
Abstract
The present study demonstrated the design and synthesis of sulfonamide-1,2,3-triazole-acetamide derivatives 11a-o and screening against urease in vitro and in silico. These compounds were designed based on reported potent urease inhibitors and optimized structurally based on substituents on acetamide moiety. In vitro studies showed that all the new compounds 11a-o (IC50 values = 0.12–4.53 µM) were more potent than stand inhibitor thiourea (IC50 value = 23.76 µM). In this regard, the most potent compounds were N-phenylacetamide derivatives 11b, 11f, and 11 h with 2-methyl, 4-methoxy, and 2-fluoro substituents, respectively. In this regard, the most potent compound 11b was 198-folds more potent than thiourea against urease. In silico studies demonstrated that this compound with the binding energy less than thiourea attached to the urease’s active site. Druglikeness, pharmacokinetics, and toxicity of compound 11b and thiourea were predicted by two credible online servers. These in silico studies showed that, in terms of druglikeness and pharmacokinetics, compound 11b was almost similar to thiourea while in term of toxicity, compound 11b was better than thiourea.
Similar content being viewed by others
Introduction
Many studies show that the source of many simple and complex digestive problems is helicobacter pylori (H. pylori) infection1. H. pylori is a gram-negative bacterium that affected up to 50% of the world’s human population2. Common treatment for H. pylori infection is a combination of several drugs3. These drugs are antibiotics such as Amoxicillin and Metronidazole and proton pump inhibitors such as Omeprazole and pantoprazole4. Due to the resistance of H. pylori to antibiotics, the mentioned drug regime dose not completely treatment the H. pylori infection. Therefore, the treatment of this infection remains as a problem and the search for new therapeutic targets for this infection is still ongoing5. The main place of residence of H. pylori is stomach, and in order to survive in the acidic environment of stomach, this bacterium needs to the function of an urease that can be converted urea into ammonia6. Therefore, urease is an attractive target for medicinal chemists because inhibition of it can help to deterioration of H. pylori7,8,9,10.
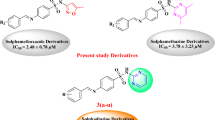
Sulfonamide is an important pharmacophore in the design of bioactive compounds with anti-microbial, anti-cancer, anti-inflammatory, and diuretic activities11. Recently, sulfonamide scaffold has been highly used in the design of urease inhibitors12. Some of sulfonamide derivatives with significant inhibitory activity against urease are showed in Fig. 113,14,15. On the other hand, 1,2,3-triazole ring was found in the several series of the potent urease inhibitors such as derivatives D and E (Fig. 1)16,17.

Synthetic urease inhibitors containing sulfonamide moiety (compounds A-C) or 1,2,3-triazole ring (compounds D and E).
To design sulfonamide-1,2,3-triazole-acetamide scaffold as a new scaffold with anti-urease property, we considered compounds A1 and D1 as the most potent compounds of template compounds A and B (Fig. 2). As can be seen in the Fig. 2, methyl substituent was fund in both template compounds, therefore, we synthesized fifteen derivatives 11a-o of sulfonamide-1,2,3-triazole-acetamide scaffold that have a methyl substituent on 4-position of phenyl ring attached to sulfonamide moiety. After that, in vitro and in silico studies were performed on the new synthesized compounds 11a-o.

Design strategy for sulfonamide-1,2,3-triazole-acetamide derivatives 11a-o as new urease inhibitors.
Results and discussion
Chemistry
Sulfonamide-1,2,3-triazole-acetamide derivatives 11a-o were prepared by click reaction18. For this purpose, firstly, propargylted derivative 5 and various azide derivatives 10a-o were synthesized by simple and efficient chemical reactions as showed in Scheme 1. 4-Methyl-N-phenyl-N-(prop-2-yn-1-yl)benzenesulfonamide (5) obtained by two steps: 1) reaction between 4-methylbenzenesulfonyl chloride (1) and aniline (2) in the presence of Et3N in THF that led to formation of 4-methyl-N-phenylbenzenesulfonamide (3) and 2) reaction between latter compound and propargyl bromide (4) in the presence of K2CO3 in DMF19. On the other hand, acetamide-azide derivatives 10a-o were synthesized by reported method in our previous work20. In the final step, compound 5 reacted with acetamide-azide derivatives 10a-o to give target compounds 11a-o.

