
Abstract
The Zika virus (ZIKV) causes acute febrile illness and can lead to complications such as Guillain-Barré syndrome and congenital disorders. As arbovirus outbreaks increase, vaccination becomes a crucial preventive strategy. Currently, no commercial vaccines are available for ZIKV, which is transmitted by mosquitoes and bodily fluids, underscoring the need for a safe vaccine that induces both systemic and mucosal immune responses. In this study, we present a ZIKV vaccine candidate utilizing virus-like particles (VLPs) technology combined with variant-specific surface proteins (VSP) from Giardia lamblia. Previous research demonstrated that these VSP act as effective adjuvants and are resistant to gastrointestinal degradation, expanding administration possibilities via orogastric routes in addition to the conventional subcutaneous route. To develop the immunogen, we engineered retrovirus-derived VLPs decorated with the ZIKV envelope glycoprotein (ZIKV-E) as the target antigen, incorporating VSPs on their surface. Immunocompetent Balb/c mice were immunized with VSP-VLPs ZIKV-E via oral and subcutaneous routes. Immune characterization revealed robust systemic and mucosal humoral responses, as well as a specific cellular activation. Moreover, a significant neutralizing capacity of serum antibodies was observed. These findings highlight the potential of the vaccine candidate to elicit a targeted immune response, achieved through different administration methods.
Similar content being viewed by others


Introduction
In recent decades there has been an unprecedented increase in epidemics caused by a variety of arthropod-borne viruses (arboviruses). Complex factors, including uncontrolled urbanization, geographic expansion of both hosts and vectors, and other environmental, social, and technological changes, have contributed to this rise1. As a result, vectors such as Aedes spp. mosquitoes expand their distribution range and spread arboviruses to different regions of the world2. The recent resurgence of various arboviruses points to a troubling trend, characterized by significant outbreaks of alphaviruses such as Venezuelan3Eastern4and Western Equine Encephalitis5,6and Chikungunya7as well as flaviviruses like Saint Louis Encephalitis8 and Dengue9 in 2024, particularly in South America. In 1947, ZIKV was first isolated from Rhesus monkeys in Africa and, for 60 years, remained in a sylvatic cycle involving mosquito vectors and non-human primates (NHPs), with occasional human infections in Africa and Asia10. Since 2007, a series of epidemics11,12,13 marked a turning point, leading to the virus’s spread in Latin America. The largest outbreak began in Brazil and later extended throughout South America and the Caribbean Islands14,15. Consequently, the World Health Organization declared a global health emergency from February 1 to November 18, 201616. Although autochthonous transmission is currently limited in Europe and North America, there have been reported cases of local transmission, as well as an increase in cases among travelers during large outbreaks17,18,19. The possibility of a shift in this scenario looms due to rising average temperatures in various countries and the adaptation of the recognized ZIKV vectors, Ae. aegypti and Ae. albopictus, to new latitudes and cooler temperatures, respectively20. At present, there is evidence of transmission in 89 countries21 and its unpredictable behavior raises concerns, as studies indicate that by 2050, over 1.3 billion additional people will be at risk of ZIKV exposure22.
In most cases, ZIKV infection is either asymptomatic or causes mild symptoms resembling those of dengue virus (DENV) in 20–25% of individuals10. These symptoms include fever, rash, joint pain, and conjunctivitis. However, the primary concern is its impact on pregnancy, as it is associated with drastic fetal complications, including microcephaly and other neurological malformations, as well as ocular and auditory problems in newborns23,24,25. Collectively referred to as congenital Zika syndrome (CZS), these issues have devastating consequences for child development. Additionally, ZIKV infection has been linked to an increased risk of Guillain-Barré syndrome (GBS) in adults, further adding to its severity26,27. The complexity of ZIKV pathogenesis is further compounded by its multiple transmission modes. Besides being spread by Aedes mosquitoes, the virus can also be transmitted through bodily fluids during sexual intercourse, blood transfusions, and potentially breastfeeding28,29,30,31. This range of transmission mechanisms undermines efforts to control and prevent the infection, pointing to the need for comprehensive strategies to address it effectively.
The genome of ZIKV consists of an approximately 10.8-kilobase positive-sense RNA that undergoes translation into a single polyprotein. This polyprotein is processed into three structural components: capsid (C), membrane (M) formed from the precursor pre-membrane (prM), and envelope (E). These structural proteins play a central role in virus assembly, host cell binding and entry, and determine the overall infectivity and tissue tropism. Additionally, the viral genome encodes seven nonstructural proteins: NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS532, which work together to facilitate genome replication, packaging, and interactions with host pathways, ultimately promoting viral proliferation. The individual structural and nonstructural proteins result from the cleavage of the complete polyprotein through the tightly regulated action of viral and host proteases33. Once assembled, the virions are enveloped in a lipid bilayer derived from the host cell membrane and decorated by 180 copies of E and M proteins. The ZIKV E glycoprotein (ZIKV GpE), which consists of three ectodomains (EDI, EDII, and EDIII), promotes binding to the receptors of target cells during the processes of attachment and viral entry34. Furthermore, the distal tip of EDII contains an internal fusion peptide that, when triggered by the low pH of endosomes, becomes exposed and inserts into the target membrane, initiating the fusion of the viral and endosomal membranes35. Such structural characteristics contribute to the functionality and pathogenicity of ZIKV, underscoring the importance of understanding its molecular makeup for effective therapeutic interventions, as these domains are targets of neutralizing antibodies (nAbs) against ZIKV35,36.
The health impacts associated with ZIKV infection position it as a significant concern, making it imperative to address the challenges that arise37. In this context, advancing the research and development of ZIKV vaccines is a key component of strategies for controlling and preventing the virus, safeguarding global public health. An effective vaccine could reduce ZIKV transmission and suppress the progression of the infection and associated complications, thereby lessening the severity of potential future outbreaks38. Correlates of protection for flaviviruses demonstrate that the generation of nAbs is indispensable for achieving immunity, as evidenced by the efficacy of authorized immunogens, including the yellow fever virus (YFV) vaccine39 and the tetravalent dengue vaccine TAK-00340. Specifically for ZIKV, GpE is regarded as a promising vaccine target because of its roles during the viral infection cycle and its capability to induce the production of nAbs35,36. Several technologies are in development to elicit an effective immune response against ZIKV, and subunit vaccines are emerging as a promising strategy. Among these, virus-like particles (VLPs) stand out for their high efficacy and safety. VLPs can mimic, at least partially, viral surface structures and lack the ability to infect and replicate, making them an attractive option for vaccine development. The VLP-based approach is likely to effectively present the native E: E dimer antigens on its surface repetitively within the vaccine components, thus bolstering its potential efficacy against ZIKV41,42. Additionally, VLP-based vaccines simplify logistics by not requiring the − 20 to -80 °C freezing conditions needed for mRNA vaccines43.
Currently, the development of ZIKV vaccines primarily focuses on parenteral administration, as the most advanced candidates in clinical trials fall into this category44,45,46. In contrast, vaccines administered through alternative routes, such as oral or nasal47,48remain relatively rare. Since ZIKV is transmitted not only through vectors but also through bodily fluids a vaccine that induces both systemic and mucosal immune responses would be highly beneficial. An integrated approach that takes these factors into account will address the virus’s multiple transmission routes and enhance global public health protection.
Previously, a vaccine platform based on VLPs decorated with variant-specific surface proteins (VSP) from the protozoan Giardia lamblia was developed in our laboratory using the influenza virus as an infection model49. These surface proteins play a crucial role in evading the immune system through antigenic variation50. Additionally, they facilitate adhesion to the intestinal surface of the host and provide protection against adverse conditions, such as extreme pH environments and exposure to proteolytic enzymes in the upper small intestine49. Thus, the study demonstrated that VSPs can withstand the harsh conditions of the digestive tract, protect heterologous antigens, and exert a robust adjuvant effect that supports both humoral and cellular immune responses.
In the present study, we demonstrate that incorporating ZIKV GpE into VSP-VLPs enabled the proper presentation of the vaccine antigen, eliciting a robust humoral and cellular immune response in Balb/c mice following both subcutaneous (sc) and oral administration, and notably induced a high titer of neutralizing antibodies.
Results
Production of VSP-VLPs ZIKV-E
Many studies have demonstrated that ZIKV GpE plays a crucial role in inducing immunity against infection35,36. Thus, we developed a construct by cloning this glycoprotein and replacing its transmembrane region by the cytoplasmic tail of the VSV-G (designated pZIKV-E, Fig. 1a). This modification was made to enhance interaction through the VSV-G transmembrane domain, which improves the proper assembly of ZIKV-E with the VSP-VLPs structure. Our previous research demonstrated that this approach effectively directs heterologous proteins to the surface of VLPs49. After transfection of VSP-expressing cell line with pZIKV-E, direct immunofluorescence technique was employed to validate the accurate expression of the vaccine antigen (Fig. 2a). Additionally, it was confirmed that the ZIKV-E expression is effective in the presence of MLV-Gag. This evaluation was conducted through a co-transfection assay (pGag and pZIKV-E) and simultaneously with the stable expression of VSP1267 in VSP-expressing cells (Fig. 2a).

