
MD simulation studies
In this study 2 protein-ligand complexes i.e. 5d-LRP6 and 5d-Dkk1 were simulated in an aqueous environment in order to augment the docking results. The RMSD and RMSF are 2 most reliable parameters to understand the simulation results. The RMSD and RMSF values of a system should be less than 2.0 Å for strong and stable protein-ligand complex70,71. The RMSD value of Protein-ligand complex of LRP6-5d complex indicates that the complex is fairly stable throughout the trajectory period with slight fluctuations, while RMSD value is lower than 2.0 Å. The RMSD graphs of both protein-ligand complex. In the same way simulation results of Dkk1-5d complex shows that protein-ligand complexes are fairly stable and average RMSD value of protein-ligand complexes are up to 2.0 Å (Fig. 11).

RMSD and RMSF graphs of (A) LRP6-5d Complex (B) Dkk1-5d.
RMSF graphs were also plotted in order to further analyze the MD simulation results (Fig. 10). The value of RMSF provides the range of variation in the C and N terminal lobe of target protein. The average RMSF value for the bulk of LRP6 amino acid residues was found to be less than 2Å whereas the Dkk1 protein residues had an average RMSF value below 2.1Å indicating optimal stability of the Dkk1 protein. On the other hand, there were minor changes in the few residues of the Dkk1 which may be attributed to the slightly hanging position.
Conclusion
A series of 10 sulfanilamide derivatives with thiazole (4,5 d) pyrimidin-2-thione based fused ring heterocyclic system were synthesized and evaluated for their biological applications. All the derivatives were characterized and confirmed by FT-IR, 1HNMR, 13CNMR and UV spectroscopy. These derivatives were also evaluated for free radical scavenging activity, DNA binding studies and Carbonic anhydrase inhibition activity. The compound 5d was proved to be the most potent derivative with lowest IC50 Value (0.00690 ± 0.1119) for Carbonic anhydrase activity. In addition to this a comprehensive set of computational studies was also performed and all the results indicated that compound 5d may serve as a candidate drug in future. It was also observed that 4-(7-(4-Hydroxy-3-methoxyphenyl)-5-thioxo-5,6-dihydrothiazolo[4,5-d]pyrimidin-2-ylamino)benzenesulfonamide (5f) has highest free radical inhibitory activity (IC50 89.1384 ± 0.0011 µg/mL) comparable to vitamin c (IC50 95.1571 ± 0.057 µg/mL). All the synthetic compounds (5a-5j) showed good drug score values which depicts their drug like behavior and may be considered as drug candidate molecules specifically compound 5d.
Data availability
All the associated data will be available on request from corresponding author.
References
Mattiuzzi, C. and Lippi, G., 2019. Current cancer epidemiology. Journal of Epidemiology and Global Health, 9(4), pp.217–222.
Avuthu, V. S. R., Allaka, T. R., Afzal, M., Kishore, P. V. V. N., Venkatesan, S., & Patel, P. R. (2024). Novel 1, 2, 3-triazole derivatives containing benzoxazinone scaffold: Synthesis, Docking study, DFT analysis and biological evaluation. Results in Chemistry, 11, 101800.
Singh, K., Bhori, M., Kasu, Y.A., Bhat, G. and Marar, T., 2018. Antioxidants as precision weapons in war against cancer chemotherapy induced toxicity–Exploring the armoury of obscurity. Saudi Pharmaceutical Journal, 26(2), pp.177–190.
Scozzafava, A., Mastrolorenzo, A. and Supuran, C.T., 2004. Modulation of carbonic anhydrase activity and its applications in therapy. Expert Opinion on Therapeutic Patents, 14(5), pp.667–702.
T Supuran, C., Di Fiore, A., Alterio, V., Maria Montib, S. and De Simone, G., 2010. Recent advances in structural studies of the carbonic anhydrase family: the crystal structure of human CA IX and CA XIII. Current Pharmaceutical Design, 16(29), pp.3246–3254.
Supuran, C.T., 2007. Carbonic anhydrases as drug targets-an overview. Current Topics in Medicinal Chemistry, 7(9), pp.825–833.
Hilvo, M., Supuran, C.T. and Parkkila, S., 2007. Characterization and Inhibition of the recently discovered carbonic anhydrase isoforms CA XIII, XIV and XV. Current Topics in Medicinal Chemistry, 7(9), pp.893–899.
