
Abstract
The relationship between porous morphology and current-time curves cannot be explained by the field-assisted dissolution theory (FADT). Double-layer structures of porous anodic alumina (PAA) with different pore sizes were obtained by multi-step anodizations. These important results cannot be interpreted by the traditional FADT theory. Here, based on the theories of ionic current and electronic current, the always controversial growth kinetics of PAA are clarified by the current-time curves. The ionic current under high electric field is the driving force for the rapid growth of oxides, resulting in the decline of the current curve. The electronic current results in the rise of the current curve, and causes oxygen bubble to form the pore embryos. After the electrolyte enters the pore bottom, the thickness of the bottom barrier layer remains unchanged. Therefore, constant electronic current maintains the oxygen evolution and oxygen bubble mold. Constant ionic current maintains the oxide growth around the oxygen bubble mold at the pore bottom, and maintains the upward growth of PAA channel in the viscous flow mode. The field-assisted dissolution rate is much less than the rate of channel growth determined by the total current.
Similar content being viewed by others

Introduction
Porous anodic oxides (e.g., Al2O3, TiO2, ZrO2, etc.) obtained by electrochemical anodization are widely used in the fields of solar cells, photocatalysis, sensors and supercapacitors1,2,3,4. However, the formation mechanisms of self-organized nanochannels in porous anodic alumina (PAA) and self-ordered nanotube structure in anodic TiO2 still remain controversial5,6,7,8,9,10. However, the dissolution reaction (Al2O3 + 6H+→2Al3++3H2O) advocated by the field-assisted dissolution theory (FADT) is still a popular mechanism11,12,13,14,15. They believe that there is a balance between oxide growth and oxide dissolution in the porous growth stage due to the anodizing current stable, however, few people have studied the relationship between current-time curve and oxide growth rate, dissolution rate13,14,15,16.
As early as 1969, Diggle et al.16 proposed many doubts about the chemical dissolution forming pores: (i) Does it decrease in thickness at the same rate as the pore diameter increases? (ii) How does dissolution rate depend on the film dimensions? (iii) What type of kinetics does the dissolution rate follow? These questions still remain unresolved in the latest review articles on PAA17,18,19,20,21 and anodic TiO2 nanotubes22,23,24,25.
According to the “top-down digging mode” of FADT, Masuda et al.26,27 invented two-step anodizing and imprinting technologies, that is, the pores of second anodization will continue to grow along the remained pits of first anodization. However, no matter in the anodizing process of aluminum or titanium, the pits left after the first anodization cannot completely guide the pore formation in the second anodization27,28,29,30,31. In fact, as early as 2006, the viscous flow growth mode of PAA, which is completely opposite to the FADT model, was discovered by Manchester group32. Moreover, the relationship between the current-time curve reflecting the whole process of anodization and the PAA morphology remains to be further studied33. Moreover, for 60 years, no article has been able to prove that oxide growth rate and field-assisted dissolution rate are equal, that is, it is impossible to prove the existence of equilibrium between oxide growth and field-assisted dissolution, let al.one prove the relationship between field-assisted dissolution and anodizing current (or electric field)12,33. As Hebert et al.34 indicated that although porous aluminum and titanium oxides have found extensive applications, pore initiation and self-ordering kinetics are not yet understood34. Based on the theories of ionic current and electronic current, we have also published some papers on the growth mechanism of anodic TiO2 nanotubes23,24,35,36. However, previous articles have hardly touched upon the growth rate and dissolution rate of porous alumina, nor have they covered the kinetic explanation of the current-time curves of Al multi-step anodizations.
In order to verify the growth mode of PAA channels is dissolved digging mode or viscous flow mode around the oxygen bubble mold35,36,37,38. In this paper, we found three interesting double-layer structures in PAA. These double-layer structures with different diameters were prepared by two-step anodizing technology. These interesting findings contradict the FADT and is consistent with the viscous flow model. In this paper, the formation mechanism and growth kinetics of double-layer PAA are explained by the viscous flow model and oxygen bubble model combining current-time curve. The present results have important implications for the growth mechanism of other porous anodic oxides (TiO2, ZrO2, SnO2, etc.).
Materials and methods
In the experiment, aluminum foils (thickness 0.2 mm, 99.99% purity) were cut into 1 cm*8 cm strips and immersed in an aqueous solution containing 5wt% H3PO4 for 30 min at 60 °C to ensure that the natural oxide film on the surface was completely removed. After the immersion, quickly removed the aluminum strips and placed them in deionized water, repeatedly washed them several times. After being dried, the aluminum strips were pasted with the insulating tape to ensure the anodizing area was 4cm2 (2 cm × 1 cm × 2 sides).
