
Abstract
Lassa virus (LASV) is a high-risk pathogen associated with severe viral hemorrhagic fever in both humans and animals. Owing to its significant treatment challenges and high infectivity, LASV is classified as a biosafety level 4 (BSL-4) pathogen. It is essential to establish a rapid LASV detection method to prevent and control the disease. To address the biosecurity threats caused by LASV, in this study, we developed a new test method for LASV detection by combining the recombinase-mediated isothermal amplification (RAA) and CRISPR-Cas13a detection technology. The detection efficiency of this method was evaluated and compared with existing methods. The results demonstrate that this new detection maintains relatively high sensitivity and specificity, while having excellent simplicity and rapidity. The sensitivity of the method for detecting the LASV can achieve a threshold of 101 copies/µL using fluorescence detection in 90 min and 102 copies/µL with lateral flow strip detection in just an hour, which only needs a simple constant temperature equipment to achieve. The application of this detection method holds substantial biosecurity significance for underdeveloped regions (e.g., West Africa), as well as for countries like China, which have a vast territory and uneven development of medical testing levels in various regions.
Similar content being viewed by others
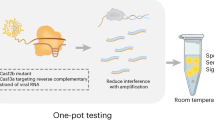
Introduction
LASV is a member of the Arenavirus genus, characterized by its single-stranded RNA genome, whose particles exhibit polytypism and are enveloped1. Lassa fever, endemic to West Africa, is caused by the LASV, which transmits between humans and animals, presenting symptoms typical of viral hemorrhagic fever. Rodents, particularly Mastomys species, serve as the primary natural hosts of the LASV2,3,4 Human infection usually results from exposure to rodent excreta or secretions, mucosal infection through broken skin, or consumption of contaminated food. Patients often exhibit severe multisystem symptoms5. A notable outbreak in Sierra Leone in 1996 had a case fatality rate of 18.16% and attracted worldwide attention; since then, Lassa fever has remained endemic in Guinea, Liberia, and other West African countries2,6. Furthermore, China faces biosecurity risks as LASV’s potential natural hosts (Rattus norvegicus, Mus musculus) are widely distributed7,8,9. Therefore, it is imperative to strengthen monitoring and prevention efforts, timely and effectively detecting the pathogen is crucial to disease control and prevention.
Currently, RT-PCR is the gold standard for virus detection; however, it is a complex technique that requires specialized equipment, extended processing times, and skilled personnel. Moreover, the sensitivity of commercially available RT-PCR detection kits remains suboptimal10. Enzyme-linked immunosorbent assay (ELISA) is another widely used method for detecting infectious agents while the sensitivity of this method is relatively low11, considerable time is required for its reagent development. These limitations highlight the urgent need for the development of a rapid, convenient, and relatively sensitive method for LASV detection.
Recombinase-mediated strand substitution nucleic acid amplification technology (RAA) represents a swift, isothermal nucleic acid amplification technique that utilizes recombinant enzymes sourced from bacterial or fungal origins. At 30–42 °C, the recombinase enzyme exhibits high-affinity binding to primer DNA, thereby forming an enzyme-primer complex. This complex facilitates the primer’s search for a complementary sequence on the template DNA, with single-stranded DNA-binding proteins aiding in the unwinding of the double-stranded template. Following this, DNA polymerase catalyzes the synthesis of a new complementary strand, leading to exponential amplification of the target nucleic acid. In comparison with conventional PCR, RAA is time-efficient, user-friendly, and minimally demanding in terms of equipment.
Additionally, CRISPR-Cas13a, a Cas protein that specifically recognizes RNA sequences, can recognize specific single-stranded RNA based on the designed crRNA and generate collateral cleavage upon recognition. This feature enables the detection of conserved RNA virus sequences: In the presence of report RNA within the detection system, the Cas13a protein can cleave Report RNA by collateral cleavage, producing detectable fluorescence and then reflecting the situation of identification. The integration of these two technologies enables rapid and easy detection of conserved pathogen sequences, facilitating effective pathogen detection (Fig. 1).