Schematic representation of the ZIKV immunogen and immunization protocol. (A) Organization of the genomic construct containing the signal peptide (SP), ectodomain Zika virus envelope (E), and the Vesicular Stomatitis Virus G glycoprotein transmembrane and cytosolic tail (VSVg TM-CT). (B) Overview of the experimental design in mice, including immunization, dosing and sampling schedules for oral and subcutaneous administration.
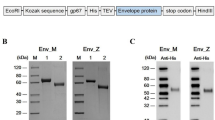
MLV-Gag expression leads to the generation of genome-free retroviral particles. When combined with the pZIKV-E construct in the VSP-expressing cell line, these particles should assemble into VSP-pseudotyped VLPs containing ZIKV-E. Upon transfection of VSP-expressing cells with pGag and pZIKV-E, the production and subsequent secretion of VSP-VLPs ZIKV-E into the supernatant was analyzed. Following the harvest and purification of the supernatants, the different components were evaluated to ensure the correct production of the immunogen. Transmission electron microscopy (TEM) served to elucidate the self-assembly of the VSP-VLPs ZIKV-E and the incorporation of the vaccine antigen on the surface of these chimeric nanoparticles. Morphological analysis revealed predominantly rounded shapes encapsulated by a lipid bilayer, with size variations ranging from 120 to 130 nm, as illustrated in Fig. 2b. To achieve a deeper understanding of the antigenic composition, ZIKV-E, MLV-Gag and VSP1267 proteins were identified through immunogold labeling (Fig. 2b, arrows). Additionally, western blotting was performed to confirm the presence of ZIKV-E on the VLPs (Fig. 3). A band was detected at approximately 58 kDa, consistent with the expected molecular weight of the full-length envelope protein. A second band of lower molecular weight was also observed. Since this band was recognized by the monoclonal anti-ZIKV-E antibody, it likely retains at least one epitope of the E protein. This signal may represent a degradation product, an incompletely processed form, or a conformational variant affecting its migration in SDS-PAGE.

Expression and assembly of VSP-VLPs ZIKV-E. (A) Fluorescence microscopy was conducted after transfecting VSP-expressing cells with the DNA constructs pZIKV-E and/or pGag. The expression of each specific protein is indicated at the top of each image, using the corresponding fluorescence color. Cells were analyzed for the expression of ZIKV-E and VSP proteins after incubating with the respective specific antibodies, followed by a fluorophore-conjugated secondary antibody. The expression of MLV-Gag was visualized by the fluorescence emitted from eYFP. In all fluorescence images, nuclei are labeled with DAPI (blue), and the yellow signal indicates co-expression. (B) Purified VSP-VLPs ZIKV-E were examined through transmission electron microscopy (TEM) and immunogold labeling, which confirmed the proper display of ZIKV-E (left), VSP (center) and MLV-Gag (right). Specific monoclonal antibodies were employed to identify the various components present in the particles. Immunogold-positive signals (black dots) corresponding to the specific markers are indicated by blue arrows.

Detection of ZIKV-E from purified VSP-VLPs by Western blot. Purified VSP-VLPs ZIKV-E and VSP-VLPs without ZIKV-E (Ctrl) were resolved by SDS-PAGE, under reducing (5% β-ME) conditions, followed by immunodetection using anti-Zika envelope-specific antibody. The protein molecular weight marker (MW) is indicated on the left. The upper band (~ 58 kDa, red arrow) corresponds to the full-length ZIKV-E protein. A lower band (blue arrow), also recognised by the antibody, is marked as a putative truncated or degraded form of ZIKV-E.
VSP-VLPs ZIKV-E are immunogenic in a murine model
Our research on the vaccine’s immune response assessed two different administration routes: sc and oral. These routes were systematically compared to an untreated control group. We conducted a thorough analysis of both humoral and cellular immune responses in immunocompetent Balb/c mice following the inoculations (Fig. 1b). Animals immunized with VSP-VLPs ZIKV-E exhibited no observable symptoms or adverse effects, including inflammation, swelling, weight loss, stress, reduced activity, or lethargy.
To study the induction of a specific humoral immune response, our research involved analyzing different isotypes of antibodies specific to ZIKV in the serum and mucosal tissues of mice vaccinated with VSP-VLPs ZIKV-E. All of them were quantified using ELISA with the ZIKV-AR isolate as antigen. To evaluate the dynamics of the predominant isotypes in serum following each dose, a kinetic study of IgG + IgM was conducted. In the group immunized via the oral route, serum IgG + IgM levels increased progressively throughout the vaccination schedule. Fourteen days after the first dose (14 d.p.i.), a marked rise in antibody levels was already observed (mean OD₄₀₅= 0.394) compared to baseline values (mean OD₄₀₅= 0.243). Following administration of the second dose on day 14, antibody levels continued to rise, reaching a mean OD₄₀₅ of 0.516 by 28 d.p.i. Finally, fourteen days after the third dose (42 d.p.i.), serum IgG + IgM levels peaked at 0.688, representing an increase of more than 2.8-fold relative to baseline (0 d.p.i.). In the group immunized via the subcutaneous route, serum IgG + IgM levels showed a moderate but steady increase over the course of the immunization schedule. A slight rise was observed fourteen days after the first dose (14 d.p.i.) (mean OD₄₀₅= 0.307) compared to baseline levels (mean OD₄₀₅= 0.268, 0 d.p.i.). Following the second dose on day 14, antibody levels increased further, reaching a mean OD₄₀₅ of 0.368 by 28 d.p.i. The response peaked at 42 d.p.i. (14 days after the third dose), with a mean OD₄₀₅ of 0.501, approximately 1.9-fold higher than baseline. The titres at the end of the kinetics were predominantly 1/640 or higher, reflecting the capacity to achieve high titres with both oral and sc administration (Fig. 4b). To identify a potential predominant T helper (Th) response profile, we analyzed the distribution of IgG subclasses induced by the vaccine. Both administration routes elicited a significant IgG1 response, while IgG2a levels remained low (Fig. 4c–d). Additionally, IgA was determined in the serum, demonstrating a significant increase through both administration routes (Fig. 4e).

Induction of anti-ZIKV systemic humoral response in mice vaccinated with VSP-VLPs ZIKV-E. (A) Mice were vaccinated orally or subcutaneously with VSP-VLPs ZIKV-E and the specific anti-ZIKV IgG plus IgM were determined in sera 14 days after each dose. (B) Serum IgG plus IgM titer was evaluated at 42 days post immunization (d.p.i.) (C) Serum levels of IgG1 subclass were measured at 42 d.p.i. (D) Serum levels of IgG2a subclass were measured at 42 d.p.i. (E) Serum IgA levels were assessed at 42 d.p.i. n = 6 from two independent experiments for VSP-VLPs ZIKV-E vaccinated groups; n = 5 for mock group. Data are shown as means ± SEM. In the antibodies kinetic response (A) data were analyzed by two-way ANOVA and Bonferroni’s multiple comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 respect to mock group at the same d.p.i.
Mucosal vaccination, administered orally in this study, not only elicits systemic immune responses, as demonstrated by serum samples, but is also expected to induce strong local responses in mucosal fluids. At the end point of the vaccination assay, vaginal washes were conducted to obtain these fluids and determine specific IgA and IgG levels against ZIKV. Vaginal IgG + IgM showed a significant increase only in the oral route and a positive trend in the sc one (Fig. 5A). Regarding mucosal IgA, the findings revealed that oral immunization with VSP-VLPs ZIKV-E significantly increased IgA levels in vaginal and faecal extracts (Fig. 5B and C, respectively).

Specific anti-ZIKV mucosal response after VSP-VLPs ZIKV-E immunization. Mice were vaccinated orally or subcutaneously with VSP-VLPs ZIKV-E and the specific anti-ZIKV antibodies were determined in mucosal samples 14 days after the last dose (42 d.p.i.). (A) IgG + IgM levels in vaginal swabs were measured. (B) Specific IgA levels in vaginal swabs. (C) Specific IgA levels in faecal samples. n = 6 from two independent experiments for VSP-VLPs ZIKV-E vaccinated groups; n = 5 for mock group. Data are shown as means ± SEM. *p < 0.05, **p < 0.01.