Four kinds of different electrolytes were used in these experiments, including 3wt% oxalic acid aqueous solution, 3wt% and 5wt% phosphoric acid aqueous solution and 10wt% ammonium phosphite aqueous solution. Placed a beaker containing 200 ml of electrolyte into a DL-3005 circulating cooling pump to ensure that the temperature of the electrolyte was constant at 20 °C. The aluminum strip was used as the anode, while the graphite strip (100 mm × 15 mm) was used as the cathode. The aluminum strips were anodized by constant voltage in the above four electrolytes, performed two or more anodizing steps on the same aluminum strip. The same test was repeated two or three times, aiming to ensure a good repeatability of the current-time curve measured by the computer system. In order to prepare double-layer PAA with different pore sizes, the experiments in this paper were divided into three groups.
The first set of experiments was to prepare PAA in H3PO4 electrolyte. The first step of the anodizing process was anodized for 320 s at a constant voltage of 40 V with 3wt% H3PO4 as the electrolyte. After the first step of anodization was completed, quickly removed the aluminum strip from the electrolyte, soaked and rinsed the aluminum strip in deionized water for 2 min to remove the residual electrolyte on it. The second anodizing step was performed after about 3 min. The second anodizing step: 5wt% H3PO4 was used as the electrolyte, and the aluminum strip after the first step anodizing was used as the anode, and the anodizing was carried out for 600 s at a constant voltage of 80 V. After the experiment was completed, the aluminum strip was soaked and rinsed in deionized water for 5 min, then the sample was dried in the air and retained for SEM characterization.
The second set of experiments was to prepare PAA in ammonium phosphite electrolyte and oxalic acid electrolyte. The first step of the anodizing process was anodized for 300 s at a constant voltage of 20 V with 10wt% ammonium phosphite as the electrolyte. After the first step of anodization was completed, quickly removed the aluminum strip from the electrolyte, soaked and rinsed the aluminum strip in deionized water for 2 min to remove the residual electrolyte on it. The second anodizing step was performed after about 3 min. The second anodizing step: 3wt% oxalic acid was used as the electrolyte, and the aluminum strip after the first step anodizing was used as the anode, and the anodizing was carried out for 300 s at a constant voltage of 50 V. After the experiment was completed, the aluminum strip was soaked and rinsed in deionized water for 5 min, then the sample was dried in the air and retained for SEM characterization.
The third set of experiments was to prepare PAA in oxalic acid electrolyte and ammonium phosphite electrolytes. The first step of the anodizing process was anodized for 330 s at a constant voltage of 40 V with 3wt% oxalic acid as the electrolyte. After the first step of anodization was completed, quickly removed the aluminum strip from the electrolyte, soaked and rinsed the aluminum strip in deionized water for 2 min to remove the residual electrolyte on it. The second anodizing step was performed after about 3 min. The second anodizing step: 10wt% ammonium phosphite was used as the electrolyte, and the aluminum strip after the first step anodizing was used as the anode, and the anodizing was carried out for 600 s at a constant voltage of 28 V. Since the current was always zero during the second step anodizing of 600 s, therefore, the third step anodizing electrolyte was not replaced, and the voltage was directly increased to 29 V until the current curve showed an upward trend, the anodizing was stopped by 130 s. The sample after the third step anodizing was replaced into 3wt% oxalic acid electrolyte for the fourth step anodizing. In the fourth step anodizing, the voltage was also 40 V, maintained at the same voltage as the first step anodizing and anodized for 600 s. After the experiment was completed, the aluminum strip was soaked and rinsed in deionized water for 5 min, then the sample was dried in the air and retained for SEM characterization.
The anodized samples of the above three groups were cut into a rectangle shape of about 1 mm×5 mm, artificially bent into an Ω shape with the help of tweezers, and affixed to the conductive adhesive on the SEM sample table. The microstructure of PAA was characterized by FESEM (Zeiss Supra 55).
Results and discussion
Figure 1 shows the surface and cross section morphologies of double-layer PAA obtained by two-step anodizing in H3PO4 electrolyte. From the surface topography of Fig. 1ab, it is difficult to tell whether these pores were formed by the dissolved digging mode, or were formed from the bottom up with a bubble mold? As can be seen from the cross section of Fig. 1c, PAA channels are divided into upper and lower layers, and their lengths are 933 nm and 874 nm, respectively. Moreover, the diameter of the upper channel is obviously smaller than that of the lower channel. Figure 1d shows that the inner diameter of the upper channel is about 28 ~ 32 nm, while the inner diameter of the lower channel is about 110 nm. That is to say, the porosity of the upper layer is much higher than that of the lower layer, which is obviously contrary to the expected results of two-step anodizing and imprinting technologies.
If the pores were digged from top to bottom in the dissolution mode, then the number of pores in the upper and lower layers should be the same. However, it is obvious from Fig. 1c that the number of pores formed by the second anodization is much smaller than that obtained by the first anodization. This shows that the formation of pores is not in accordance with the digging mode of field-assisted dissolution. This fact completely negates the digging mode of field-assisted dissolution from top to bottom. Then, the growth of PAA channels can only conform to the “bottom-up” viscous flow mode.