Recent advancements in CRISPR-based diagnostics have generated detection schemes based on various CRISPR subsystems12,13,14 particularly SHERLOCK (specific high sensitivity enzymatic reporter unlocking technology), provide promising alternative solutions15,16,17,18,19,20. This approach, which leverages CRISPR-Cas13a and (RT)-RAA technology, allows for rapid and accurate detection of LASV, significantly reducing the need for specialized instruments and shortening the testing process. Importantly, it enables pathogen detection under low condition of equipment while maintaining relatively high sensitivity and specificity.

Schematic diagram of the detection principle.
In this study, we combined RAA technology with CRISPR-Cas13a fluorescence detection technology and lateral flow test strips developed in our laboratory to create a simple, sensitive, and low-equipment rapid pathogen detection technology. Despite being unable to get genuine positive samples for effect evaluation due to limitations in the distribution of cases, the current results still reflect the advantages of this approach to some extent. This method aims to enable timely and rapid detection of LASV, then it will reduce the difficulty of disease surveillance in the corresponding epidemic areas, and provide guidance for the prevention and control of possible disease transmission, so as to make a certain contribution to the cause of world public health.
Materials and methods
Materials
Gene sequences and reference materials
Currently, the absence of an epidemic trend of the Lassa virus in China has made it difficult to directly access its nucleic acid. In this study, LASV plasmid was designed based on the construction of a viral sequence. Following the relevant selection criteria, the corresponding region of the nucleoprotein within the relatively conserved S segment of LASV (NC_004292.1) was synthesized and ligated into the pUC57 vector to serve as the positive plasmid for subsequent testing. Additionally, five crRNA targeting sequences within the conserved region were selected to establish a foundation for future detection efforts. For the specificity test, nucleic acids from other pathogens were extracted from the pathogen samples derived from the corresponding inactivated viruses stored in our laboratory.
Main reagents and devices
NTP mix (art. No. N0466S), and T7 transcription kit (art. No. E2050S) and T7 RNA polymerase (art. No. E2050S), and RNase inhibitors (art. No. E2050S) were purchased from the New England Biological Laboratory (NEB) of the United States, the RT-RAA amplification kit (basic type) (art No. S003ZC) was purchased from Hangzhou ZhongCe Biotechnology Co., Ltd. Cas13a (art. No. db005) protein was purchased from Nanjing Genscript Biotechnology Co., Ltd. The RNaseAlert™ QC System v2; art. No. 4,479,769 was purchased from Thermo Fisher company of the United States. In addition, 2× Super Pfx MasterMix (art. No. CW2965M) was purchased from Jiangsu Cowin Biotechnology Co., Ltd. The PCR Instrument Applied Biosystems, Thermo Scientific, USA; fluorescence quantitative PCR instrument of Mastercycler-realplex4 was produced by Eppendorf (Germany).
Design and synthesis of crrna, (RT)-RAA primers and positive plasmids
After downloading and analyzing 563 distinct LASV genomes from the NCBI database, we identified a relatively conserved segment within the nucleoprotein region as the target for amplification. Subsequently, nine (RT)-RAA primers targeting the target plasmids were designed and synthesized to amplify the target plasmids. For subsequent transcription, a T7 promoter sequence was appended to the 5′ end of the forward primer. Additionally, five crRNAs and the amplification primers of the crRNAs were designed between the RAA primers21. The primers and crRNAs were synthesized by Tianyihuiyuan (Beijing) Biotechnology Co., Ltd. Following PCR amplification, vitro transcription, and purification, the synthesized crRNA was stored for subsequent use. The procedure was conducted as follows: the crDNA and primers listed in Table 1 were diluted to a concentration of 10 µM prior to PCR amplification. The reaction mixture per unit was composed of 100 µL of 2× Super Pfx Master Mix, 8 µL each of forward and reverse primers, 8 µL of template, and 76 µL of double-distilled water (ddH2O). The components were thoroughly mixed, and the PCR amplification was carried out under the following conditions: an initial denaturation at 95 °C for 5 min, followed by 35 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 45 s, with a final extension at 72 °C for 10 min. Following amplification, the resultant DNA products were purified using a mixture of Tris-buffered phenol and chloroform. This mixture was prepared by combining equal volumes of Tris-buffered phenol and chloroform, followed by vigorous shaking and centrifugation to separate the phases, after which the supernatant was discarded. Subsequently, 600 µL of the prepared mixture was added to the PCR product, shaken to ensure thorough mixing, and centrifuged at 12,000 rpm for 5 min. The supernatant obtained was then combined with absolute ethanol in a 3:7 ratio, followed by centrifugation at 12,000 rpm for 10 min, after which the supernatant was discarded. The products were washed with 200 µL of 70% ethanol and subjected to centrifugation at 12,000 rpm for 10 min, finally, the supernatant was discarded, and this washing step was repeated twice. The products were dried in the air for about 10 min, and 50 µL of water was added to fully dissolve the products. The products were then transcribed and purified using the T7 transcription kit to obtain the crRNA for subsequent experiments, and the obtained crRNA was stored at − 80 °C for later use.