Synthetic procedure for sulfonamide-1,2,3-triazole-acetamide derivatives 11a-o.
The obtained conditions for the first reaction step were selected based on our optimization results and reported work by Dehghanpour Kalan et al. at 202219. Some observed results are summarized in Table 1. As can be seen in Table 1, three types of bases (K2CO3, Et3N, and Piperidine) and two types of solvents (THF and DMF) were evaluated under different reaction conditions in terms of temperature and time. The best results were obtained with Et3N as base and THF as solvent (entries 6, 8, and 9 in Table 1). Therefore, we chose the mildest conditions to carry out the reaction. This condition was entry 6 in Table 1 that at RT led to the highest yield achieved in 1 h (95%). Replacement of Et3N in entry 6 with K2CO3 and piperidine, in the cases of entry 1 and entry 13, decreased yields to 45% and 35%, respectively.
A plausible mechanism for formation of sulfonamide derivatives 11 is showed in Scheme 2.

A plausible mechanism for formation of compounds 11.
In vitro anti-urease activity
In vitro urease inhibitory activity of the new sulfonamide-1,2,3-triazole-acetamide derivatives 11a-o was evaluated against jack bean urease (JBU) from canavalia ensiformis (Table 2). For this evaluation, thiourea, that is a known urease inhibitor, was used as positive control. All new compounds 11a-o with IC50 values < 4.53 µM were more potent than thiourea with IC50 value of 23.76 µM. In this regard, the most potent compound among the new sulfonamide-1,2,3-triazole-acetamide derivatives (compound 11b) with IC50 value of 0.12 µM was 198-folds more potent than thiourea.
Structure-activities relationships (SARs)
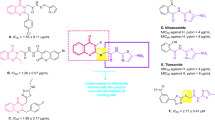
Structurally, with the exception of N-benzylacetamide derivative 11n and N-phenethylacetamide derivative 11o, rest derivatives 11a-m were derivatives of N-phenylacetamide (Scheme 1 and Table 2). Diagram of SARs of compounds 11a-o was depicted in Fig. 3.
As can be seen in Fig. 3a; Table 2, the most potent compound among the N-phenylacetamide derivatives and the all synthesized compounds was N−2-methylphenylacetamide derivative 11b. Replacement of methyl substituent of compound 11b with 2-fluoro substituent led to produce of the third potent compound 11 h. In general, the order of anti-urease activity in 2-substituted compounds was methyl > F > Cl. This pattern showed that electron donating substituents in 2-position of phenyl ring acted better than electron withdrawing substituents. On the other hand, changing the position of the methyl group of compound 11b of 2-position to 4-position and or adding a methyl group to 3-position of this compound, as in the cases of compounds 11c and 11d, leads to a significant decrease in anti-urease activity. The second potent compound against JBU was 4-methoxy derivative 11f. As can be seen in Fig. 3a, order of anti-urease activity in the 4-substituented compounds was methoxy > Cl > methyl > F > ethyl. This order showed that in 4-position, type of substituent as well as electron proprieties affected anti-urease activity. Moreover, adding a methoxy substituent on 2-position of phenyl ring of compound 11f, as in the case of 2,4-dimethoxy derivative 11 g, anti-urease activity diminished to 3.7 folds.
The comparison of IC50 values of N-phenylacetamide derivative 11a with N-benzylacetamide derivative 11n and N-phenethylacetamide derivative 11o revealed that anti-urease activity decreased with adding methylene groups (Fig. 3b).

Diagram of SARs of N-phenylacetamide derivatives (a) and comparison of N-phenylacetamide derivative 11a with N-benzylacetamide derivative 11n and N-phenethylacetamide derivative 11o (b).
Comparison of new compounds 11a-o with the used templates
Survey on IC50 values of the template compounds A1, D1, and the most potent compound in the present work (compound 11b) demonstrated that our new compound was more potent than used templates (Fig. 4)13,16.

The comparison of the most potent compound 11b with the used templates.