SEM images show the PAA obtained by two-step anodizations in H3PO4 electrolyte, (a) (b) surface morphology, (c)(d) cross section morphology.
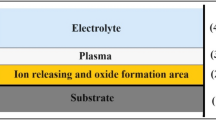
The viscous flow model was first discovered by the Manchester group, but the dynamics of the viscous flow are much debated32,33. Like the field-assisted dissolution model, the field-assisted flow model cannot be correlated with current-time curves (Fig. 2ab). Hebert et al.39,40,41 proposed that flow is driven by nonuniform compressive stress near the oxide-solution interface, resulting from anion adsorption on surface growth sites. The stress-driven ionic transport toward the pore walls is envisioned to assist pore formation39,40,41. However, the oxygen bubble model believes that the oxide growth at the pore base flows to the surrounding pore walls because of the existence of oxygen bubble molds at the pore bottoms. It is because of the existence of oxygen bubble at the pore bottom that the viscous flow of oxides has to flow to the pore walls around the oxygen bubble molds35,36,37,38. The idea that oxygen bubbles lead to the formation of porous embryos is also accepted by many researchers42,43,44,45,46,47,48. Here, in combination with the current-time curve (Fig. 2a), the growth kinetics of barrier oxide and oxygen bubble are illustrated below (Fig. 3).

The current-time curves anodized in the (a) first anodization at 40 V in 3wt% H3PO4 electrolyte, (b) second anodization at 80 V in 5wt% H3PO4 electrolyte.
In stage I of Fig. 2, the total current rapidly declines and the ionic current dominates, decreasing rapidly as the thickness of the initially formed barrier oxide increases, which conforms to the ionic current formula of the high electric field model (HEFM)42. It is well known that the ionic current may be expressed simply as Jion=Aexp(BE)42,49,50,51,52 where Jion is ionic current, A and B are temperature dependent constants, and E is the field strength (E = Uapp/d), Uapp is the applied voltage, d is the thickness of barrier oxide. Diggle et al.16 have clearly pointed out that in the early stage of anodization, high electric field leads to ionic conductance. With the increase of oxide thickness, the electric field and ionic conductance will decrease, while the electronic conductance will dominate16. The increase of current-time curves in Fig. 2a cannot be clarified by FADT and field-assisted flow model32,33. Albella et al.53 proposed that theoretical expressions of electronic current is Je= j0exp(αd), where α is the impact ionization coefficient of avalanche, d is the thickness of barrier oxide, and j0 is the primary electronic current. Thus, the rapid decrease in the current at stage I in Fig. 2a reflects the decrease in the ionic current. On the contrary, in the stage II, the increase of the total current indicates that the electronic conductance is dominant, and the electronic current leads to the increase of the total current. The barrier oxides grown at this stage are shown in Fig. 3(I). However, since the electric field is always present, the barrier oxide will inevitably be incorporated by electrolyte anions, and the oxide film will become an anionic contaminated alumina (ACA) layer near the electrolyte and a barrier oxide near the metal substrate54,55,56. It is because of the existence of ACA layer and anion discharge that the electronic current is generated, and the electronic current becomes the driving force of oxygen evolution (O2).
In the stage II of Fig. 2a, the barrier oxide continues to grow due to the presence of ionic currents, while the presence of electronic currents leads to the formation of oxygen bubbles. Wu et al.57,58 found that the double-layer pore wall structure of titanium oxide nanotubes has many small holes, and these nanopores are formed by oxygen bubbles. They demonstrated that the location of oxygen evolution is right at the interface between the ACA layer and the barrier oxide layer57,58. The location of oxygen evolution is at the interface between the ACA and the barrier oxide as shown in Fig. 3(II). Because the oxygen bubbles have to withstand the outside atmospheric pressure, electrolyte pressure, and ACA pressure, the naked eye rarely sees the overflow of oxygen bubbles. Oxygen bubbles under high pressure cannot immediately overflow outside the oxide film, so the oxygen bubbles act as a gas bubble mold above the barrier oxide. In this way, the newly grown barrier oxides can only grow upward around the oxygen bubble mold to form the pore walls, thus forming the pore embryo of PAA, corresponding to stage II in Fig. 3. The thickness of the barrier oxide is the critical thickness generated by the electronic current corresponding to the anodizing voltage of 40 V. Because only the barrier oxide reaches this critical thickness, can sufficient electronic current be generated (Je= j0exp(αd)).

Schematic diagram of the PAA growing kinetic model in which ionic current leads to oxide growth and electronic current promotes oxygen bubble formation.