(RT)-RAA amplification
Following dilution, the synthesized (RT)-RAA primers were adjusted to a concentration of 10 µM in accordance with the kit instructions, utilizing an appropriate concentration of plasmids as the template to set up the (RT)-RAA system. Before use, the reaction unit containing the dry powder was briefly centrifuged. Subsequently, 2 µL of primers (with a final concentration of 400 nM), 5 µL of template, and 25 µL of Buffer A were added to the reaction unit to fully dissolve the dry powder. Next, 2.5 µL of Buffer B was applied to the cover of the reaction tube, which was then inverted to ensure thorough mixing. After a brief centrifugation, the reaction unit was incubated at 39 °C.
Visualization detection by agarose gel electrophoresis
Preparation of Agarose Gel: A total of 1.5 g (1.5%) of agarose was accurately weighted by an electronic balance and transferred into a conical flask. Subsequently, 100 mL of 1× TAE buffer solution was measured by a graduated cylinder and added to the flask. The mixture was thoroughly agitated to ensure homogeneity and then heated in a microwave for 4 min. After heating, the solution was allowed to cool to a temperature range of 50 –60 °C. At this point, 10 µL of 10000× DNA dye was incorporated into the solution. The mixture was then poured into a casting tray, and a comb was inserted to form wells. Agarose Gel was left to solidify at room temperature. Sample Loading: A volume of 5 µL of a 500 bp DNA marker was carefully loaded into the wells of the agarose gel. Additionally, 2 µL of 6× DNA Loading Buffer was mixed with 10 µL of amplification products prior to loading into the gel. The voltage was adjusted to 125 V, and the electrophoresis was started for 30 min. Observation: After 30 min, the gel was removed and put into an AlphaImager HP gel-imager to observe the results.
CRISPR fluorescence detection
For a detailed description of this method, please refer to22. For each experimental group, the reaction system was configured in the reaction unit with the following components: 1.5 µL of crRNA, 0.5 µL of T7 transcriptase, 2 µL of NTP, 2.5 µL of Report RNA, 1 µL of LwaCas13a, 5 µL of (RT)-RAA amplification product, 1 µL of RNase Inhibitor, 0.25 µL of MgCl2, and 0.5 µL of HEPES (20 mM HEPES, 140 mM NaCl, 5 mM KCl, 1.2 mM MgCl2 and 1.5 mM CaCl2, pH 7.2–7.6), with ddH2O added to achieve a total volume of 25 µL. The reaction was conducted using a CFX 96 Touch Roche fluorescence quantitative PCR instrument under the following conditions: at a temperature of 37 °C and detection on the FAM channel with an excitation wavelength of 490 nm and an emission wavelength of 520 nm. Fluorescence measurements were taken every 2 min over a period of 30 min to continuously detect the changes in fluorescence intensity in the system.
CRISPR detection by lateral flow strip method
The experimental protocol was conducted as follows: the reaction mixture comprised 4 µL of NTP Mix, 2 µL of inhibitor, 2 µL of LwaCas13a, 1 µL of T7, 0.5 µL of MgCl2, 1 µL of HEPES (20 mM HEPES,140 mM NaCl,5 mM KCl,1.2 mM MgCl2 and 1.5 mM CaCl2, pH 7.2–7.6), 26.5 µL of ddH2O, 5 µL of Report RNA (ERASE), 3 µL of crRNA, and 5 µL of plasmid, consistent with previous experiments. For each experimental group, the reagents were sequentially added to the reaction tube, thoroughly mixed by shaking, and subsequently incubated in a metal bath at 39 °C for 30 min. Following incubation, 40 ~ 50 µL of the reaction products were transferred to the sampling chamber of the CRISPR test card equipped with a lateral flow strip. The reaction system was then applied to the test paper for 3–5 min before the results were determined.