Pastorekova, S., Parkkila, S. and Zavada, J., 2006. Tumor-associated carbonic anhydrases and their clinical significance. Advances in Clinical Chemistry, 42, pp.167–216.
Benej, M., Pastorekova, S. and Pastorek, J., 2014. Carbonic anhydrase IX: regulation and role in cancer. Carbonic anhydrase: mechanism, regulation, links to disease, and industrial applications, pp.199–219.
Winum, J.Y., Rami, M., Scozzafava, A., Montero, J.L. and Supuran, C., 2008. Carbonic anhydrase IX: A new druggable target for the design of antitumor agents. Medicinal Research Reviews, 28(3), pp.445–463.
Facchin, C., 2019. Simultaneous in vivo imaging of tumor vascularization and metabolism (Doctoral dissertation, Université Paris Cité).
Niehrs, C., 2006. Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene, 25(57), pp.7469–7481.
Patel, S., Barkell, A.M., Gupta, D., Strong, S.L., Bruton, S., Muskett, F.W., Addis, P.W., Renshaw, P.S., Slocombe, P.M., Doyle, C. and Clargo, A., 2018. Structural and functional analysis of Dickkopf 4 (Dkk4): new insights into Dkk evolution and regulation of Wnt signaling by Dkk and Kremen proteins. Journal of Biological Chemistry, 293(31), pp.12149–12166.
Jiang, H., Zhang, Z., Yu, Y., Chu, H.Y., Yu, S., Yao, S., Zhang, G. and Zhang, B.T., 2022. Drug discovery of DKK1 inhibitors. Frontiers in pharmacology, 13, p.847387.
Glantschnig, H., Hampton, R.A., Lu, P., Zhao, J.Z., Vitelli, S., Huang, L., Haytko, P., Cusick, T., Ireland, C., Jarantow, S.W. and Ernst, R., 2010. Generation and selection of novel fully human monoclonal antibodies that neutralize Dickkopf-1 (DKK1) inhibitory function in vitro and increase bone mass in vivo. Journal of Biological Chemistry, 285(51), pp.40135–40147.
Rusu, A., Moga, I.M., Uncu, L. and Hancu, G., 2023. The role of five-membered heterocycles in the molecular structure of antibacterial drugs used in therapy. Pharmaceutics, 15(11), p.2554.
Baran, W., Adamek, E., Ziemiańska, J. and Sobczak, A., 2011. Effects of the presence of sulfonamides in the environment and their influence on human health. Journal of Hazardous Materials, 196, pp.1–15.
Apaydın, S. and Török, M., 2019. Sulfonamide derivatives as multi-target agents for complex diseases. Bioorganic & Medicinal Chemistry Letters, 29(16), pp.2042–2050.
Petri, W.A., 2006. Sulfonamides, trimethoprim, sulfamethoxazole, quinolones, and agents for urinary tract infections. Goodman & Gilman’s the pharmacological basis of therapeutics. New York (NY): McGraw Hill, pp.1111–1125.
Prescott, J.F., 2013. Sulfonamides, diaminopyrimidines, and their combinations. Antimicrobial therapy in veterinary medicine, pp.279–294.
Supuran, C.T., Innocenti, A., Mastrolorenzo, A. and Scozzafava, A., 2004. Antiviral sulfonamide derivatives. Mini Reviews in Medicinal Chemistry, 4(2), pp.189–200.
El-Gaby, M., A Ammar, Y., IH El-Qaliei, M., F Hussein, M. and A Faraghally, F., 2020. Sulfonamides: synthesis and the recent applications in medicinal chemistry. Egyptian Journal of Chemistry, 63(12), pp.5289–5327.
Marshall Jr, E.K., 1939. Bacterial chemotherapy: the Pharmacology of sulfanilamide. Physiological Reviews, 19(2), pp.240–269.
Brackett, C.C., Singh, H. and Block, J.H., 2004. Likelihood and mechanisms of cross-allergenicity between sulfonamide antibiotics and other drugs containing a sulfonamide functional group. Pharmacotherapy: the Journal of Human Pharmacology and Drug therapy, 24(7), pp.856–870.
Connor, E.E., 1998. Sulfonamide antibiotics. Primary Care Update for Ob/gyns, 5(1), pp.32–35.