VSP-VLPs ZIKV-E triggers cellular immune responses
To measure the T cell responses induced by immunization, spleens were collected and different cytokines indicating Th1/Th2/Th17 profiles were measured. The immunized animals demonstrated a strong cellular immune response, as shown by the production of specific cytokines in response to ZIKV stimulation of splenocytes (Fig. 6). Among the findings, IL-10 levels did not differ significantly between groups. Although IL-4 showed statistically significant differences, the absolute values were low and not biologically meaningful. Although IL-2 and IL-6 levels did not reach statistical significance, an upward trend was observed in both vaccinated groups compared to the mock group. A marked increase in IL-17 A release was detected exclusively in the orally immunized group. IFN-γ levels, as well as the IFN-γ/IL-10 ratio, were significantly elevated in both immunized groups. Meanwhile, TNF-α exhibited a trend toward increased levels in the oral group.

Induction of anti-ZIKV systemic cellular immune response in mice vaccinated with VSP-VLPs ZIKV-E. Balb/c mice were vaccinated orally or subcutaneously with VSP-VLPs ZIKV-E and the cellular immune response against ZIKV was evaluated 14 days after the last dose (42 d.p.i.) by the measurement of cytokines in splenocyte supernatants. n = 6 from two independent experiments for VSP-VLPs ZIKV-E vaccinated groups; n = 5 for mock group. Data are shown as means ± SEM. *p < 0.05, **p < 0.01.
Antibodies elicited by the VSP-VLPs ZIKV-E vaccine neutralize ZIKV
The neutralization assay is widely regarded as the “gold standard” for assessing circulating levels of nAbs against flaviviruses. To this end, we employed the MNT50 and MNT80 assays with minor modifications51,52 (Fig. 7A and B). These techniques measure the serum titre required to neutralize 50% or 80% of the virus, respectively. In our study, we specifically evaluated the neutralizing efficacy of sera derived from both oral and sc administration of the VSP-VLPs ZIKV-E vaccine against the ZIKV-AR. All animals that received the vaccine orally successfully generated antibodies capable of neutralizing at least 50% of the virus, with 5 out of 6 achieving 80% neutralization. In parallel, for sc administration, 4 out of 6 animals demonstrated neutralization of both 50% and 80% of the virus.

VSP-VLPs ZIKV-E immunization increases neutralizing antibodies. Balb/c mice were vaccinated orally or subcutaneously at days 0, 14 and 28 with VSP-VLPs ZIKV-E. Neutralizing antibody endpoint titers were determined in serum samples collected 14 days after the final dose using ZIKV-AR. The neutralization titer was defined as the serum dilution that reduces the cytopathic effect by 50% (MNT50) (A) or 80% (MNT80) (B). A total of n = 6 animals were analyzed from two independent experiments in the VSP-VLPs ZIKV-E vaccinated groups, while n = 5 animals were analyzed from the mock group. Data are shown as means ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001.
Discussion
The severe consequences of ZIKV, such as congenital CZS and GBS53,54coupled with its diverse transmission pathways—including biological fluids and intrauterine routes—highlight the urgent need for effective preventive strategies. Intensive research is currently focused on various drug and vaccine candidates. Among these, TAK-426, a purified inactivated Zika vaccine, has shown promising results in Phase II clinical trials55. The VSP-VLPs platform was previously developed in our laboratory using influenza virus antigens as a model49. This study not only demonstrated the platform’s efficacy against this specific virus but also highlighted its significant potential for various applications in vaccine development against other enveloped viruses, paving the way for future research. Building on this foundation, we adapted these chimeric particles to another class of enveloped viruses, specifically flaviviruses.
Various types of VLPs exist, and for this study, we selected retrovirus-derived VLPs composed of MLV-Gag. This choice has been made based on their ability to assemble with the VSP49resulting in the VSP-VLPs, which provide an adjuvant and protective effect to the platform. In contrast, the development of VLPs as immunogens against flaviviruses typically relies on the use of their own structural proteins, and only a few studies have explored retrovirus-derived VLPs41,56. The approach proposed in our work presents a challenge in ensuring immunogenicity, as the particle is formed using MLV-Gag instead of the native structural proteins of the virus. To assess the feasibility of co-expression, we performed co-transfection assays, which demonstrated the localization of ZIKV-E and MLV-Gag at the periphery of the cells, as indicated by the fluorescent signal. This finding suggests a localized concentration of both proteins in the plasma membrane, which serves as the primary site for VLPs assembly (Fig. 2a). Moreover, the correct budding of complete particles was validated by immunoelectron microscopy of purified VSP-VLPs (Fig. 2b). Western blotting further confirmed that the ZIKV-E antigen displayed on these particles had a molecular weight consistent with the ZIKV GpE fused to the cytosolic tail and transmembrane domain of VSV-G (~ 58 kDa, Fig. 3), thereby also verifying the identity of the antigen. Concerning particle size, TEM images indicated dimensions that align with those reported for other flaviviral VLPs assembled with MLV-Gag56. These VLPs, larger than native ZIKV particles34owe their size to the structural properties of the MLV-Gag protein, since the retroviral capsid domain is crucial in determining virion morphology56,57,58,59. The TEM images also revealed the presence of VSP1267 and ZIKV-E within an intact lipid membrane (Fig. 2b). Altogether, these observations confirm that the VSP-VLPs can effectively incorporate ZIKV-E as an antigen, allowing us to evaluate its potential as a vaccine candidate against ZIKV.
Characterizing the response to an experimental vaccine requires the use of animal models that represent the repertoire of interactions between the administered antigen and the components of the innate and adaptive immune systems. For this reason, we used immunocompetent adult BALB/c mice, as has been done in similar studies60,61. Here, we analyzed the immune response generated after inoculating VSP-VLPs ZIKV-E through two administration routes: the classic sc and the oral route, which we specifically explored to leverage the distinctive capabilities of the platform (Fig. 1b).
Specific total antibodies against ZIKV provide an overall measure of the humoral immune response, serving as an indicator of the magnitude and breadth of antibody production following vaccination. Based on the kinetic study showing an increase in serum concentrations of IgG and IgM antibodies after each dose, our research underscores the necessity of administering three doses to achieve a substantial immune response (Fig. 4a). This aligns with findings from another mucosally administered ZIKV vaccine model62. In addition to the rise in total antibodies, a significant increase in IgG1 levels was observed (Fig. 4c), a subclass typically associated with Th2-skewed immune response63,64. This IgG1-dominant profile is consistent with findings from other studies evaluating ZIKV-specific vaccines52,65and may reflect common immunological features associated with subunit-based approaches.
Understanding cellular immunity is key to revealing the integral immune response triggered by vaccines. Significant parallels are observed between serological findings and the diverse functional responses elicited by T lymphocytes. However, the serological profile observed in our study (Fig. 4c and d), characterized by a predominance of IgG1, contrasts with the cytokine data obtained from restimulated splenocytes, where no biologically meaningful increase in IL-4 (a signature Th2 cytokine) was detected. Although IL-4 levels were slightly but significantly higher in the vaccinated group (~ 15 pg/mL) compared to controls (~ 14.4 pg/mL), the minimal absolute difference suggests that the response lacks functional relevance. This discrepancy may indicate that the IgG1-skewed humoral response does not necessarily reflect a canonical Th2 polarization at the cellular level. In this context, T cells play a pivotal role in controlling flavivirus infections by enhancing viral clearance and limiting disease severity, highlighting the importance of evaluating cellular immune responses beyond antibody profiles alone66,67,68. For instance, T-cell responses are key to controlling DENV infections and reducing the risks associated with antibody-dependent enhancement (ADE)69. In the context of immunizations, a robust T-cell response plays a clear role in protection following the administration of the yellow fever virus vaccine YFV-17D70. Cytokines shape the immune profile by regulating T-cell responses, making them a focus of study in both natural infection and vaccine development. In this regard, interferon-gamma (IFN-γ) induction is a major contributor to the establishment of a Th1 profile. Its production is particularly significant during immunization, as demonstrated by its role in generating robust immune responses to the YFV-17D vaccine, thereby enhancing its immunogenicity71,72. Several studies have demonstrated that CD8 + T cell populations can effectively control ZIKV infection and reduce mortality, primarily through the production of IFN-γ73,74. Likewise, the development of effective vaccines against ZIKV relies on the role of IFN-γ, among other parameters75. This is further supported by findings showing that passive immunization with serum alone was insufficient to protect mice, as viral clearance occurred only in those animals where the production of IFN-γ and TNF-α was combined with humoral immunity76. Our findings revealed that VSP-VLPs ZIKV-E triggered IFN-γ production in both the oral and sc administration groups (Fig. 6) and, although not statistically significant, TNF-α levels also showed a trend toward elevation in the orally immunized group.