Probe Q-PCR detection
According to the specified protocol, the reaction mixture should be prepared as follows: utilize 10 µL of TaqMan™ Fast Advanced Master Mix, 0.5 µL of primers and probe each at a concentration of 10 µM, 3 µL of plasmid, and 5.5 µL of ddH2O, totaling 20 µL. Thermal cycling conditions were as follows: an initial denaturation at 95 °C for 5 min, followed by 35 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 45 s, concluding with a final extension at 72 °C for 10 min. For each experimental group, add the reagents to the reaction tube, ensure thorough mixing by shaking, and proceed with amplification using the Bio-Rad fluorescence quantitative PCR system. Upon completion of the reaction, evaluate the results based on the Cq values to draw conclusions.
Data analysis and result interpretation
The quantitative data were repeated three times in each group and averaged for independent samples test, the analysis of variance was used when comparing multiple sets of data with each other (spss20.0 software https://www.ibm.com/spss). The results of test is judged in accordance with the “easy-readout and sensitive-enhanced (ERASE) strip” method23, the presence of just “C” lines on the test lateral flow strip was considered positive; the presence of both “T” and “C” lines on the test lateral flow strip was considered negative. No “C” line on the test lateral flow strip was considered as failure of the test lateral flow strip, and the test lateral flow strip should be replaced and redone.
Results
Screening of (RT)-RAA primers and CrRNAs
We aligned 563 genomes of LASV and selected conserved sequences in nucleoprotein, then, 3 upstream primers and 3 downstream primers for (RT)-RAA were designed according to the conserved sequences (Fig. 2A), as a total of 9 kinds of combination. (RT)-RAA amplification of conserved sequence plasmids was performed using 9 primer combinations respectively.

Results of primers and fluorescence of different crRNAs.
The following data were repeated three times in each group (A) Screening and evaluation of crRNA;(B) Detected with agarose electrophoresis of 105 copies/µL concentration of template amplified by all primers(The unit of the mark is bp; M: Marker, E: Experimental group, N: Negative control); (C) The IntDen analysis of the agarose gel electrophoresis band 1 by ImageJ software; (D) Gradient detection (105copies/µL-101copies/µL) with agarose electrophoresis of 105 copies/µL concentration of template amplified by selected primers; (E) The IntDen analysis of the agarose gel electrophoresis band 2 by ImageJ software; (F) The fluorescence detection results of CRISPR-Cas13a system with different crRNAs targeting LASV genome; (G) The fluorescence intensity of two selected crRNAs at 30 min after reaction start.
The results indicated that the combination of primers F2R1, F2R3, F1R1, and F2R2 successfully amplified the target band, which was visually detectable, and amplified bands of F2R1 and F2R3 were more pronounced (Fig. 2B). The amplified bands were subsequently analyzed in grayscale using ImageJ software(Image J version1.54 g https://imagej.net/ij/). Quantitative analysis revealed that the band integration density amplified by the F2R1 primers was approximately 1.12 times that of F2R3, 1.96 times that of F1R1, and 1.56 times that of F2R2 (Fig. 2C). Due to the minimal gap between F2R1 and F2R3, we diluted the substrate concentration and conducted tests to ensure the accuracy of the results. The template plasmid was diluted in a gradient ranging from 10^5 copies/µL to 10^1 copies/µL, and RT-RAA was performed using the two primer pairs obtained from the initial screening. The amplification products were subsequently purified and verified through agarose gel electrophoresis (Fig. 2D). Gray scale analysis revealed that the product amplified by F2R1 was 1.31 times that of F2R3 (Fig. 2E). Consequently, the RT-RAA primer was provisionally designated as F2R1.