Basireddy, A., Basireddy, S., Allaka, T., Afzal, M., & Kishore, P. V. V. N. (2024). New Tetrazole-Annulated Pyrazolyl–Pyrimidine derivatives as antimycobacterial targets: design, synthesis, molecular docking, and ADME profiling. Russian Journal of General Chemistry, 94(8), 2008–2017.
Tomaselli, D., Lucidi, A., Rotili, D. and Mai, A., 2020. Epigenetic polypharmacology: A new frontier for epi-drug discovery. Medicinal Research Reviews, 40(1), pp.190–244.
Zhou, J., Jiang, X., He, S., Jiang, H., Feng, F., Liu, W., Qu, W. and Sun, H., 2019. Rational design of multitarget-directed ligands: strategies and emerging paradigms. Journal of Medicinal Chemistry, 62(20), pp.8881–8914.
Marinova, G. and Batchvarov, V., 2011. Evaluation of the methods for determination of the free radical scavenging activity by DPPH. Bulgarian Journal of Agricultural Science, 17(1), pp.11–24.
Gulcin, İ. and Alwasel, S.H., 2023. DPPH radical scavenging assay. Processes, 11(8), p.2248.
Merz Jr, K.M., Murcko, M.A. and Kollman, P.A., 1991. Inhibition of carbonic anhydrase. Journal of the American Chemical Society, 113(12), pp.4484–4490.
Supuran, C.T., Scozzafava, A. and Casini, A., 2003. Carbonic anhydrase inhibitors. Medicinal Research Reviews, 23(2), pp.146–189.
Boger, D.L., Fink, B.E., Brunette, S.R., Tse, W.C. and Hedrick, M.P., 2001. A simple, high-resolution method for Establishing DNA binding affinity and sequence selectivity. Journal of the American Chemical Society, 123(25), pp.5878–5891.
Labarca, C. and Paigen, K., 1980. A simple, rapid, and sensitive DNA assay procedure. Analytical Biochemistry, 102(2), pp.344–352.
Levison, P.R., Dennis, J.W., Jones, K.D., Philpott, R.W., Taylor, S.L. and Grimm, V., 1998. New approaches in the binding of DNA for clinical applications. Clinical Chemistry, 44(9), pp.2060–2061.
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H. Gaussian 09, Rev. Gaussian Inc, Wallingford. 2009.
Dennington, R.D.I.I., Keith, T.A. and Millam, J.M., 2016. GaussView, Version 6.0. 16. Semichem Inc Shawnee Mission KS.
Trott, O. and Olson, A.J., 2010. AutoDock vina: improving the speed and accuracy of Docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), pp.455–461.
Bilal, M.S., Ejaz, S.A., Zargar, S., Akhtar, N., Wani, T.A., Riaz, N., Aborode, A.T., Siddique, F., Altwaijry, N., Alkahtani, H.M. and Umar, H.I., 2022. Computational investigation of 1, 3, 4 oxadiazole derivatives as lead inhibitors of VEGFR 2 in comparison with EGFR: Density functional theory, molecular docking and molecular dynamics simulation studies. Biomolecules, 12(11), p.1612.
Abida Ejaz, S., Sajjad Bilal, M., Aziz, M., Wani, T.A., Zargar, S., Fayyaz, A., Hassan, S., Ahmed, A., Al Kahtani, H.M. and Siddique, F., 2023. Computational exploration of Fluorocyclopentenyl-purines and‐pyrimidines derivatives as potential inhibitors of epidermal growth factor receptor (EGFR) for the treatment of breast Cancer. Chemistry & Biodiversity, 20(12), p.e202301190.
Ejaz, S.A., Aziz, M., Fawzy Ramadan, M., Fayyaz, A. and Bilal, M.S., 2023. Pharmacophore-based virtual screening and in-silico explorations of biomolecules (curcumin derivatives) of curcuma longa as potential lead inhibitors of ERBB and VEGFR-2 for the treatment of colorectal cancer. Molecules, 28(10), p.4044.