Stage III in Fig. 2a corresponds to Fig. 3(III). Sufficient electronic current leads to the continuous expansion of oxygen bubbles, and sufficient ionic current leads to the upward growth of barrier oxide. In this way, high-pressure oxygen bubbles emerge one after another in the pore embryos as shown in Fig. 3(III), and external bulk electrolyte also penetrates into the pore embryos. When the electrolyte in all channels penetrates into the bottom of the channels, the current curve enters stage III (Fig. 2a). Since both ionic current and electronic current are related to the thickness of the barrier oxide as shown in Fig. 3, the ionic current and electronic current are basically constant. Therefore, in stage III of Fig. 2a, the total current is basically constant because the barrier oxide thickness at the bottom is constant as shown in Fig. 3(III). At this stage, sufficient electronic current ensures the oxygen evolution at the pore bottom, and sufficient pressure of the oxygen bubble mold ensures that the barrier oxide layer at the pore bottom forms a hemispherical bottom35,36,37. Sufficient ionic current (radial distribution) at the pore bottom maintains the upward growth of the barrier oxide around the bubble mold, which promotes the upward growth of the PAA channel as shown in Fig. 3(III).
Thompson et al.59,60 reported the existence of oxygen bubbles in the growth of porous anodic alumina, and proved that the pressure of oxygen bubbles was about 100 MPa. The size of oxygen bubbles was about tens of nanometers. It is also pointed out that the existence of oxygen bubbles will prevent the migration of ions, and the oxygen evolution also leads to the reduction of the growth efficiency of oxides59,60.
According to the anodizing current theory, the HEFM model and the oxygen bubble model in Fig. 3, it is easy to explain that the lower layer PAA with large pore size grown by the second anodization in Fig. 1cd. The anodizing voltage of the first step is 40 V, and the barrier oxide layer at the bottom of the upper layer PAA is about 45 nm. After the anodizing voltage is increased to 80 V in the second step, the original thickness (~ 45 nm) of the barrier oxide layer at the bottom of upper layer PAA is quickly increased to new barrier oxide layer with a thickness (~ 92 nm) in the stage I in Fig. 2b. Yu et al.61 reported that the new barrier oxide grows simultaneously at two interfaces at the pore bottom of the PAA, namely, the electrolyte/oxide interface and the oxide/Al interface61. Therefore, the anodizing current of the stage I in second anodizing also rapidly drops to its lowest point (Fig. 2b). That is, the anodizing voltage of 80 V corresponds to the new critical thickness (around 92 nm in Fig. 1c). At this time, the electronic current is generated during the second anodization, but the duration of the stage II (~ 280 s shown in Fig. 2b) is much longer than that of the first anodization (~ 50 s shown in Fig. 2a). In other words, the formation duration of the porous embryos in the second anodization is much longer than that of in the first anodization. This is due to the longer formation duration of the ACA layer and the longer time for oxygen bubbles to open the overlying layer remained from the first anodization. If the porous channels of second PAA layer are formed according to the dissolved digging mode, there is no difference in the time of digging the pore embryos in the stage II as shown in Fig. 2a and b.
Figure 2b shows that the second PAA channels were not really connected until 340 s after the second anodization beginning, and the electrolyte entered the second PAA channels and the anodizing current entered the stable growth stage (III) of the second PAA channels. By contrast, the two stable currents in the stage III in Figs. 2ab are 45 mA and 10 mA, respectively. And the stable growth durations of PAA in the stage III are about 225 s and 260 s, respectively. Therefore, the calculated result should be that the height of the first PAA layer should be about 4 times the height of the second PAA layer. In fact, the height of the first PAA layer is 933 nm, the second PAA layer is 874 nm. These interesting findings indicate that the PAA growth of the second layer is also promoted during the growth of the stage II in Fig. 2b, and the growth of the second PAA layer is also promoted due to the extension of the time of the stage II and the volume expansion of oxygen bubbles62,63. Similarly interesting results are found in both Figs. 4 and 6.
Figure 4 shows the double-layer PAA channels obtained by anodizing in ammonium phosphite electrolyte and oxalic acid electrolyte. Figure 5 shows the current-time curve of the two anodizing processes. Figures 4 cd show that the pore diameter of the lower PAA channel is significantly larger than that of the upper PAA channel. This is exactly the same as in Fig. 1cd obtained in H3PO4 electrolyte, and is also counterevidence to the “top-down digging mode” of FADT. As can be seen in Fig. 4b, the pore diameter in the top surface layer is less than 20 nm. After anodizing in two electrolytes, the surface pores did not expand, that is, although the total height of the two layers of PAA channels exceeded 1400 nm, the surface pores did not expand due to field-assisted dissolution. This would answer the doubt questions raised by Diggle et al.16 (i) Does it decrease in thickness at the same rate as the pore diameter increases? Neither the shortening of PAA channels nor the enlargement of pore diameters were caused by the field-assisted dissolution reaction. The results show that there is almost no so-called field-assisted dissolution reaction in the ammonium phosphite electrolyte, because the pH value of this electrolyte is about 6.0, and its acidity is much less than oxalic acid solution and phosphoric acid solution. (ii) How does dissolution rate depend on the film dimensions? Although many articles claim that pores are formed by the field-assisted dissolution, few articles study the dissolution rate of alumina. The Manchester group reported that the electrolyte dissolved the oxide at a rate of less than 4 nm/min64,65. This dissolution rate is much less than the growth rate of porous channels (1400 nm/10 min = 140 nm/min) in Fig. 4c. It can be seen from the surface morphology of Fig. 4ab that the dissolution rate of oxides does not depend on the height (1400 nm in Fig. 4c) of porous channels. (iii) What type of kinetics does the dissolution rate follow? The dynamics of the dissolution rate are ambiguous because the so-called field-assisted dissolutions are not directly related to either the electric field or the anodizing current12,42. As can be seen from Fig. 5a, for ammonium phosphite electrolyte with very weak solubility, the current-time curve also indicates that electronic current generated and PAA channels with very small pore size can be formed due to the oxygen bubble mold. The channel height of the second PAA layer in Fig. 4c is also greater than that of the first PAA layer, because the anodizing current in oxalic acid in Fig. 5b is greater than that in ammonium phosphite. The size of the anodizing current is related to the voltage level, and has nothing to do with the solubility of the electrolyte.