To identify crRNA with superior targeting capabilities, a high concentration of plasmid was utilized to enhance the observation of data differences. The template plasmid, at a concentration of 107 copies/µL, was amplified using the (RT)-RAA primer F2R1. This amplified product was subsequently incorporated into the CRISPR-Cas13a reaction system for the CRISPR fluorescence detection with various crRNAs. The results indicated that the average fluorescence detection value for crRNA1 was 1.96 times, 1.45 times, and 1.13 times higher than that of crRNA2 at 20, 40, and 60 min, respectively. These findings demonstrate that the detection efficiency of crRNA1 was significantly superior to that of crRNA2, particularly at the earlier and middle time points. The other crRNAs basically had no target recognition ability (Fig. 2F,G). Therefore, the CRISPR-Cas13a detection system containing crRNA1 was selected for the detection of LASV.
Confirmation of (RT)-RAA primers and determination of detection sensitivity
The amplification effect of (RT)-RAA played a crucial role in enhancing detection sensitivity. To ensure the accuracy of RAA primer screening results, after selecting the appropriate crRNA, the sensitivity of the detection system and the screening outcomes of the (RT)-RAA primers were reconfirmed using the CRISPR system containing the selected crRNA. This was achieved by performing a 10-fold serial dilution of the viral plasmid templates (diluted to 102 copies/µL, 101 copies/µL, and 100 copies/µL). The plasmid templates were then amplified using nine different (RT)-RAA primers, followed by CRISPR fluorescence detection of the results.

Results of fluorescence of different primers and Q-PCR.
The following data were repeated three times in each group (A) Fluorescence detection results of a CRISPR-Cas13a system with different (RT)-RAA primers targeting LASV at plasmid concentration of 102copies/µL; (B) Fluorescence detection results of a CRISPR-Cas13a system with different (RT)-RAA primers targeting LASV at plasmid concentration of 101copies/µL; (C) Fluorescence detection results of a CRISPR-Cas13a system with different (RT)-RAA primers targeting LASV at plasmid concentration of 100copies/µL;(F) Fluorescence detection results of probe Q-PCR with LASV at different plasmid concentration; (G) The fluorescence values of the three optimal primer plateaus at different concentrations.
The results demonstrated that the three primer pairs, F2R1, F2R3, and F3R2, effectively amplified detectable products at a plasmid concentration of 101 copies/µL rather than 100 copies/µL (Fig. 3B,C,E,G). Specifically, the F2R1 amplification product exhibited relatively high fluorescence values at plasmid concentrations of 102 copies/µL and 101 copies/µL, with fluorescence values measured at 13,936 and 4,932, respectively, after 30 min of reaction (Fig. 3A,B,D,E,G). In contrast, the F2R3 primer pair yielded a low fluorescence value of 2,964 in the detection of the 101 copies/µL plasmid system (Fig. 3B,E,G); F3R2 rapidly reached a low fluorescence value plateau when detecting a plasmid system at a concentration of 102 copies/µL (Fig. 3A,D,G), which could easily be mistaken for negative samples in practical applications. Consequently, F2R1 was selected as the final (RT)-RAA amplification primer. Concurrently, the sensitivity of the CRISPR detection system for LASV samples was determined to be 101 copies/µL (Fig. 3B,C). To compare the Q-PCR detection method with the CRISPR-based detection method, primers and probes were designed, and probe Q-PCR, which could be better recognized, was employed for amplification and detection. The results showed that the sensitivity of probe Q-PCR for LASV detection was also 101 copies/µL (Fig. 3F).
LASV CRISPR nucleic acid detection test lateral flow strip sensitivity and specific test
Easy-readout and sensitive-enhanced (ERASE) strip is an innovative tool for CRISPR detection developed by our group, which is easy to read and can avoid the miscalculation to a certain extent. Its principle involves Gold-labeled FAM-biotinylated reporter molecules flow to the test capture band, and redundant gold nanoparticles flow to the control capture band. Upon recognition of the target RNA, the crRNA/Cas13a complex cleaves the reporter molecule, allowing the passage by the test band (Fig. 4).

Principle of ERASE strip.