Bohurudeen, S. B., Ambala, A., Allaka, T. R., Ahmed, M. Z., Thirunavukkarasu, B., Tirumalareddy, R., & Venkatesan, S. (2025). Novel benzothiophene–tethered 1, 3, 4–oxadiazoles as potent antimicrobial targets: design, synthesis, biological evaluation, DFT exploration and in Silico Docking study. Journal of Molecular Structure, 1322, 140251.
https//www.rcsb.org/1V9E (last accessed 01 April 2024)
https://www.rcsb.org/3S2K (last accessed 01 April 2024)
Pettersen, E.F., Goddard, T.D., Huang, C.C., Couch, G.S., Greenblatt, D.M., Meng, E.C. and Ferrin, T.E., 2004. UCSF Chimera—a visualization system for exploratory research and analysis. Journal of Computational Chemistry, 25(13), pp.1605–1612.
Lovell, S.C., Davis, I.W., Arendall III, W.B., De Bakker, P.I., Word, J.M., Prisant, M.G., Richardson, J.S. and Richardson, D.C., 2003. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins: Structure, Function, and Bioinformatics, 50(3), pp.437–450.
Chen, V.B., Arendall, W.B., Headd, J.J., Keedy, D.A., Immormino, R.M., Kapral, G.J., Murray, L.W., Richardson, J.S. and Richardson, D.C., 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica Section D: Biological Crystallography, 66(1), pp.12–21.
Visualizer, D.S., 2005. Discovery Studio Visualizer. 2. Accelrys software inc.
ChemDraw Ultra 12.0 0 (Copyright) 1986 to 2009 by CambridgeSoft Corp. [Cambridge, Mass., U.S.A.].
O’Boyle, N.M., Banck, M., James, C.A., Morley, C., Vandermeersch, T. and Hutchison, G.R., 2011. Open babel: an open chemical toolbox. Journal of Cheminformatics, 3, pp.1–14.
Schrödinger, L., 2015. The PyMOL Molecular Graphics System, Version 1.8. (No Title).
Kozakov, D., Hall, D.R., Xia, B., Porter, K.A., Padhorny, D., Yueh, C., Beglov, D. and Vajda, S., 2017. The cluspro web server for protein–protein Docking. Nature Protocols, 12(2), pp.255–278.
Laskowski, R.A., 2001. PDBsum: summaries and analyses of PDB structures. Nucleic Acids Research, 29(1), pp.221–222.
Ahmed, A., Aziz, M., Ejaz, S.A., Channar, P.A., Saeed, A., Zargar, S., Wani, T.A., Hamad, A., Abbas, Q., Raza, H. and Kim, S.J., 2022. Design, synthesis, kinetic analysis and pharmacophore-directed discovery of 3-ethylaniline hybrid imino-thiazolidinone as potential inhibitor of carbonic anhydrase II: an emerging biological target for treatment of cancer. Biomolecules, 12(11), p.1696.
Qadri, T., Aziz, M., Channar, P.A., Ejaz, S.A., Hussain, M., Attaullah, H.M., Ujan, R., Hussain, Z., Zehra, T., Saeed, A. and Shah, M.R., 2023. Synthesis, biological evaluation and in Silico investigations of benzotriazole derivatives as potential inhibitors of NIMA related kinase. RSC Advances, 13(48), pp.33826–33843.
Aksu, K., Özgeriş, B., Taslimi, P., Naderi, A., Gülçin, İ. and Göksu, S., 2016. Antioxidant activity, acetylcholinesterase, and carbonic anhydrase inhibitory properties of novel Ureas derived from phenethylamines. Archiv Der Pharmazie, 349(12), pp.944–954.
Hussain, R.A., Badshah, A., Tahir, M.N., Lal, B. and Khan, I.A., 2013. Synthesis, chemical characterisation, and DNA binding studies of ferrocene-incorporated Selenoureas. Australian Journal of Chemistry, 66(6), pp.626–634.
Patujo, J., Badshah, A., Hussain, R.A., Tahir, M.N., Tahir, S. and Rauf, M.K., 2015. Synthesis, chemical characterization, DNA interaction and antioxidant, studies of new nitrosubstituted thioureas and their copper complexes. Journal of the Chinese Chemical Society, 62(11), pp.1020–1027.
Reichmann, M.E., Rice, S.A., Thomas, C.A. and Doty, P., 1954. A further examination of the molecular weight and size of desoxypentose nucleic acid. Journal of the American Chemical Society, 76(11), pp.3047–3053.
Chattaraj, P.K. and Roy, D.R., 2007. Update 1 of: electrophilicity index. Chemical Reviews, 107(9), pp.PR46-PR74.