If the occurrence of field-assisted dissolution reaction is closely related to the non-uniformity of the surface morphology, then the existence of gaps between several particles (the ellipse mark in Fig. 4b) and the surface will cause the so-called gap corrosion. Because the electric field distribution at the gap is extremely uneven, under the action of extremely uneven tip electric field, serious dissolution corrosion should be formed around the tiny particles in Fig. 4b. However, SEM images showed that no so-called field-assisted dissolution occurred around the four tiny particles. This fact also confirms the Manchester group’s claim that the dissolution rate of alumina is weak (1 ~ 4 nm/min)64,65. The surface morphology of PAA in the more acidic oxalic acid solution did not expand the pore size due to field-assisted dissolution as shown in Fig. 6d.

SEM images show the PAA obtained by anodizing in 10wt% ammonium phosphite electrolyte and 3wt% oxalic acid electrolyte, (a) (b) surface morphology, (c) (d) cross section morphologies.

The corresponding current-time curves, (a) first anodization at 20 V in 10wt% ammonium phosphite, (b) second anodization at 50 V in 3wt% oxalic acid.
Figure 6 shows the SEM photos of the double-layer PAA obtained in oxalic acid electrolyte. Figure 7 shows the four-step anodizing process of the formation of double-layer PAA channels. The dividing line between the upper and lower PAA layer can be clearly seen in Fig. 6ab. Near the interface in Fig. 6b, it is shown that the upper and lower channels are misaligned with each other (elliptical marks). Both the upper and lower layers are obtained under the same anodizing conditions (3wt% oxalic acid electrolyte and constant voltage of 40 V). Interestingly, under the same voltage in the same electrolyte, the lower PAA channel did not grow down from the bottom of the upper channel. This shows that the pore left by the first anodization has no guiding effect on the pore formation of the second anodization. The visible surface pores in Fig. 6cd are all less than 30 nm in diameter. If the field-assisted dissolution rate is 2 nm/min64,65, after 27 min of anodizing duration, the diameter of the surface pore should also be above 54 nm. This proves from the opposite side that the field-assisted dissolution is weak both in lengthways and in radial directions. Moreover, it can be seen from the surface of Fig. 6cd that the influence of dissolution corrosion on the surface morphology during the whole anodizing process can be ignored66.

SEM images show the PAA obtained by four-step anodization in ammonium phosphite electrolyte and oxalic acid electrolyte, (a) (b) cross section morphologies, (c) (d) surface morphologies.

Four-step anodizing corresponding current-time curves, (a) first anodization at 40 V in 3wt% oxalic acid, (b) second anodization at 28 V in 10wt% ammonium phosphite, (c) third anodization at 29 V in 10wt% ammonium phosphite, (d) forth anodization at 40 V in 3wt% oxalic acid.
Since FADT cannot associate current-time curve with PAA topography33 many literatures ignore the analysis of current-time curve. More important information about the critical electric field can be obtained from the analysis of the current-time curve in Fig. 7. Figure 7a shows the current-time curve corresponding to the upper PAA channels, because the anodizing voltage is 40 V, so the thickness of the barrier layer is about 40 nm, but after entering the ammonium phosphite electrolyte, at 28 V, the anodizing current is not generated during the anodizing process for 600 s. This means that the electric field at this time (28 V/40 nm = 0.7 V/nm = 7 × 108 V/m) is too small to generate an electronic current at all. But we think that since there is an electric field, new ACA layer can still be created. After increasing the voltage to 29 V during the third anodization, the electronic current rose to 11 mA in 130 s. This indicates that for ammonium phosphite electrolyte, the critical electric field strength to generate electronic current is about 7.25 × 108 V/m (29 V/40 nm = 0.725 V/nm). At this time, it goes back to the oxalic acid electrolyte to continue the fourth anodization, and the anodizing voltage also continues to maintain 40 V. At this time, the electronic current reaches the maximum electronic current (~ 18 mA) in about 50 s, which is 25 s earlier than the maximum electronic current in Fig. 7a, because the ACA layer is also formed in the second and third anodizing processes. The accumulation of anions is a prerequisite for the generation of electronic current35,52.