In order to shorten the inspection process, reduce the need for equipment for inspection, and thus realize the rapid visualization of inspection results, the plasmid intended for detection by LASV was serially diluted to concentrations of 103 copies/µL, 102 copies/µL, 101 copies/µL, and 100 copies/µL. Subsequently, the lateral flow strip CRISPR detection assay was conducted. The “C” line and “T” line appeared after the plasmid concentration of 101 copies/µL and 100 copies/µL templates was added to the test lateral flow strip, so the test result was judged to be negative. The plasmid concentrations of 103 copies/µL and 102 copies/µL were applied to the test lateral flow strip, resulting in the appearance of only the “C” line and the absence of the “T” line, thereby indicating positive test results. Consequently, the sensitivity of the LASV lateral flow strip detection was determined to be 102 copies/µL (Fig. 5A,B). When compared to the probe Q-PCR detection method and the CRISPR fluorescence detection method, the sensitivity of this approach is reduced by an order of magnitude; however, it offers significantly enhanced convenience. On the other hand, to ensure that our tests are not confounded by other pathogens, we utilized nucleic acids from 12 different pathogens, each diluted to a concentration of 103 copies/µL, for the CRISPR detection test paper method. The lateral flow strips, when tested with the addition of nucleic acids from these 12 pathogens as well as a negative control (no pathogen nucleic acids), exhibited both “C” and “T” lines simultaneously, so the test results were judged as negative; The test lateral flow strip with LASV nucleic acid showed only a “C” line and no “T” line, and the test results were judged positive (Fig. 5C,D). These results indicated that the lateral flow strip CRISPR assay had high specificity.

Result of lateral flow strip detection.
The following data were repeated three times in each group (A) Sensitivity evaluation results of LASV CRISPR lateral flow strip nucleic acid detection (B) The heat map of the Sensitivity lateral flow strip detection of LASV. (C) Specificity evaluation results of LASV CRISPR lateral flow strip nucleic acid detection. CHIKV: Chikungunya virus; JEV: Japanese encephalitis virus SA14-14-2; TBEV: Forest encephalitis virus senzhang; HTNV: Hantaan virus vaccine strains; YFV: Yellow fever virus 17D; HAdV 4 ~ 7: Human adenovirus 4 ~ 7; DENV: Dengue virus; ZIKV: Zika virus; HIBV: influenza B; SA: Staphylococcus aureus; NC: Negative control. (D) The heat map of the Specificity lateral flow strip detection of LASV.
Discussion
Since 2023, Lassa virus (LASV) has re-emerged in Nigeria24. Currently, the detection of the virus primarily relies on Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and immunofluorescence techniques, among others. These methods, however, require advanced technical expertise, sophisticated testing equipment, and stringent laboratory conditions25. Regions most affected by the virus, such as West Africa, often lack the necessary infrastructure to meet these requirements due to underdevelopment26, and because of China’s vast territory, unbalanced economic development between regions, the test coverage scope and timeliness cannot be guaranteed. At the same time, even if a centralized testing point is set up, in the process of sample transport, the instability of RNA may also compromise test accuracy27. Therefore, developing a simple, portable, and low-cost LASV rapid detection method applicable across diverse regions holds significant practical value. This advancement would contribute to global biosafety efforts6.
In this study, the CRISPR-Cas13a system was integrated with (RT)-RAA isothermal amplification technology and the “sensitive-enhanced (ERASE)” CRISPR test lateral flow strip to establish a virus nucleic acid-based lateral flow strip CRISPR detection technology for LASV. This method achieves detection within 1 h, improving efficiency compared to conventional RT-PCR10. Importantly, this method significantly reduces the need for equipment, which does not require special nucleic acid detection equipment, only a thermostatic device and ERASE strips are needed to complete the relevant nucleic acid detection on the spot, such devices are usually lightweight and portable, in extreme cases, the isothermal amplification process can even be tried to perform in the hands of the inspector and completely freeing itself from the limitations of the device, on the other hand, it is not limited by the detection flux. While exhibiting excellent convenience and speed, it also maintains a relatively high sensitivity: it can detect 102copies/µL samples.
Despite the notable advantages of this method compared to traditional testing protocols, there remains potential for further development. Due to the workload, we have not yet been able to detect other conserved loci in the whole genome of LASV, and perhaps with the development of bioinformatics technology, we can predict the detectability of conserved loci, and then conduct comprehensive screening for multiple targets.