A broader cytokine response was also evident, with significantly elevated IL-17 A levels in the oral group (Fig. 6). IL-17 A is a pleiotropic cytokine that exerts functions in the contexts of infections, inflammation, and autoimmune diseases77,78. Within the proinflammatory microenvironment, IL-17 A plays multiple roles, sometimes contributing to an antiviral immune response, while at other times promoting the development of severe disease during infection. These divergent effects can be observed even among related viruses within the Flavivirus genus. For example, in humans infected with DENV, IL-17 A overexpression is indirectly associated with the development of severe forms of the disease79,80. Conversely, in humans and mice infected with West Nile virus, IL-17 A facilitates viral clearance through CD8 + T cell-mediated cytotoxicity81. IL-17 A, along with some of its downstream ligands, significantly increases during the acute phase of ZIKV infection in humans82playing a role in the neuroinflammatory processes that lead to microcephaly83. Further detailed studies are needed to fully understand the mechanisms by which this cytokine might contribute to the protection elicited by this vaccine, especially considering that a similar cytokine profile was observed when the VSP-VLPs platform was used in an oral vaccine against the influenza virus49. Likewise, a notable increase in IL-17 was reported following the oral vaccination of gerbils against Giardia lamblia using a cocktail of VSPs from this protozoan84. These findings suggest that VSPs may play a significant adjuvant role in inducing immune responses that enhance the observed cellular profile. Based on the findings of other authors and our results, it is suggested that this interaction is crucial for fostering an effective immune response, especially in infections where Th17 cells play a key role in inflammation and in the clearance of pathogens at mucosal surfaces85,86. Furthermore, IL-17-producing Th17 cells have been implicated in the IgA class switch in plasma cells, optimizing secretory IgA production87,88. On the other hand, Th17 cells differentiate in part due to IL-6 activity89even, some studies have linked increases in IL-17 A with elevated levels of IL-6 during DENV and ZIKV infections90,91. Although IL-6 levels did not reach statistical significance in our experimental setting, both vaccinated groups exhibited a trend toward increased levels compared to the mock group, despite the high variability within the data (Fig. 6). Altogether, our results do not support a clearly defined immune polarization pattern but rather suggest that the vaccine induces a mixed or polyfunctional immune activation, providing a valuable foundation for future studies aimed at elucidating the underlying mechanisms.
In developing immunogens against ZIKV, it is important to ensure the classical protection correlates for flaviviruses, while also acknowledging the role of ZIKV sexual transmission, which accounts for approximately 3–5% of infections92,93. Although there are few studies focused on protection correlates for this route of transmission, some have shown significant advances. Particularly, one study demonstrates that the passive transfer of ZIKV-specific IgG significantly reduces intravaginal infection in previously unexposed mice94. This result illustrates the relevance of IgG, the predominant isotype in vaginal secretions in both mice and humans, which has proven effective in preventing the dissemination of the virus in the reproductive tract95. Additionally, other studies have investigated how mucosal vaccines against ZIKV induce the production of IgA, contributing to an effective immune response62. We observed that both immunoglobulins showed a significant increase following oral immunization (Fig. 5a and b), suggesting that oral vaccination is a promising approach with immunogenic effects on the vaginal mucosa, potentially limiting viral replication in this area. In the context of sc inoculation, only one immunoglobulin exhibited a statistically significant increase: IgA (Fig. 5b). It is also relevant to consider the role of other mucosal sites in the immune response, particularly given that immunization through the rectal mucosa with an attenuated ZIKV strain has been previously demonstrated to result in effective protection against a lethal sc challenge47. This immunization also elicited robust memory CD4 + and CD8 + T cell responses, as well as ZIKV-specific nAbs47. We specifically assessed the production of ZIKV-specific IgA antibodies in the intestinal mucosa for both routes of administration and observed statistically significant results (Fig. 5c). These findings provide insight into how local immune responses in mucosal tissues can be induced through vaccination to protect against ZIKV sexual transmission or through fluids.
The infectivity and virulence of ZIKV rely largely on the structural properties of GpE. In particular, the configuration of its different domains is important for triggering neutralizing antibody responses35,36which is one of the main objectives in vaccine design for achieving effective infection control65. Although this work does not provide information on the precise structure adopted by ZIKV GpE in VSP-VLPs -since we did not conduct structural studies or apply stabilizing mutations to the protein, as seen in other research96,97,98- we can still draw some conclusions. The conformation observed in retrovirus-derived VLPs is likely adequate for proper epitope exposure, which may explain the induction of high titres of nAbs following either oral or sc administration (Fig. 7). The observed titres exceeded the protective threshold of 1:10 by a considerable margin, which is commonly used as a reference point for flavivirus vaccines44,99. This suggests that the potential efficacy of the vaccine could be comparable to that of licensed flavivirus vaccines.
Despite significant advancements in research, developing a safe and effective vaccine against ZIKV remains a major challenge due to the ongoing risk of outbreaks and the lack of immunization strategies. To address this, efforts should prioritize regions with high virus circulation, ensuring they have the necessary resources for research, development, and the implementation of preventive measures. The endemic nature of ZIKV in various regions, along with the challenges it poses for travel medicine, highlights the need to develop effective vaccines and implement both local and regional tools to combat the disease. Local research and production support timely responses, strengthens healthcare infrastructure, and improves preparedness for future public health crises. In this regard, the socio-economic situation of developing countries gives efforts like the present work a particular perspective, within a context of historical technological dependence on central nations when addressing our public health emergencies.
Our research demonstrates that the VSP-VLPs ZIKV-E vaccine is effective through both administration routes; oral delivery, in fact, has an even greater effect on the immune system. Notably, this approach leverages technology that may be particularly well-suited for immunocompromised individuals and pregnant women, who are at higher risk of severe outcomes from Zika infection. These vulnerable populations should be key targets in vaccine development due to their increased susceptibility. Attenuated virus vaccines are generally not recommended for these groups, as they require significant precautions, making the VSP-VLPs technology a potentially safer and more suitable option among other alternatives. The efficacy of the VSP-VLPs platform against ZIKV underscores its versatility, making it a strong candidate for broader and more accessible immunization strategies.
The further development and the eventual implementation of this platform will require careful consideration of various factors. The demonstrated preclinical robustness not only supports the pursuit of trials in lethal murine models, such as AG129 or IFNAR−/−100,101,102, which are susceptible to viral challenges, but also establishes a framework for investigating different protective pathways. Also, a detailed characterization of the structure and functionality of the VSP used in this study should be achieved. Previous studies have suggested that the resistance of this protein to the different proteolytic environments is attributed, at least partly, to the CxxC motifs in the VSP49; however, many questions remain regarding the structure of this protein and how it confers its adjuvant and protective effects for use in vaccines. Finally, establishing a safe profile for the vaccine is one of the key challenges in developing the platform. Various studies have demonstrated the necessity of introducing strategies that involve structural mutations of the flaviviral antigen96,97,98 to prevent the production of sub neutralizing cross-reactive antibodies which could lead to antibody-dependent enhancement of infection (ADE).
In a global context where emerging and re-emerging viral infections pose an increasing threat, this vaccine represents a contribution to situated and time-sensitive efforts to mitigate these challenges. Rather than considering vaccine development an arms race, we emphasize the importance of context-specific responses that address immediate public health needs, while also acknowledging the necessity for deeper, systemic reflections on the historical and structural roots of many issues, including those caused by viral diseases.
Materials and methods
Genes and plasmid constructs
The ectodomain and part of the stem of the ZIKV GpE gene were amplified from a PCR product containing the sequence of the structural proteins of the ZIKV-71,516 human isolate from Argentina, kindly provided by Dr. Andrea Gamarnik from the Instituto de Investigaciones Bioquímicas de Buenos Aires (IIBBA). The forward primer was designed to incorporate a signal peptide from the final transmembrane domain of the membrane protein (VIYLVMILLIAPAYS), ensuring the proper folding and secretion of the E protein from the lumen of the endoplasmic reticulum103. The sequences of the oligonucleotides are as follows: forward primer (Fw) - TCTCTGAAGCTTACCATGGTCATATACTTGGTCATGATACTG, and reverse primer (Rv) - CGCGGATCCTGATTTGAAAGCTGCTCCAAAAAT. To be amplified by PCR, Q5® High-Fidelity DNA Polymerase (New England Biolabs, M0491S) was used following the manufacturer’s instructions. The PCR product was subcloned into the pcDNA3.1 + expression vector (Invitrogen) between the BamHI and HindIII restriction sites. Instead of using the ZIKV GpE transmembrane domain, the transmembrane domain of the vesicular stomatitis virus glycoprotein (VSV-G) was employed (Fig. 1a)49. The glycoprotein resulting from this construct was named ZIKV-E. The cDNA sequence encoding the Gag capsid protein of the Murine Leukemia Virus (MLV-Gag) (UniProt: P0DOG8.1) was cloned into the phCMV expression vector with enhanced yellow fluorescent protein (eYFP), resulting in pGag49. The plasmids were amplified in competent E. coli DH5-α bacteria and subsequently purified using an EndoFree Plasmid Midi Kit (Qiagen®).