Conclusions
The traditional field-assisted dissolution theory holds that PAA pores are formed in a top-down digging mode, so two-step anodizing and imprinting technologies are proposed. Here, double-layer PAA with different pore sizes was prepared by two-step and multi-step anodizing processes. It is proved that the pore left by the first anodization has no guiding effect on the pore formation of the second anodization. These facts negate the top-down digging mode and confirm that PAA pores grow from the bottom-up in a viscous flow model. The three stages of the current-time curve cannot be explained reasonably by FADT and the viscous flow model. In this paper, the growth dynamics of double-layer PAA channels are carefully analyzed. The analysis shows that the current drop of current-time curve is caused by ionic current forming barrier oxide, and the rise of current is caused by electronic current, which leads to the formation of oxygen bubble mold and pore embryos. In the third stage (III) of the current-time curve, since the ionic current is inversely proportional to the thickness of the barrier oxide, and the electronic current is directly proportional to the thickness of the barrier oxide at the pore bottom, it is determined from both positive and negative aspects that under a given anodizing voltage, there is a corresponding critical thickness, and under this critical thickness, there is a critical electric field strength to keep the ionic current and electronic current basically constant. The two currents maintain oxide growth and oxygen evolution, respectively, and the two currents coexist to promote the orderly growth of PAA channels. These new viewpoints have important significance for understanding the growth mechanism and growth dynamics of other porous anodic oxides (TiO2, ZrO2, SnO2, etc.).
Data availability
All data generated or analysed during this study are included in this published article.
References
Liu, D., Wang, X., Liu, D., Zhao, J. & Zhao Remarkable fluorescence enhancement of acridine orange and Rhodamine B through immobilization on zirconia nanotube array film and its application on nitrite sensing. J. Mater. Sci. Technol. 208, 26–37 (2025).
Li, P. Z. et al. Formation mechanism of nanopores in dense films of anodic alumina. Trans. Nonferrous Met. Soc. China. 34, 2918–2927 (2024).
Li, B. W. et al. Morphology and photocurrent response of TiO2 nanotubes prepared in electrolytes containing different content of polyethylene glycol. Ceram. Int. 50, 49862–49870 (2024).
Xu, Q., Yin, J., Zhang, J. & Zhang, Y. Preparation and optical characterization of 3D-TiO2 thin films with brilliant colors. Appl. Surf. Sci. 679, 161315 (2025).
Li, Z. B., Pan, J., Bian, H. D., Lu, J. & Li, Y. Y. New explanation on formation mechanism of anodic TiO2 nanotubes. Mater. Sci. Eng., B. 286, 115985 (2022).
Iwai, M., Kikuchi, T. & Suzuki, R. O. Selfordered nanospike porous alumina fabricated under a new regime by an anodizing process in alkaline media. Sci. Rep. 11, 7240 (2021).
Su, Z. X. & Zhou, W. Z. Formation mechanism of porous anodic aluminium and titanium oxides. Adv. Mater. 20, 3663–3667 (2008).
Asoh, H., Ota, S. & Hagiwara, K. Communication–Anodization of aluminum in phosphoric acid containing glycerol at 30℃. J. Electrochem. Soc. 171, 033502 (2024).
Sun, X. D. et al. A new study on formation mechanism of ordered porous anodized metal oxides. Rare Met. 43, 5410–5418 (2024).
Qin, L. Y. et al. Influence of micro-arc oxidation on the microstructure and dielectric properties of anodic aluminum oxide. Sci. Rep. 14, 23673 (2024).
Mebed, A. M. & Abd-Elnaiem, A. M. A thermodynamic Understanding of horizontal pores formation in anodized doped aluminum with alloying elements. J. Electroanal. Chem. 829, 138–147 (2018).
Oh, J. & Thompson, C. V. The role of electric field in pore formation during aluminum anodization. Electrochim. Acta. 56, 4044–4051 (2011).
Lee, W. & Park, S. J. Porous anodic aluminum oxide: anodization and templated synthesis of functional nanostructures. Chem. Rev. 114, 7487–7556 (2014).
Małgorzata, N. Self-ordered porous anodic alumina with large pore intervals: review on experimental and theoretical research. J. Electrochem. Soc. 169, 123503 (2022).
Furneaux, R. C., Rigby, W. R. & Davidson, A. P. The formation of controlled-porosity membranes from anodically oxidized aluminum. Nature 337, 147–149 (1989).
Diggle, J. W., Downie, T. C. & Goulding, C. W. Anodic oxide films on aluminum. Chem. Rev. 69, 365–405 (1969).