Data availability
The data that support the findings of this study are available from the corresponding author, [Yz F], upon reasonable request.
Abbreviations
- LASV:
-
Lassa virus
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- Cas:
-
CRISPR associated protein
- RAA:
-
Recombinase-aided amplification
- crRNA:
-
CRISPR RNA
- qPCR:
-
Quantitative real-time PCR
- HAdV:
-
Human adenovirus
- DENV:
-
Dengue virus
- ZIKV:
-
Zika virus
- TBEV:
-
Forest encephalitis virus
- JEV:
-
Japanese encephalitis virus
- HIBV:
-
influenza B
- SA:
-
Staphylococcus aureus
- CHIKV:
-
Chikungunya virus
- YFV:
-
Yellow fever virus
- HTNV:
-
Hantaan virus
References
Günther, S. & Lenz, O. LASV. Crit. Rev. Clin. Lab. Sci. 41(4), 339–390. https://doi.org/10.1080/10408360490497456 (2004).
Schmaljohn, C. & Safronetz, D. Editorial overview: LASV. Curr. Opin. Virol. 37, vii–ix. https://doi.org/10.1016/j.coviro.2019.09.001 (2019).
Smither, A. R. & Bell-Kareem, A. R. Ecology of Lassa virus. Curr. Top. Microbiol. Immunol. 440, 67–86. https://doi.org/10.1007/82_2020_231 (2023).
Mylne, A. Q. et al. Mapping the zoonotic niche of Lassa fever in Africa. Trans. R. Soc. Trop. Med. Hyg. 109 (8), 483–492. https://doi.org/10.1093/trstmh/trv047 (2015).
Asogun, D. A., Günther, S., Akpede, G. O., Ihekweazu, C. & Zumla, A. Lassa fever: Epidemiology, clinical features, diagnosis, management and prevention. Infect. Dis. Clin. N. Am. 33(4), 933–951. https://doi.org/10.1016/j.idc.2019.08.002 (2019).
Shaffer, J. G. et al. Viral hemorrhagic fever consortium. Data set on Lassa fever in post-conflict Sierra Leone. Data Brief. 23, 103673. https://doi.org/10.1016/j.dib.2019.01.021 (2019).
You, F. F. et al. Kobuviruses carried by Rattus norvegicus in Guangdong, China. BMC Microbiol. 20 (1), 94. https://doi.org/10.1186/s12866-020-01767-x (2020).
Zou, Y. et al. The detection of Toxoplasma gondii in Wild Rats (Rattus norvegicus) on Mink Farms in Shandong Province, Eastern China. Vector Borne Zoonotic Dis. 22(3), 199–204. https://doi.org/10.1089/vbz.2021.0087 (2022).
Yan, C. et al. Prevalence and genotyping of Toxoplasma gondii in naturally-infected synanthropic rats (Rattus norvegicus) and mice (Mus musculus) in Eastern China. Parasit. Vectors. 7, 591. https://doi.org/10.1186/s13071-014-0591-6 (2014).
Luo, X. L. et al. Comparative evaluation of standard RT-PCR assays and commercial real-time RT-PCR kits for detection of Lassa virus. Microbiol. Spectr. 11 (2), e0501122. https://doi.org/10.1128/spectrum.05011-22 (2023).
Peeling, R. W., Heymann, D. L., Teo, Y. Y. & Garcia, P. J. Diagnostics for COVID-19: moving from pandemic response to control. Lancet (London England). 399 (10326), 757–768. https://doi.org/10.1016/S0140-6736(21)02346-1 (2022).
Zeping Yang, B. et al. Photoactivatable CRISPR/Cas9 lateral flow strip platform for one-pot rapid detection of squamous cell carcinoma antigen DNA in blood. Sens. Actuators B Chem. 434, 137607. https://doi.org/10.1016/j.snb.2025.137607 (2025).
Gu, X. et al. Visual detection of HPV16 using a photoactivatable CRISPR-Cas12 system. Chem. Commun. (Camb). 61(22), 4383–4386. https://doi.org/10.1039/d5cc00369e (2025).