Cells and virus
African Green Monkey kidney cells (Vero E6) were obtained from ATCC®. The Flip-In™ -293 system (Invitrogen, R75007) was used to generate a cell line that stably expresses the extracellular portion of the VSP 1267 of G. lamblia (GenBank: M63966.1/VSP1267) fused to the transmembrane domain and the cytoplasmic tail of the VSV-G, hereafter referred to as VSP-expressing cells49. Both cell lines were maintained at 37 °C in a 5% CO2 environment, in Dulbecco’s modified Eagle medium (DMEM, Gibco) supplemented with 2 mM L-glutamine, Penicillin-Streptomycin-Amphotericin B (Antibiotic-Antimycotic 100X, Gibco), and 10% heat-inactivated fetal bovine serum (FBS, Gibco). Regular testing was conducted on all cell lines, which consistently showed negative results for Mycoplasma spp. Spleen cells were cultured in plates containing RPMI 1640, 10% FBS, Penicillin-Streptomycin-Amphotericin B Suspension (Antibiotic-Antimycotic 100X, Gibco). The ZIKV clinical isolate INEVH116141 (ZIKV-AR), kindly provided by Dr. Carlos Bueno from the Instituto de Química Biológica de la Facultad de Ciencias Exactas y Naturales (IQUIBICEN), was propagated by inoculating a monolayer of Vero E6 cells at a MOI of 0.01. Supernatants were harvested at 72 and 120 h post-infection. Virus stocks were titrated by focus-forming assays in Vero E6 cells and stored at − 80 °C for subsequent use in microneutralization assays. All procedures involving the manipulation of infectious viral particles were carried out under strict biosafety level 2 conditions with enhanced practices.
Mice
Female immunocompetent BALB/c mice, aged 8 to 10 weeks, were employed in the experiments, bred and housed under specific pathogen-free (SPF) conditions in the CIDIE vivarium. Mice were euthanized following an experimental endpoint criterion. After induction of anesthesia by isoflurane inhalation (2%), euthanasia was performed by cervical dislocation. All animal procedures were approved by the Institutional Animal Care and Use Committee of the Catholic University of Córdoba (CICUAL-UCC; 15/2019), ensuring the ethical and transparent reporting of in vivo research, in compliance with the ARRIVE guidelines.
Immunofluorescence
After 48 h of plasmid transfection, the cells were fixed and permeabilized by incubation in acetone and methanol solution at 4 °C. Subsequently, specific antibodies were used for labeling: monoclonal antibody (mAb) 7F5 for VSP1267 (1:500)49 and anti-ZIKV GpE mAb (R&D Systems, MAB10009,1:300). Goat anti-mouse IgG Alexa Fluor™ 546 (Thermo Fisher, A-11030, 1:3000) and goat anti-mouse polyvalent immunoglobulins FITC (Sigma-Aldrich, F1010, 1:3000) were used for immunofluorescence detection. DNA staining was performed with DAPI, and images were captured using a Hamamatsu ORCA ER-II camera mounted on a Leica IRBE inverted fluorescence microscope (N.A. 1.40).
VSP-VLPs ZIKV-E production
VSP-VLPs ZIKV-E were produced by transient transfection of VSP-expressing cells with the vectors pGag and pZIKV-E, using polyethylenimine, PEI MAX® (Polysciences, 24765), as the transfection reagent. Cell cultures at 70% confluence were transfected in T175 flasks with 70 µg of total DNA per flask at a PEI: DNA mass ratio of 3:1. The supernatant was harvested 72 h after transfection, and then clarified by centrifugation at 3,200 × g for 30 min at 4 °C. Afterward, it was filtered through a 0.45 μm pore size membrane and concentrated 20x in a centrifugal filter device Centricon® Plus-70–100 K (Millipore, UFC710008).
Purification of virus and VSP-VLPs ZIKV-E
The viral stocks and the supernatants containing VSP-VLPs ZIKV-E were ultracentrifuged through a 20% sucrose cushion in an SW41T Beckman rotor (100,000 × g, 4 h, at 4 °C). Pellets were resuspended in sterile TNE buffer (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 0.1 mM EDTA), and total protein content was quantified using the Bradford assay.
Western blotting
The identity of ZIKV-E from VSP-VLPs ZIKV-E particles was confirmed by SDS-PAGE and Western blotting. Samples were conditioned by dissolving them in a reducing buffer containing 62.5 mM Tris-HCl (pH 6.8), 10% glycerol, 2% SDS, and 5% β-mercaptoethanol, with 0.01% bromophenol blue added as a loading dye. Following this, the samples were incubated at 95 °C for 5 min to denature the proteins and to break the disulfide bonds. The samples were resolved by 12% SDS-PAGE and transferred onto PVDF membranes (GVS, 7064555). The membranes were then incubated with the specific primary anti-ZIKV GpE mAb (R&D Systems, MAB10009, 1:300). For detection, an alkaline phosphatase-conjugated goat anti-mouse IgG secondary antibody (Molecular Probes, G-21060, 1:2000) was used, along with the BCIP/NBT substrate.
Transmission electron microscopy (TEM)
The ultracentrifuged VSP-VLPs ZIKV-E were prepared for TEM using negative staining and immunogold labeling by resuspending them in 2% w/v paraformaldehyde, and 7 µL of preparation was dropped onto formvar-carbon-coated electron microscopy grids and post-fixed in 1% w/v glutaraldehyde. The grid was washed with distilled water, and then the samples were contrasted in 2% w/v uranyl oxalate (pH:7) followed by 2% w/v methyl cellulose for negative contrasting. Then, fixed VSP-VLPs ZIKV-E were incubated with the anti-ZIKV GpE mAb (R&D Systems, MAB10009, 1/100), 7F5 mAb (1/100), and R187 mAb (ATCC® CRL-1912™, 1/1000). The immunoreactive sites were labeled with rabbit anti-mouse secondary antibody conjugated to 15-nm colloidal gold particles (Electron Microscopy Sciences, Hatfield, PA, USA, 1:20) or protein A conjugated to 15-nm colloidal gold particles (1:15). The images of VSP-VLPs ZIKV-E were captured using an NS 15 AMT camera on a Hitachi HT 7800 electron microscope operated at 80 kV.
Evaluation of immunogenicity in mice
Female BALB/c mice (n = 6 each) were randomly allocated to two different experimental groups (n = 6 each). One group underwent sc immunization, while the other group was immunized orally, receiving three doses with a two-week interval, with doses of 20 µg (sc) and 120 µg (oral) per mouse (Fig. 1b). Mice receiving oral immunization fasted for 4 h before administration. A control group (mock, n = 5) received PBS via the subcutaneous route, following the same immunization schedule. Two weeks after the final booster immunization, animals from all groups were euthanized, and serum samples, along with spleen, fecal samples, and vaginal washes, were collected for multiple immunogenicity assays.
Isolation of splenocytes and cytokine quantification
Cell suspensions from the spleen were generated and cultured on 96-well plates for 72 h in the presence of PBS or ZIKV-AR104. The levels of cytokines in the supernatants were measured using the CBA Mouse Th1/Th2/Th17 kit (BD Biosciences, 560485), following the provided protocol. This kit allows the simultaneous quantification of seven cytokines: IL-2, IL-4, IL-6, IFN-γ, TNF, IL-17 A, and IL-10.
Fluid collection
Blood was collected via submandibular puncture the day before each immunization and on the study endpoint day. After coagulation, the samples were centrifuged 6,000 × g for 15 min at 4 °C, and the sera were harvested, aliquoted, and stored at − 80 °C until needed. To obtain vaginal wash samples, fluid was collected from the vaginas of female mice through a lavage procedure performed three times with 35 µl of sterile PBS containing protease inhibitors. These washes were carried out both before immunization and at the endpoint of the study. Subsequently, the samples were subjected to centrifugation at 1,500 × g for 15 min at 4 °C, and the resulting supernatants were collected and stored at − 80 °C until further use. Fecal pellets were resuspended in PBS containing protease inhibitor (Complete® Protease Inhibitor Cocktail; Roche), at a ratio of 0.1 g per 500 µl. After 30 min on ice, the mixture was centrifuged at 10,000 × g and the supernatant was stored at − 80 °C.