Docking study
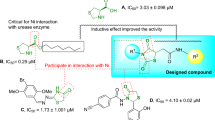
A docking study was conducted to evaluate the interactions between the new synthesized compounds and the urease’s active site. An appropriate crystallographic structure of JBU was selected of the protein data bank (PDB). Original inhibitor of the selected crystallographic structure was acetohydroxamic acid (AHA) and our used inhibitor was thiourea. Therefore, firstly, interaction modes of these inhibitors were studied. As can be seen in Fig. 5, AHA established hydrogen bonds with residues His409, Asp633, Ala440, and KCX490. This inhibitor formed an unfavorable interaction with Ni901. AHA also established weak van der Waals interactions with residues Thr441, Ala636, Met637, His545, His492, Ni902, Arg439, Gly550, and Arg609. The binding energy (BE) of AHA in the active site of JBU was − 4.060 Kcal/mole.
Thiourea as the standard inhibitor established four hydrogen bonds with residues Ala440, Asp633, Gly550, and His492 (Fig. 5). The latter residue also formed a π-Sulfur interaction with thionyl unit of thiourea. Thiourea also formed two unfavorable interactions with Ni2+ cations with codes of 901 and 902. Several weak van der Waals interactions between thiourea and residues Thr441, Ala636, Met637, His545, His519, KCX490, and His409 were also observed. BE of thiourea in the active site of JBU was − 3.306 Kcal/mole.

Interaction modes of the standard inhibitors AHA and Thiourea in the JBU’s active site.
In the next step, thiourea as standard inhibitor and the most potent compound 11b were placed in the urease’s active site and the obtained superimposed structure was showed in Fig. 6.

Superimposed structure of thiourea (gray) and the most potent compound 11b (orang) in the active site of JBU.
Interaction mode of the most potent compound 11b was showed in Fig. 7. This compound established two conventional hydrogen bonds with Arg439 and a non-classical hydrogen bond with residue CME592 (Fig. 7). Compound 11b also formed a π-π interaction with His593. In the interaction mode of this compound several hydrophobic interactions with residues Ala636, His593, Ala440, and Ala436 and weak van der Waals interactions with residues Ni902, Met637, Gly550, His519, His492, Asp494, His594, and Gly437 were also observed. The BE of compound 11b in the active site of JBU was − 7.528 Kcal/mole.

Interaction mode of compound 11b in the active site of JBU.
As can be seen Table 2, IC50 values of compounds 11f and 11 h were approximately same. The interaction modes of these compounds were shown in Fig. 8. Compound 11f formed two conventional hydrogen bonds with residues Arg439 and Arg609 and non-classical hydrogen bonds with Asp494 and Glu493. This compound established the fallowing π interactions with JBU’s active site: a π-cation with Arg439, a π-anion with Asp494, a π-π with His593, and a π-sigma with Trp495. Compound 11f also formed hydrophobic interactions with Ala436 and Ala440 and van der Waals interactions with residues CME592, His492, Gly437, His519, Met637, Thr441, and Ala636. The BE of this compound was − 7.43 Kcal/mole. Compound 11 h formed two conventional hydrogen bonds Arg439 and Ala636 and a non-classical hydrogen bond with CME592. In term of number of π-interactions, compound 11 h formed an additional π-interaction in comparison to compound 11f. This additional interaction was a π-sulfur interaction between 1,2,3-triazole ring of compound 11 h and residue CME592. In addition, compound 11 h formed a halogen interaction via its fluoro substituent. Moreover, compound 11 h in comparison to compound 11f formed an additional hydrophobic interaction with Ala636. Compound 11 h also formed van der Waals interactions with residues His492, Asp633, Gln635, Gly437, His519, Met637, Thr441, and Arg609. The BE of compound 11 h was − 7.609 Kcal/mole.
The general review on the obtained results showed that the new synthesized compounds 11b, 11f, and 11 h were attached to the urease’s active site easier than standard inhibitors thiourea and AHA.

Interaction modes of compounds 11f and 11 h in the active site of JBU.
In Silico druglikness, pharmacokinetics, and toxicity study
In silico druglikness, pharmacokinetics, and toxicity predictions of thiourea as the standard urease inhibitor and compound 11b as the most potent new compounds performed by SwissADME and PreADMET web tools21. The obtained result were shown in Table 3. As can be seen in this table, both studied compounds fallowed of rule of five (Lipinski rule) and Veber rule as two famous druglikness rules.