Guo, B. et al. Test strip coupled Cas12a-assisted signal amplification strategy for sensitive detection of uracil-DNA glycosylase. Lab Chip. 24(7), 1987–1995. https://doi.org/10.1039/d4lc00096j (2024).
Myhrvold, C. et al. Field-deployable viral diagnostics using CRISPR-Cas13. Science. 360 (6387), 444–448. https://doi.org/10.1126/science.aas8836 (2018).
Wang, B. et al. Cas12aVDet: A CRISPR/Cas12a-based platform for rapid and visual nucleic acid detection. Anal. Chem. 91(19), 12156–12161. https://doi.org/10.1021/acs.analchem.9b01526 (2019).
Broughton, J. P. et al. CRISPR-Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 38 (7), 870–874. https://doi.org/10.1038/s41587-020-0513-4 (2020).
Dai, Y. et al. Exploring the trans-cleavage activity of CRISPR-Cas12a (cpf1) for the development of a universal electrochemical biosensor. Angew. Chem. Int. Ed. Engl. 58 (48), 17399–17405. https://doi.org/10.1002/anie.201910772 (2019).
Guo, L. et al. SARS-CoV-2 detection with CRISPR diagnostics. Cell. Discov. 6, 34. https://doi.org/10.1038/s41421-020-0174-y (2020).
Barnes, K. G. et al. Deployable CRISPR-Cas13a diagnostic tools to detect and report Ebola and Lassa virus cases in real-time. Nat. Commun. 11 (1), 4131. https://doi.org/10.1038/s41467-020-17994-9 (2020).
Li, H. et al. CRISPR-Cas13a cleavage of dengue virus NS3 gene efficiently inhibits viral replication. Mol. Ther. Nucleic Acids. 19, 1460–1469 (2020).
Wang, S. Research on high-sensitivity detection technology of hepatitis B virus DNA and YMDD resistance mutation based on CRISPR technology.Acad. Milit. Sci. (2019).
Tian, Y. et al. CRISPR/Cas13a-assisted accurate and portable hepatitis D virus RNA detection. Emerg. Microbes Infect. 12 (2), 2276337 (2023).
Naeem, A. et al. Re-emergence of Lassa fever in nigeria: A new challenge for public health authorities. Health Sci. Rep. 6 (10), e1628. https://doi.org/10.1002/hsr2.1628 (2023).
Boisen, M. L. et al. Field evaluation of a Pan-Lassa rapid diagnostic test during the 2018 Nigerian Lassa fever outbreak. Sci. Rep. 10 (1), 8724. https://doi.org/10.1038/s41598-020-65736-0 (2020).
Wonderly, B. et al. Comparative performance of four rapid Ebola antigen-detection lateral flow immunoassays during the 2014–2016 Ebola epidemic in West Africa. PLoS One. 14 (3), e0212113. https://doi.org/10.1371/journal.pone.0212113 (2019).
Forsgren, E., Locke, B., Semberg, E., Laugen, A. T. & Miranda, J. R. Sample preservation, transport and processing strategies for honeybee RNA extraction: influence on RNA yield, quality, target quantification and data normalization. J. Virol. Methods. 246, 81–89 (2017).
Funding
This work was funded by The National Key Research and Development Program of China, grant number 2023YFC2605100.
Author information
Authors and Affiliations
Contributions
W.T., Y.Y. wrote the main manuscript text; M.N. reviewed the text; W.T. and M.N. prepared figures; X.D. summarized the data; H.L., Y.S. and Y.F. guided the conduct of the experiment.
Corresponding authors
Ethics declarations
Competing interests
Yunzhi Fa, Hao Li, Tong Wei, Yansong Sun, Xue Dong, Yujie Yan has patent pending , If there are other authors, they declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Animal ethics
No approval of research ethics committees was required to accomplish the goals of this study because experimental work was conducted with no animal.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wei, T., Yan, Y., Niu, M. et al. A rapid LASV detection method based on CRISPR-Cas13a and recombinase aided amplification with special lateral-flow test strips. Sci Rep 15, 20640 (2025). https://doi.org/10.1038/s41598-025-07071-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-07071-w