Enzyme-linked immunosorbent assay (ELISA)
ELISA was employed to quantify antibody levels specific to ZIKV, including total IgG + IgM, IgG1, IgG2a, and IgA. This ELISA protocol was adapted from previously established laboratory methods104. ZIKV-AR viral particles were purified by ultracentrifugation through a 20% sucrose cushion using a Beckman SW41T rotor (100,000 × g, 4 h, 4 °C), inactivated at 60 °C for 30 min, and quantified using the Bradford assay. Flat-bottom 96-well polystyrene plates were coated overnight at 4 °C with inactivated ZIKV-AR particles at a final concentration of 10 µg/mL, diluted in carbonate-bicarbonate buffer (1.5 g/L sodium carbonate and 2.93 g/L sodium bicarbonate in distilled water, pH 9.6). After coating, plates were washed with PBS-Tween 0.05% and blocked for 2 h at room temperature (RT) with assay buffer consisting of PBS supplemented with 10% fetal bovine serum (FBS). Samples were diluted in assay buffer and incubated in the plates for 90 min at RT. Mouse serum and fecal samples were diluted 1:10, whereas vaginal samples were diluted 1:2 prior to incubation. Plates were washed five times with PBS-Tween before incubation with secondary antibodies, also diluted in assay buffer, for 90 min at RT. After a final washing step (five times with PBS-Tween and once with PBS), the enzymatic reactions were developed. For total IgG + IgM detection, the secondary antibody used was goat anti-mouse IgG + IgM (Jackson ImmunoResearch, 115-006-068, 1:2000), and p-nitrophenyl phosphate (pNPP; Sigma, S0942-50TAB) was used as the substrate for alkaline phosphatase. Plates were incubated for 15 min in the dark, and the reaction was stopped with 3 N NaOH. Optical density (OD) was measured at 405 nm using a microplate reader. For IgG1 and IgG2a detection, HRP-conjugated goat anti-mouse IgG1 (Thermo Fisher, A10551, 1:4000) and IgG2a (Thermo Fisher, 046220, 1:1000) antibodies were used. TMB (BD Biosciences) was added as the HRP substrate and incubated for 15 min in the dark. The reaction was stopped with 1 N sulfuric acid, and OD was measured at 450 nm. For IgA detection, a biotin-conjugated anti-mouse IgA antibody (BD Pharmingen™, Clone C10-1, 556978, 1:5000) and streptavidin-HRP (BD Biosciences, 51-9002208, 1:1300) were employed. TMB was used as the substrate, and the reaction was stopped with 2 N sulfuric acid. Absorbance was recorded at 450 nm. For serum titration, serial dilutions ranging from 1:20 to 1:640 were performed. The endpoint titer was defined as the highest dilution yielding an OD value greater than or equal to the cutoff. The cutoff was calculated as the mean OD of negative control sera plus two standard deviations (cutoff = 0.212).
Microneutralization assay (MNT)
The MNT assay was performed with modifications according to a previously described method51,52. Briefly, serum samples were heat-inactivated at 56 °C for 30 min. Serial dilutions of the serum were performed and incubated for 1 h at 37 °C with 1000 plaque forming units (PFU) of the ZIKV-AR isolate in DMEM with 10% fetal bovine serum (FBS). Subsequently, the virus-serum mixture was added to duplicate wells of confluent Vero cells in 96-well plates and incubated at 37 °C for 5 days. At the end of this period, the cells were fixed and stained with 10% (w/v) crystal violet in 10% (v/v) formalin. The cytopathic effect (CPE) was evaluated visually; a well was considered to exhibit CPE if any damage was observed in the cell monolayer. The neutralization titre was defined as the serum dilution that reduced the CPE by 50% or 80% (MNT50 or MNT80). In parallel, the plates were scanned to measure the absorbance of the medium at 595 nm after washing away the excess dye and applying ethanol to dissolve it51. A lower CPE resulted in greater absorbance, indicating that the cells were able to stain more effectively due to reduced damage. The neutralizing titre was determined as the inverse dilution of serum at which the absorbance at 595 nm fell below 50% of the established threshold (≥ 50% inhibition) or 80% (≥ 80% inhibition)51.
Statistics
Normality of the data was assessed using the Shapiro–Wilk test. For datasets that followed a normal distribution, statistical comparisons were performed using one-way ANOVA followed by Tukey’s post hoc test. For datasets that did not meet the assumption of normality, the non-parametric Kruskal–Wallis test was used, followed by Dunn’s multiple comparisons test when appropriate. For kinetic response analyses, data were analyzed using two-way ANOVA followed by Bonferroni’s multiple comparisons test. A p-value < 0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism version 8.0.2. All data points, with no exclusion of outliers, were included in the analyses.
Data availability
Data is provided within the manuscript or supplementary information files.
References
Baker, R. E. et al. Infectious disease in an era of global change. Nat. Rev. Microbiol. 20, 193–205 (2022).
Pierson, T. C. & Diamond, M. S. The continued threat of emerging flaviviruses. Nat. Microbiol. 5, 796–812 (2020).
Carrera, J. P. et al. Clinical and serological findings of Madariaga and Venezuelan equine encephalitis viral infections: A follow-up study 5 years after an outbreak in Panama. Open. Forum Infect. Dis. 7, ofaa359 (2020).
Lindsey, N. P., Martin, S. W., Staples, J. E. & Fischer, M. Notes from the field: multistate outbreak of Eastern equine encephalitis Virus — United states, 2019. Morb Mortal. Wkly. Rep. 69, 50–51 (2020).
Encefalitis equina del oeste - Argentina. https://www.who.int/es/emergencies/disease-outbreak-news/item/2023-DON499.
Campos, A. S. et al. Molecular epidemiology of Western equine encephalitis virus in brazil, 2023–2024. MedRxiv Prepr Serv. Health Sci. (2024). 2024.04.15.24305848.
Souza, W. M. et al. Chikungunya: a decade of burden in the Americas. Lancet Reg. Health – Am 30 (2024).
Moraes, M. M. et al. Detection of saint Louis encephalitis virus in two Brazilian States. J. Med. Virol. 94, 776–781 (2022).
Dengue emergency in. The americas: time for a new continental eradication plan. Lancet Reg. Health - Am. 22, 100539 (2023).
Pielnaa, P. et al. Zika virus-spread, epidemiology, genome, transmission cycle, clinical manifestation, associated challenges, vaccine and antiviral drug development. Virology 543, 34–42 (2020).
Duffy, M. R. et al. Zika virus outbreak on Yap island, federated States of Micronesia. N Engl. J. Med. 360, 2536–2543 (2009).
Musso, D. et al. Zika virus in French Polynesia 2013–14: anatomy of a completed outbreak. Lancet Infect. Dis. 18, e172–e182 (2018).
Cao-Lormeau, V. M. et al. Zika virus, French polynesia, South pacific, 2013. Emerg. Infect. Dis. 20, 1085–1086 (2014).
Zanluca, C. et al. First report of autochthonous transmission of Zika virus in Brazil. Mem. Inst. Oswaldo Cruz. 110, 569–572 (2015).
Faria, N. R. et al. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature 546, 406–410 (2017).
WHO Director-General summarizes the outcome of the Emergency Committee. regarding clusters of microcephaly and Guillain-Barré syndrome. https://www.who.int/news/item/01-02-2016-who-director-general-summarizes-the-outcome-of-the-emergency-committee-regarding-clusters-of-microcephaly-and-guillain-barré-syndrome.
Durand, G. A. et al. Vector-Borne transmission of the Zika virus Asian genotype in Europe. Viruses 12, 296 (2020).
Dinh, L., Chowell, G., Mizumoto, K. & Nishiura, H. Estimating the subcritical transmissibility of the Zika outbreak in the state of florida, USA, 2016. Theor. Biol. Med. Model. 13, 20 (2016).
Wilder-Smith, A., Chang, C. R. & Leong, W. Y. Zika in travellers 1947–2017: a systematic review. J. Travel Med. 25, (2018).
Kraemer, M. U. G. et al. Past and future spread of the arbovirus vectors Aedes aegypti and Aedes albopictus. Nat. Microbiol. 4, 854–863 (2019).
Sah, R., Mohanty, A., Paul, D. & Padhi, B. K. Recent outbreak of Zika virus in India amid ongoing COVID-19 and Monkeypox outbreak: A call for action. Int. J. Surg. Lond. Engl. 109, 601–603 (2023).
Ryan, S. J. et al. Warming temperatures could expose more than 1.3 billion new people to Zika virus risk by 2050. Glob Change Biol. 27, 84–93 (2021).
Araújo, T. V. B. et al. Association between Zika virus infection and microcephaly in brazil, January to may, 2016: preliminary report of a case-control study. Lancet Infect. Dis. 16, 1356–1363 (2016).
de Freitas, P. Ocular findings in infants with microcephaly associated with presumed Zika virus congenital infection in salvador, Brazil. JAMA Ophthalmol. 134, 529–535 (2016).