In term of pharmacokinetics, gastrointestinal (GI) absorption of thiourea and compound 11b was high and both these compounds had not permeability to blood-brain barrier (BBB) (Table 3). Moreover, these compounds were not substrates for P-glycoprotein (P-gp) that is a drug efflux pump. Furthermore, as can be seen in Table 3, thiourea did not inhibit any of the main isoforms of cytochrome P450 (CYP) enzyme while compound 11b inhibited the fallowing isoforms of the latter enzyme: CYP2C19, CYP2C9, and CYP3A4. It should be noted that CYP enzymes play a pivotal role in drug metabolism.
In silico toxicity study on thiourea and compound 11b predicted that compound 11b was not mutagen and carcinogen while thiourea was mutagen and had carcinogen effect on rat. In term of cardiotoxicity, two studied compounds had low risk.
The bioavailability radars of thiourea and compound 11b were showed in Fig. 9 and these radars indicated that our new compound 11b was largely placed in pink area (the optimal range for oral bioavailability) while thiourea dramatically violated of this area22.

Bioavailability radar of thiourea and compound 11b. Saturation (INSATU); Flexibility (FLEX); Polarity (POLAR); Lipophilicity (LIPO); Molecular weight (SIZE); Solubility (INSOLU).
Experimental
Synthesis of 4-methyl-N-phenylbenzenesulfonamide (3)
A solution of 4-methylbenzenesulfonyl chloride (1, 10 mmol), aniline (2, 10 mmol), and Et3N (10.1 mmol) in THF (50 ml) was stirred at room temperature (RT) for 1 h. Then, THF was evaporated and ethyl acetate (50 ml) was added to the residue. The mixture was extracted using water (3 × 30 ml). The organic phase was separated and dried by Na2SO4 and ethyl acetate was evaporated under reduced pressure. The residue was pure 4-methyl-N-phenylbenzenesulfonamide (3).
Synthesis of 4-Methyl-N-phenyl-N-(prop-2-yn-1-yl)benzenesulfonamide (5)
A mixture of 4-methyl-N-phenylbenzenesulfonamide (3, 10 mmol), propargyl bromide (4, 10 mmol), and K2CO3 (10.1 mmol) in DMF (50 mL) was stirred at room temperature (RT) for 8 h, and after that, the reaction mixture was poured in the cold water and 4-methyl-N-phenyl-N-(prop-2-yn-1-yl)benzenesulfonamide (5) as target compound in this step was separated by filtration.
General procedure for synthesis of sulfonamide-1,2,3-triazole-acetamide derivatives 11a-o
A mixture of compound 5 (1 mmol), sodium ascorbate, and CuSO4·5H2O (7 mol %) was added to in situ prepared acetamide-azide derivatives 10a-o (1 mmol), and the obtained mixture was stirred at RT for 18–24 h20. After that, reaction mixture was poured into crushed ice and precipitated products 11a-o were filtered off, washed with water, and purified by recrystallization in ethanol.
2-(4-(((4-methyl-N-phenylphenyl)sulfonamido)methyl)−1 H-1,2,3-triazol-1-yl)-N-phenylacetamide (11a).
Yield: 83%; Light yellow solid; mp: 172–175 °C; IR (KBr, vmax) 3310 (NH), 3025 (CH Aromatic), 2970(CH Aliphatic), 1665(C = O) Cm−1; 1H NMR (301 MHz, DMSO) δ 10.45 (s, 1H), 7.98 (s, 1H), 7.58 (d, J = 8.0 Hz, 2 H), 7.53 (d, J = 7.9 Hz, 2 H), 7.43–7.36 (m, 3 H), 7.35–7.28 (m, 4 H), 7.15–7.08 (m, 3 H), 5.27 (s, 2 H), 4.92 (s, 2 H), 2.41 (s, 3 H); 13C NMR (76 MHz, DMSO) δ 164.56, 144.04, 142.88, 139.36, 138.88, 135.57, 130.20, 129.38, 128.86, 128.23, 127.85, 126.11, 124.24, 119.68, 52.63, 46.47, 21.52. Anal. Calcd for C24H23N5O3S: C, 62.46; H, 5.02; N 15.17; Found: C, 62.61; H, 5.14; N 15.03.