Duan, L.L., Feng, G.Q. and Zhang, Q.G., 2016. Large-scale molecular dynamics simulation: Effect of polarization on thrombin-ligand binding energy. Scientific reports, 6(1), p.31488.
Huang, Y., Rong, C., Zhang, R. and Liu, S., 2017. Evaluating frontier orbital energy and HOMO/LUMO gap with descriptors from density functional reactivity theory. Journal of Molecular Modeling, 23, pp.1–12.
Ziegler, T., 1991. Approximate density functional theory as a practical tool in molecular energetics and dynamics. Chemical Reviews, 91(5), pp.651–667.
Vainio, M.J., Puranen, J.S. and Johnson, M.S., 2009. ShaEP: molecular overlay based on shape and electrostatic potential.
Fattah, T.A., Saeed, A., Channar, P.A., Larik, F.A., Hassan, M., Raza, H., Abbas, Q. and Seo, S.Y., 2018. Synthesis and molecular Docking studies of (e)-4-(substituted-benzylideneamino)-2 h-chromen-2-one derivatives: entry to new carbonic anhydrase class of inhibitors. Drug Research, 68(07), pp.378–386.
Abbasi, M.A., Raza, H., Siddiqui, S.Z., Shah, S.A.A., Hassan, M. and Seo, S.Y., 2019. Synthesis of novel N-(1, 3-thiazol-2-yl) benzamide clubbed oxadiazole scaffolds: Urease inhibition, Lipinski rule and molecular docking analyses. Bioorganic chemistry, 83, pp.63–75.
Walters, W.P., 2012. Going further than lipinski’s rule in drug design. Expert Opinion on Drug Discovery, 7(2), pp.99–107.
Caron, G. and Ermondi, G., 2016. Molecular descriptors for Polarity: the need for going beyond Polar surface area. Future Medicinal Chemistry, 8(17), pp.2013–2016.
Kotecha, J., Shah, S., Rathod, I. and Subbaiah, G., 2008. Prediction of oral absorption in humans by experimental immobilized artificial membrane chromatography indices and physicochemical descriptors. International Journal of Pharmaceutics, 360(1–2), pp.96–106.
Pantsar, T. and Poso, A., 2018. Binding affinity via docking: fact and fiction. Molecules, 23(8), p.1899.
da Fonseca, A.M., Caluaco, B.J., Madureira, J.M.C., Cabongo, S.Q., Gaieta, E.M., Djata, F., Colares, R.P., Neto, M.M., Fernandes, C.F.C., Marinho, G.S. and Dos Santos, H.S., 2023. Screening of potential inhibitors targeting the main protease structure of SARS-CoV-2 via molecular docking, and approach with molecular dynamics, RMSD, RMSF, H-bond, SASA and MMGBSA. Molecular Biotechnology, pp.1–15.
Acknowledgements
The authors extend their appreciation to their ongoing Research Funding Program project number (ORF-2025-357), King Saud University, Riyadh Saudi Arabia for funding this research. The funding body played potential role in the design of the study, purchasing, sample characterization, analysis, interpretation of data, and in writing the manuscript.
Funding
This research was funded by King Saud University, Riyadh Saudi Arabia under researchers supporting Research Funding Program project number (ORF-2025-357) King Saud University, Riyadh Saudi Arabia.
Author information
Authors and Affiliations
Contributions
The study plan was designed and supervised by S.A. Ejaz. The whole experimental work was carried out by Muhammad Sajjad Bilal following the directions of Pervaiz Ali Channar and Aamer saeed. The data curation and analysis was performed by Muhammad Sajjad Bilal, Rabil Ujan and Rehsma Sahito.The manuscript write-up was carried out by Muhammad Sajjad Bilal, Seema Zargar, Tanveer A. Wani, Sadia Naseem, and Qamar abbas under the guidance of S.A. Ejaz. The complete manuscript was finally edited and approved by S.A Ejaz.
Corresponding authors
Ethics declarations
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bilal, M.S., Ejaz, S.A., Naseem, S. et al. Synthesis, in vitro evaluation and computational modelling of benzene sulfonamide derivatives as Dickkopf 1 inhibitors for anticancer drug development. Sci Rep 15, 21049 (2025). https://doi.org/10.1038/s41598-025-06890-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-06890-1