Brasil, P. et al. Zika virus infection in pregnant women in Rio de Janeiro. N Engl. J. Med. 375, 2321–2334 (2016).
da Silva, I. R. F., Frontera, J. A., Bispo de Filippis, A. M., do Nascimento, O. J. M. & for the RIO-GBS-ZIKV Research Group. Neurologic complications associated with the Zika virus in Brazilian adults. JAMA Neurol. 74, 1190–1198 (2017).
Oehler, E. et al. Zika virus infection complicated by Guillain-Barré syndrome – case report, French polynesia, December 2013. Eurosurveillance 19, 20720 (2014).
Musso, D. et al. Potential sexual transmission of Zika virus. Emerg. Infect. Dis. 21, 359–361 (2015).
Barzon, L. et al. Isolation of infectious Zika virus from saliva and prolonged viral RNA shedding in a traveller returning from the Dominican Republic to italy, January 2016. Eurosurveillance 21, 30159 (2016).
Foy, B. D. et al. Probable non-vector-borne transmission of Zika virus, colorado, USA. Emerg. Infect. Dis. 17, 880–882 (2011).
Magnus, M. M. et al. Risk of Zika virus transmission by blood donations in Brazil. Hematol. Transfus. Cell. Ther. 40, 250–254 (2018).
Sirohi, D. et al. The 3.8 Å resolution cryo-EM structure of Zika virus. Science 352, 467–470 (2016).
Sager, G., Gabaglio, S., Sztul, E. & Belov, G. A. Role of host cell secretory machinery in Zika virus life cycle. Viruses 10, 559 (2018).
Sirohi, D. & Kuhn, R. J. Zika virus structure, maturation, and receptors. J. Infect. Dis. 216, S935–S944 (2017).
Zhao, H. et al. Structural basis of Zika virus-specific antibody protection. Cell 166, 1016–1027 (2016).
Hasan, S. S. et al. A human antibody against Zika virus crosslinks the E protein to prevent infection. Nat. Commun. 8, 14722 (2017).
Richner, J. M. & Diamond, M. S. Zika virus vaccines: immune response, current status, and future challenges. Curr. Opin. Immunol. 53, 130–136 (2018).
Lessler, J. et al. Assessing the global threat from Zika virus. Science 353, aaf8160 (2016).
Mason, R. A., Tauraso, N. M., Spertzel, R. O. & Ginn, R. K. Yellow fever vaccine: direct challenge of monkeys given graded doses of 17D vaccine. Appl. Microbiol. 25, 539–544 (1973).
Tricou, V. et al. Long-term efficacy and safety of a tetravalent dengue vaccine (TAK-003): 4·5-year results from a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Glob Health. 12, e257–e270 (2024).
Boigard, H. et al. Zika virus-like particle (VLP) based vaccine. PLoS Negl. Trop. Dis. 11, e0005608 (2017).
Lin, H. H., Yip, B. S., Huang, L. M. & Wu, S. C. Zika virus structural biology and progress in vaccine development. Biotechnol. Adv. 36, 47–53 (2018).
Kheirvari, M., Liu, H. & Tumban, E. Virus-like particle vaccines and platforms for vaccine development. Viruses 15, 1109 (2023).
Koren, M. A. et al. Safety and immunogenicity of a purified inactivated Zika virus vaccine candidate in adults primed with a Japanese encephalitis virus or yellow fever virus vaccine in the USA: a phase 1, randomised, double-blind, placebo-controlled clinical trial. Lancet Infect. Dis. 23, 1175–1185 (2023).
Stephenson, K. E. et al. Safety and immunogenicity of a Zika purified inactivated virus vaccine given via standard, accelerated, or shortened schedules: a single-centre, double-blind, sequential-group, randomised, placebo-controlled, phase 1 trial. Lancet Infect. Dis. 20, 1061–1070 (2020).
Essink, B. et al. The safety and immunogenicity of two Zika virus mRNA vaccine candidates in healthy flavivirus baseline seropositive and seronegative adults: the results of two randomised, placebo-controlled, dose-ranging, phase 1 clinical trials. Lancet Infect. Dis. 23, 621–633 (2023).
Bacon, A. et al. Generation of a thermostable, oral Zika vaccine that protects against virus challenge in non-human primates. Vaccine 41, 2524–2533 (2023).
Hsieh, M. S. et al. Intranasal vaccination with recombinant antigen-FLIPr fusion protein alone induces long-lasting systemic antibody responses and broad T cell responses. Front. Immunol. 12, 751883 (2021).
Serradell, M. C. et al. Efficient oral vaccination by bioengineering virus-like particles with protozoan surface proteins. Nat. Commun. 10, 361 (2019).
Nash, T. E. Antigenic variation in Giardia lamblia and the host’s immune response. Philos. Trans. R Soc. Lond. B Biol. Sci. 352, 1369–1375 (1997).
Haga, K., Chen, Z., Himeno, M., Majima, R. & Moi, M. L. (eds) (Nancy), Utility of an in-vitro micro-neutralizing test in comparison to a plaque reduction neutralization test for dengue virus, japanese encephalitis virus, and zika virus serology and drug screening. Pathogens. 13, 8 (2024).
Salvo, M. A., Kingstad-Bakke, B., Salas-Quinchucua, C., Camacho, E. & Osorio, J. E. Zika virus like particles elicit protective antibodies in mice. PLoS Negl. Trop. Dis. 12, e0006210 (2018).
Mlakar, J. et al. Zika virus associated with microcephaly. N Engl. J. Med. 374, 951–958 (2016).
Styczynski, A. R. et al. Increased rates of Guillain-Barré syndrome associated with Zika virus outbreak in the Salvador metropolitan area, Brazil. PLoS Negl. Trop. Dis. 11, e0005869 (2017).
Acosta, C. J. et al. Persistence of immunogenicity of a purified inactivated Zika virus vaccine candidate in healthy adults: 2 years of follow-up compared with natural infection. J. Infect. Dis. 227, 1303–1312 (2023).
Chua, A. J. et al. A novel platform for virus-like particle-display of flaviviral envelope domain III: induction of dengue and West nile virus neutralizing antibodies. Virol. J. 10, 129 (2013).
Adamson, C. S. et al. A block in virus-like particle maturation following assembly of murine leukaemia virus in insect cells. Virology 314, 488–496 (2003).
Gashti, A. B. et al. Production, purification and immunogenicity of gag virus-like particles carrying SARS-CoV-2 components. Vaccine 42, 40–52 (2024).
Ako-Adjei, D., Johnson, M. C. & Vogt, V. M. The retroviral capsid domain dictates virion size, morphology, and coassembly of gag into Virus-Like particles. J. Virol. 79, 13463 (2005).
Larocca, R. A. et al. Vaccine protection against Zika virus from Brazil. Nature 536, 474–478 (2016).
Pardi, N. et al. Zika virus protection by a single low dose nucleoside modified mRNA vaccination. Nature 543, 248–251 (2017).
Chen, C. H. et al. Intranasal immunization with Zika virus envelope domain III-Flagellin fusion protein elicits systemic and mucosal immune responses and protection against subcutaneous and intravaginal virus challenges. Pharmaceutics 14, 1014 (2022).
Mosmann, T. R. & Coffman, R. L. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 7, 145–173 (1989).
Stevens, T. L. et al. Regulation of antibody isotype secretion by subsets of antigen-specific helper T cells. Nature 334, 255–258 (1988).
Yang, M., Dent, M., Lai, H., Sun, H. & Chen, Q. Immunization of Zika virus envelope protein domain III induces specific and neutralizing immune responses against Zika virus. Vaccine 35, 4287–4294 (2017).
Sanchez Vargas, L. A., Mathew, A. & Rothman, L. A. T lymphocyte responses to flaviviruses — diverse cell populations affect tendency toward protection and disease. Curr. Opin. Virol. 43, 28–34 (2020).
Lima, N. S., Rolland, M., Modjarrad, K. & Trautmann, L. T. Cell immunity and Zika virus vaccine development. Trends Immunol. 38, 594–605 (2017).
Huang, H. et al. CD8⁺ T cell immune response in immunocompetent mice during Zika virus infection. J. Virol. 91, e01028–17 (2017).
Weiskopf, D. & Sette, A. T-Cell immunity to infection with dengue virus in humans. Front. Immunol. 5, (2014).
Watson, A. M. & Klimstra, W. B. T cell-mediated immunity towards yellow fever virus and useful animal models. Viruses 9, 77 (2017).