2-(4-(((4-methyl-N-phenylphenyl)sulfonamido)methyl)−1 H-1,2,3-triazol-1-yl)-N-(o-tolyl)acetamide (11b).
Yield: 80%; Light yellow solid; mp: 172–175 °C; IR (KBr, vmax) 3325(NH), 3010(CH Aromatic), 2965 (CH Aliphatic) 1661 (C = O) Cm−1; 1H NMR (301 MHz, DMSO) δ 9.81 (s, 1H), 8.00 (s, 1H), 7.54 (d, J = 7.4 Hz, 2 H), 7.48–7.36 (m, 3 H), 7.34–7.08 (m, 8 H), 5.34 (s, 2 H), 4.93 (s, 2 H), 2.41 (s, 3 H), 2.24 (s, 3 H); 13C NMR (76 MHz, DMSO) δ 164.77, 144.07, 142.90, 139.33, 135.97, 135.52, 132.10, 130.93, 130.21, 129.38, 128.86, 128.25, 127.86, 126.55, 126.09, 126.06, 125.23, 52.39, 46.48, 21.52, 18.28. Anal. Calcd for C25H25N5O3S: C, 63.14; H, 5.30; N, 14.73, Found: C, 63.29; H, 5.43; N, 14.59.
2-(4-(((4-methyl-N-phenylphenyl)sulfonamido)methyl)−1 H-1,2,3-triazol-1-yl)-N-(p-tolyl)acetamide (11c).
Yield: 84%; Light yellow solid; mp: 177–180 °C; IR (KBr, vmax) 3330 (NH), 3015 (CH Aromatic), 2870 (CH Aliphatic), 1666 (C = O) Cm−1; 1H NMR (301 MHz, DMSO) δ 10.37 (s, 1H), 7.97 (s, 1H), 7.53 (d, J = 8.2 Hz, 2 H), 7.46 (d, J = 8.1 Hz, 2 H), 7.40 (d, J = 8.1 Hz, 2 H), 7.34–7.26 (m, 3 H), 7.19–7.05 (m, 4 H), 5.24 (s, 2 H), 4.91 (s, 2 H), 2.41 (s, 3 H), 2.27 (s, 3 H); 13C NMR (76 MHz, DMSO) δ 164.29, 144.07, 142.88, 139.33, 136.32, 135.52, 133.28, 130.21, 129.77, 129.38, 128.85, 128.26, 127.84, 126.12, 119.70, 52.60, 46.46, 21.51, 20.91. Anal. Calcd for C25H25N5O3S: C, 63.14; H, 5.30; N, 14.85; Found: C, 63.31; H, 5.41; N, 14.73.
N-(2,3-dimethylphenyl)−2-(4-(((4-methyl-N-phenylphenyl)sulfonamido)methyl)−1 H-1,2,3-triazol-1-yl)acetamide (11d).
Yield: 83%; Light yellow solid; mp: 166–169 °C; IR (KBr, vmax) 3290(NH), 3000(CH Aromatic), 2935 (CH Aliphatic),1659(C = O) Cm−1; 1H NMR (301 MHz, DMSO) δ 9.95 (s, 1H), 8.01 (s, 1H), 7.55 (d, J = 7.9 Hz, 2 H), 7.39 (d, J = 7.9 Hz, 2 H), 7.34–7.27 (m, 3 H), 7.21 (d, J = 7.3 Hz, 1H), 7.14 (d, J = 6.2 Hz, 2 H), 7.07 (d, J = 8.3 Hz, 2 H), 5.35 (s, 2 H), 4.95 (s, 2 H), 2.40 (s, 3 H), 2.26 (s, 3 H), 2.12 (s, 3 H); 13C NMR (76 MHz, DMSO) δ 164.84, 144.06, 142.88, 139.36, 137.63, 135.76, 135.55, 131.55, 130.20, 129.37, 128.87, 128.24, 127.87, 127.76, 126.09, 125.78, 123.76, 52.39, 46.50, 21.51, 20.61, 14.50. Anal. Calcd for C26H27N5O3S: C, 63.78; H, 5.56; N, 14.30, Found: C, 63.93; H, 5.68; N, 14.12.
N-(4-ethylphenyl)−2-(4-(((4-methyl-N-phenylphenyl)sulfonamido)methyl)−1 H-1,2,3-triazol-1-yl)acetamide (11e).