Korzekwa, J. Modification of the structure and properties of oxide layers on aluminum alloys: A review. Reviews Adv. Mater. Sci. 62, 20230108 (2023).
Mohapatra, B. D. & Sulka, G. D. Review of anodic tantalum oxide nanostructures: from morphological design to emerging applications. ACS Appl. Nano Mater. 7, 13865–13892 (2024).
Vera-Londono, L., Ruiz-Clavijo, A. & Caballero-Calero, O. Martín-González, M. Understanding the thermal conductivity variations in nanoporous anodic aluminum oxide. Nanoscale Adv. 2, 4591–4603 (2020).
Heinschke, S. & Schneider, J. J. On the nature of self-organization of porosity during aluminum anodization. J. Phys. Chem. C. 126, 709–715 (2022).
Roslyakov, I. V., Sotnichuk, E. O., Sotnichuk, S. V., Kushnir, S. E. & Napolskii, K. S. Kinetic and crystallographic control of self–ordering of pores in anodic aluminium oxide. J. Solid State Electrochem. 29, 1341–1373 (2025).
Suliali, N., Mbulanga, C., Goosen, W., Betz, R. & Botha, J. Numerical modelling of anodization reaction kinetics for TiO2 nanotubular-film growth in NH4F-based electrolytes. Electrochim. Acta. 337, 135791 (2020).
Li, C. Y. et al. A review: research progress on the formation mechanism of porous anodic oxides. Nanoscale Adv. 4, 322–333 (2022).
Li, B. et al. Relationship between the growth rate of nanotubes and the current – time curve of the anodizing process. J. Phys. Chem. C. 128, 12296–12302 (2024).
Albu, S. P. et al. Formation of double-walled TiO2 nanotubes and robust anatase membranes. Adv. Mater. 20, 4135–4139 (2008).
Masuda, H. & Fukuda, K. Ordered metal nanohole arrays made by a two-step replication of honeycomb structures of anodic alumina. Science 268, 1466–1468 (1995).
Masuda, H. et al. Ordered mosaic nanocomposites in anodic porous alumina. Adv. Mater. 15, 161–164 (2003).
Li, Y., Ling, Z. Y., Chen, S. & Wang, J. Fabrication of novel porous anodic alumina membranes by two-step hard anodization. Nanotechnology 19, 225604 (2008).
Su, Z. X. & Zhou, W. Z. Formation, morphology control and applications of anodic TiO2 nanotube arrays. J. Mater. Chem. 21, 8955–8970 (2011).
Jin, R. et al. Formation mechanism of lotus-root-shaped nanostructure during two-step anodization. Electrochim. Acta. 188, 421–427 (2016).
Zeng, H. P. et al. A comparative study of two-step anodization with one-step anodization at constant voltage. Nanotechnology 34, 065603 (2023).
Garcia-Vergara, S. J., Skeldon, P., Thompson, G. E. & Habazaki, H. A flow model of porous anodic film growth on aluminum. Electrochim. Acta. 52, 681–687 (2006).
Houser, J. E. & Hebert, K. R. The role of viscous flow of oxide in the growth of self-ordered porous anodic alumina films. Nat. Mater. 8, 415–420 (2009).
Hebert, K. R., Albu, S., Paramasivam, I. & Schmuki, P. Morphological instability leading to formation of porous anodic oxide films. Nat. Mater. 11, 162–166 (2012).
Zhu, X. F. et al. Oxygen bubble mould effect: serrated nanopore formation and porous alumina growth. Monatshefte Für Chemie-Chemical Monthly. 139, 999–1003 (2008).
Zhuang, Y. et al. Real role of fluoride ions in the growth of anodic TiO2 nanotubes. J. Phys. Chem. C. 128, 5741–5748 (2024).
Chen, W., Lai, M., Tsai, K., Liu, C. & Wang, Y. Spontaneous formation of ordered nanobubbles in anodic tungsten oxide during anodization. J. Phys. Chem. C. 115, 18406–18411 (2011).
Gong, T. et al. Evidence of oxygen bubbles forming nanotube embryos in porous anodic oxides. Nanoscale Adv. 3, 4659–4668 (2021).
Mishra, P. & Hebert, K. R. Self-organization of anodic aluminum oxide layers by a flow mechanism. Electrochim. Acta. 340, 135879 (2020).
Dou, Q., Shrotriya, P., Li, W. & Hebert, K. R. Stress-generating electrochemical reactions during the initial growth of anodic titanium dioxide nanotube layers. Electrochim. Acta. 295, 418–426 (2019).
Qin, J. R., Zhou, X. R. & Curioni, M. Investigation of oxide growth during hard anodizing using in-situ and ex-situ EIS measurements. Surf. Coat. Technol. 470, 129851 (2023).
Broens, M. I. et al. TiO2 nanotube arrays grown in ethylene glycol-based media containing fluoride: Understanding the effect of early anodization stages on the morphology. J. Electroanal. Chem. 935, 117314 (2023).