Neves, P. C. C., Santos, J. R., Tubarão, L. N., Bonaldo, M. C. & Galler, R. Early IFN-gamma production after YF 17D vaccine virus immunization in mice and its association with adaptive immune responses. PLoS ONE. 8, e81953 (2013).
Lam, L. K. M., Watson, A. M., Ryman, K. D. & Klimstra, W. B. Gamma-interferon exerts a critical early restriction on replication and dissemination of yellow fever virus vaccine strain 17D-204. Npj Vaccines. 3, 1–10 (2018).
Elong Ngono, A. et al. Mapping and role of the CD8 + T cell response during primary Zika virus infection in mice. Cell. Host Microbe. 21, 35–46 (2017).
Hassert, M., Harris, M. G., Brien, J. D. & Pinto, A. K. Identification of protective CD8 T cell responses in a mouse model of Zika virus infection. Front. Immunol. 10, (2019).
Chin, W. X. et al. A single-dose live attenuated chimeric vaccine candidate against Zika virus. NPJ Vaccines. 6, 20 (2021).
Medina-Magües, L. G. et al. mRNA vaccine protects against Zika virus. Vaccines 9, 1464 (2021).
Amatya, N., Garg, A. V. & Gaffen, S. L. IL-17 signaling: the Yin and the Yang. Trends Immunol. 38, 310–322 (2017).
McGeachy, M. J., Cua, D. J. & Gaffen, S. L. The IL-17 family of cytokines in health and disease. Immunity 50, 892–906 (2019).
Jain, A., Pandey, N., Garg, R. K. & Kumar, R. IL-17 level in patients with dengue virus infection & its association with severity of illness. J. Clin. Immunol. 33, 613–618 (2013).
Becquart, P. et al. Acute dengue virus 2 infection in Gabonese patients is associated with an early innate immune response, including strong interferon alpha production. BMC Infect. Dis. 10, 356 (2010).
Acharya, D. et al. Interleukin-17A promotes CD8 + T cell cytotoxicity to facilitate West nile virus clearance. J. Virol. 91, e01529–e01516 (2016).
Zuñiga, J. et al. A unique immune signature of serum cytokine and chemokine dynamics in patients with Zika virus infection from a tropical region in Southern Mexico. Int. J. Infect. Dis. 94, 4–11 (2020).
Azevedo, R. S. S. et al. In situ immune response and mechanisms of cell damage in central nervous system of fatal cases microcephaly by Zika virus. Sci. Rep. 8, 1 (2018).
Serradell, M. C. et al. Vaccination of domestic animals with a novel oral vaccine prevents Giardia infections, alleviates signs of giardiasis and reduces transmission to humans. NPJ Vaccines. 1, 16018 (2016).
Stewart, E. L. et al. Lung IL-17A-producing CD4 + T cells correlate with protection after intrapulmonary vaccination with differentially adjuvanted tuberculosis vaccines. Vaccines 12, 128 (2024).
Derrick, S. C., Yang, A. & Cowley, S. Enhanced efficacy of BCG vaccine formulated in adjuvant is dependent on IL-17A expression. Tuberculosis 148, 102540 (2024).
Raeven, R. H. M. et al. Molecular and cellular signatures underlying superior immunity against Bordetella pertussis upon pulmonary vaccination. Mucosal Immunol. 11, 1009 (2018).
Solans, L. et al. IL-17-dependent SIgA-mediated protection against nasal Bordetella pertussis infection by live attenuated BPZE1 vaccine. Mucosal Immunol. 11, 1753–1762 (2018).
Kimura, A., Naka, T. & Kishimoto, T. IL-6-dependent and -independent pathways in the development of interleukin 17-producing T helper cells. Proc. Natl. Acad. Sci. 104, 12099–12104 (2007).
Restrepo, B. N. et al. Serum levels of cytokines in two ethnic groups with dengue virus infection. Am. J. Trop. Med. Hyg. 79, 673–677 (2008).
Fialho, E. M. S. et al. Maternal Th17 profile after Zika virus infection is involved in congenital Zika syndrome development in children. Viruses 15, 1320 (2023).
Maxian, O., Neufeld, A., Talis, E. J., Childs, L. M. & Blackwood, J. C. Zika virus dynamics: when does sexual transmission matter? Epidemics 21, 48–55 (2017).
Gao, D. et al. Prevention and control of Zika as a Mosquito-Borne and sexually transmitted disease: A mathematical modeling analysis. Sci. Rep. 6, 28070 (2016).
Scott, J. M. et al. Cellular and humoral immunity protect against vaginal Zika virus infection in mice. J. Virol. 92, e00038–e00018 (2018).
Kumamoto, Y. & Iwasaki, A. Unique features of antiviral immune system of the vaginal mucosa. Curr. Opin. Immunol. 24, 411–416 (2012).
Thompson, D. et al. Zika virus-like particle vaccine fusion loop mutation increases production yield but fails to protect AG129 mice against Zika virus challenge. PLoS Negl. Trop. Dis. 16, e0010588 (2022).
Berneck, B. S., Rockstroh, A., Fertey, J., Grunwald, T. & Ulbert, S. A recombinant Zika virus envelope protein with mutations in the conserved fusion loop leads to reduced antibody cross-reactivity upon vaccination. Vaccines 8, 603 (2020).
Urakami, A. et al. An envelope-modified tetravalent dengue virus-like-particle vaccine has implications for flavivirus vaccine design. J. Virol. 91, e01181–17 (2017).
Vannice, K. S. et al. Demonstrating vaccine effectiveness during a waning epidemic: A WHO/NIH meeting report on approaches to development and licensure of Zika vaccine candidates. Vaccine 37, 863–868 (2019).
Sumathy, K. et al. Protective efficacy of Zika vaccine in AG129 mouse model. Sci. Rep. 7, 46375 (2017).
Vang, L. et al. Zika virus-like particle vaccine protects AG129 mice and rhesus macaques against Zika virus. PLoS Negl. Trop. Dis. 15, e0009195 (2021).
Tebas, P. et al. ZIKA-001: safety and immunogenicity of an engineered DNA vaccine against Zika virus infection. N Engl. J. Med. 385, e35 (2021).
Slon-Campos, J. L. et al. A protective Zika virus E-dimer-based subunit vaccine engineered to abrogate antibody-dependent enhancement of dengue infection. Nat. Immunol. 20, 1291–1298 (2019).
Rupil, L. L., del Carmen Serradell, M. & Luján, H. D. Production of oral vaccines based on virus-Like particles pseudotyped with protozoan-surface proteins. In Vaccine Design: Methods and Protocols, Volume 1. Vaccines for Human Diseases (ed Thomas, S.) 503–537. (Springer US, 2022).
Acknowledgements
M.C.S., G.A.L. and L.L.R. are Members of the Scientific Researcher’s Career of the National Council for Scientific and Technical Research of Argentina (CONICET). L.A.F. and F.M. are recipients of fellowships from the CONICET. This work was supported by grants from the CONICET, the Catholic University of Córdoba and the Agency for Promotion of Science and Technology of Argentina. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank Dr. Damián Peralta, belonging to the support staff career of the CONICET, for his contribution in purifying and validating the molecular constructs used in this work, and Felicitas Antonino for her participation during her internship as an undergraduate student in the Biotechnology program at the Faculty of Chemical Sciences of the National University of Córdoba. We are grateful also to Dr. Andrea Gamarnik and Dr. Carlos Bueno for providing the ZIKV structural proteins genomic material and the clinical isolate INEVH116141, respectively. We thank Dr. Laura Cervi (CIBICI-CONICET-UNC) for kindly sharing a biotin-conjugated anti-mouse IgA available in her laboratory. Finally, special thanks to Dr. Liliana del Valle Sosa and Dr. Natacha Zlocowski (INICSA-CONICET-UNC) for their contributions to the electron microscopy assays.
Author information
Authors and Affiliations
Contributions
L.A.F. designed and performed most of the experiments. L.L.R. collaborated with the experiments related to the evaluation of immunogenicity in mice. F.M. helped to amplify and concentrate the ZIKV stocks, and to perform the neutralizing antibodies assays. G.A.L. and M.C.S. designed, collaborated in the execution and supervised the experiments. L.A.F., G.A.L. and M.C.S. assisted in the analysis of the results and wrote the paper. All authors read and commented on the manuscript.
Corresponding author
Ethics declarations
Ethics statement
This research strictly followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The well-being of all animals and the management of animal facilities were supervised by CICUAL-UCC. The study protocol was approved by CICUAL-UCC, guaranteeing adherence to ethical guidelines.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fassola, L.A., Rupil, L.L., Martínez, F. et al. Induction of systemic and mucosal immune response against Zika virus by vaccination with non-infectious chimeric VLPs. Sci Rep 15, 24834 (2025). https://doi.org/10.1038/s41598-025-07059-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-07059-6