Yield: 80%; Light yellow solid; mp: 149–152 °C; IR (KBr, vmax) 3305(NH), 3010(CH Aromatic), 2930(CH Aliphatic),1660(C = O) Cm−1; 1H NMR (301 MHz, DMSO) δ 10.40 (s, 1H), 7.99 (s, 1H), 7.58–7.46 (m, 4 H), 7.40 (d, J = 7.9 Hz, 2 H), 7.35–7.24 (m, 3 H), 7.18 (d, J = 8.1 Hz, 2 H), 7.16–7.07 (m, 2 H), 5.27 (s, 2 H), 4.93 (s, 2 H), 2.62–2.54 (m, 2 H), 2.41 (s, 3 H), 1.16 (t, 3 H); 13C NMR (76 MHz, DMSO) δ 164.32, 144.04, 142.87, 139.66, 139.37, 136.58, 135.55, 130.20, 129.37, 128.86, 128.57, 128.23, 127.86, 126.12, 119.77, 52.62, 46.48, 28.07, 21.51, 16.12. Anal. Calcd for C26H27N5O3S: C, 63.78; H, 5.56; N, 14.30, Found: C, 63.94; H, 5.64; N, 14.16.
N-(4-methoxyphenyl)−2-(4-(((4-methyl-N-phenylphenyl)sulfonamido)methyl)−1 H-1,2,3-triazol-1-yl)acetamide (11f).
Yield: 80%; Yellow solid; mp: 179–182 °C; IR (KBr, vmax) 3310(NH), 3050(CH Aromatic), 2980 (CH Aliphatic) 1664 (C = O) Cm−1; 1H NMR (301 MHz, DMSO) δ 10.33 (s, 1H), 7.98 (s, 1H), 7.58–7.45 (m, 4 H), 7.40 (d, J = 8.0 Hz, 2 H), 7.36–7.23 (m, 3 H), 7.19–7.07 (m, 2 H), 6.93 (d, J = 9.1 Hz, 2 H), 5.24 (s, 2 H), 4.93 (s, 2 H), 3.74 (s, 3 H), 2.41 (s, 3 H); 13C NMR (76 MHz, DMSO) δ 164.03, 156.02, 144.04, 142.87, 139.36, 135.55, 131.99, 130.20, 129.37, 128.86, 128.23, 127.86, 126.09, 121.25, 114.48, 55.63, 52.57, 46.47, 21.52. Anal. Calcd for C25H25N5O4S: C, 61.09; H, 5.13; N, 14.25; Found: C, 61.18; H, 5.26; N, 14.09.
N-(2,4-dimethoxyphenyl)−2-(4-(((4-methyl-N-phenylphenyl)sulfonamido)methyl)−1 H-1,2,3-triazol-1-yl)acetamide (11 g).
Yield: 81%; Light yellow solid; mp: 173–176 °C; IR (KBr, vmax) 3315(NH), 3010(CH Aromatic), 2940(CH Aliphatic), 1658(C = O) Cm−1; 1H NMR (301 MHz, DMSO) δ 9.59 (s, 1H), 7.95 (s, 1H), 7.71 (d, J = 8.8 Hz, 1H), 7.53 (d, J = 7.9 Hz, 2 H), 7.40 (d, J = 8.0 Hz, 2 H), 7.34–7.27 (m, 3 H), 7.11 (d, J = 8.1 Hz, 2 H), 6.66 (d, J = 2.6 Hz, 1H), 6.55–6.45 (m, 1H), 5.31 (s, 2 H), 4.91 (s, 2 H), 3.85 (s, 3 H), 3.76 (s, 3 H), 2.41 (s, 3 H); 13C NMR (76 MHz, DMSO) δ 164.48, 157.51, 151.80, 144.04, 142.80, 139.34, 135.54, 130.20, 129.36, 128.85, 128.23, 127.85, 126.09, 123.82, 120.04, 104.58, 99.35, 56.24, 55.78, 52.50, 46.45, 21.52. Anal. Calcd for C26H27N5O5S: C, 59.87; H, 5.22; N, 13.43, Found: C, 59.98; H, 5.31; N, 13.24.
N-(2-fluorophenyl)−2-(4-(((4-methyl-N-phenylphenyl)sulfonamido)methyl)−1 H-1,2,3-triazol-1-yl)acetamide (11 h).