Cao, J. et al. Morphology evolution of the anodized Tin oxide film during early formation stages at relatively high constant potential. Surf. Coat. Technol. 388, 125592 (2020).
Ruiz-Clavijo, A. & Caballero-Calero, O. Martín-González, M. Revisiting anodic alumina templates: from fabrication to applications. Nanoscale 13, 2227–2265 (2021).
Broens, M. I. et al. The keys to avoid undesired structure defects in nanotubular TiO2 films prepared by electrochemical anodization. Ceram. Int. 46, 13599–13606 (2020).
Li, X. et al. The growth rate of nanotubes and the quantity of charge during anodization. Electrochem. Commun. 135, 107184 (2022).
Gao, J. et al. Recent progress of self-supported metal oxide nano-porous arrays in energy storage applications. Small 19, 2302786 (2023).
Heinschke, S. & Schneider, J. J. On the prediction of well-ordered porous anodic alumina films. J. Phys. Chem. C. 124, 11913–11921 (2020).
Wang, S. et al. Determining the sparking voltage of working electrolytes. J. Electrochem. Soc. 170, 093504 (2023).
Patermarakis, G. & Moussoutzanis, K. Electrochemical kinetic study on the growth of porous anodic oxide films on aluminum. Electrochim. Acta. 40, 699–708 (1995).
Thompson, G. E. Porous anodic alumina: fabrication, characterization and applications. Thin Solid Films. 297, 192–201 (1997).
Zhu, X. F. et al. Electronic current and the formation of nanopores in porous anodic alumina. Nanotechnology 20, 475303 (2009).
Albella, J. M., Montero, I. & Martinez-Duart, J. M. A theory of avalanche breakdown during anodic oxidation. Electrochim. Acta. 32, 255–258 (1987).
Thompson, G. E. & Wood, G. C. Porous anodic film formation on aluminum. Nature 290, 203–232 (1981).
Hashimoto, H., Fujita, Y., Yazawa, K. & Asoh, H. Effects of anion incorporation on the local structure of porous-type amorphous alumina prepared by anodization: NMR study of al coordination numbers. Results Phys. 44, 106177 (2023).
Brudzisz, A. M., Gizinski, D. & Stepniowski, W. J. Incorporation of ions into nanostructured anodic oxides–mechanism and functionalities. Molecules 26, 6378 (2021).
Wu, L. et al. A novel double-layer nanotube structure fabricated in high concentration H3PO4 and fluoride-containing mixed electrolyte without annealing. Chem. Phys. Lett. 716, 167–170 (2019).
Yu, M. et al. Cavities between the double walls of nanotubes: evidence of oxygen evolution beneath an anion-contaminated layer. Electrochem. Commun. 90, 34–38 (2018).
Garcia-Vergara, S. J. et al. Incorporation of gold into porous anodic alumina formed on an Al-Au alloy. J. Electrochem. Soc. 155, C333–C339 (2008).
Zhou, X. et al. Morphological development of oxygen bubbles in anodic alumina. J. Electrochem. Soc. 147, 1747–1750 (2000).
Yu, M. et al. Studies of oxide growth location on anodization of al and Ti provide evidence against the field-assisted dissolution and field-assisted ejection theories. Electrochem. Commun. 87, 76–80 (2018).
Zhang, K. et al. Rapid growth of TiO2 nanotubes under the compact oxide layer: evidence against the digging manner of dissolution reaction. Electrochem. Commun. 103, 88–93 (2019).
Zhang, Z. et al. TiO2 nanotube arrays with a volume expansion factor greater than 2.0: evidence against the field-assisted ejection theory. Electrochem. Commun. 114, 106717 (2020).
Baron-Wiechec, A. et al. Tracer study of pore initiation in anodic alumina formed in phosphoric acid. Electrochim. Acta. 113, 302–312 (2013).
Özgür Çapraz, Ö., Shrotriya, P., Skeldon, P., Thompson, G. E. & Hebert, K. R. Role of oxide stress in the initial growth of self-organized porous aluminum oxide. Electrochim. Acta. 167, 404–411 (2015).
He, C. et al. A three-layer zirconia structure composed of nanotubes, dense layer and nanotubes: evidence against the FADT. Ceram. Int. 50, 30906–30911 (2024).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 51777097, 51577093), the National Undergraduate Training Program for Innovation and Entrepreneurship (202510288026Z).
Author information
Authors and Affiliations
Contributions
Pengze Li: Investigation, Writing - review & editing. Liyang Qin: Methodology, Investigation. Bowen Li: Investigation, Writing - original draft. Yijing Tang: Conceptualization. Lianyu Chen: Investigation. Ye Song: Supervision, Formal analysis. Xufei Zhu: Project administration.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, P., Qin, L., Li, B. et al. Double-layer PAA with different pore sizes and its growth kinetics based on anodizing current curves. Sci Rep 15, 21818 (2025). https://doi.org/10.1038/s41598-025-06899-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-06899-6