Yield: 79%; Light yellow solid; mp: 168–171 °C; IR (KBr, vmax) 3335(NH), 3045(CH Aromatic), 2950 (CH Aliphatic) 1668 (C = O) Cm−1; 1H NMR (301 MHz, DMSO) δ 10.29 (s, 1H), 7.99 (s, 1H), 7.95–7.88 (m, 1H), 7.53 (d, J = 7.9 Hz, 2 H), 7.40 (d, J = 7.9 Hz, 2 H), 7.35–7.25 (m, 4 H), 7.22–7.16 (m, 2 H), 7.13–7.07 (m, 2 H), 5.37 (s, 2 H), 4.92 (s, 2 H), 2.41 (s, 3 H); 13C NMR (76 MHz, DMSO) δ 165.20, 155.53, 152.29, 144.04, 139.33, 135.54, 130.20, 129.37, 128.86, 128.24, 127.85, 126.20, 126.10, 126.01, 125.86, 125.01, 124.97, 124.22, 116.21, 115.96, 52.41, 46.47, 21.52. Anal. Calcd for C24H22FN5O3S: C, 60.11; H, 4.62; N, 14.60, Found: C, 60.26; H, 4.78; N, 14.46.
N-(4-fluorophenyl)−2-(4-(((4-methyl-N-phenylphenyl)sulfonamido)methyl)−1 H-1,2,3-triazol-1-yl)acetamide (11i).
Yield: 83%; Light yellow solid; mp: 184–187 °C; IR (KBr, vmax) 3340(NH), 3010(CH Aromatic), 2960(CH Aliphatic),1665(C = O) Cm−1; 1H NMR (301 MHz, DMSO) δ 10.54 (s, 1H), 8.00 (s, 1H), 7.69–7.59 (m, 2 H), 7.54 (d, J = 8.2 Hz, 2 H), 7.39 (d, J = 7.9 Hz, 2 H), 7.33–7.26 (m, 3 H), 7.19 (t, J = 8.9 Hz, 2 H), 7.13 (d, 2 H), 5.29 (s, 2 H), 4.93 (s, 2 H), 2.40 (s, 3 H); 13C NMR (76 MHz, DMSO) δ 164.53, 160.34, 157.16, 144.07, 142.94, 139.35, 135.52, 135.27, 135.23, 130.20, 129.38, 128.87, 128.25, 127.85, 126.14, 121.59, 121.49, 116.13, 115.84, 52.58, 46.49, 21.50. Anal. Calcd for C24H22FN5O3S: C, 60.11; H, 4.62; N, 14.60, Found: C, 60.24; H, 4.77; N, 14.42.
N-(2-chlorophenyl)−2-(4-(((4-methyl-N-phenylphenyl)sulfonamido)methyl)−1 H-1,2,3-triazol-1-yl)acetamide (11j).
Yield: 82%; Yellow solid; mp: 155–158 °C; IR (KBr, vmax) 3320(NH), 3030(CH Aromatic), 2970 (CH Aliphatic) 1669 (C = O) Cm−1; 1H NMR (301 MHz, DMSO) δ 10.07 (s, 1H), 8.03 (s, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.60–7.49 (m, 3 H), 7.40 (d, J = 8.1 Hz, 2 H), 7.36–7.20 (m, 5 H), 7.14 (d, J = 6.3 Hz, 2 H), 5.43 (s, 2 H), 4.95 (s, 2 H), 2.40 (s, 3 H); 13C NMR (76 MHz, DMSO) δ 165.30, 144.06, 143.00, 139.37, 135.57, 134.66, 130.20, 130.10, 129.38, 128.88, 128.25, 128.03, 127.87, 127.15, 126.71, 126.28, 126.21, 52.43, 46.51, 21.52. Anal. Calcd for C24H22ClN5O3S: C, 58.12; H, 4.47; N, 14.12, Found: C, 58.29; H, 4.63; N, 14.03.
N-(3-chlorophenyl)−2-(4-(((4-methyl-N-phenylphenyl)sulfonamido)methyl)−1 H-1,2,3-triazol-1-yl)acetamide (11k).
Yield: 84%; Yellow solid; mp: 160–163 °C; IR (KBr, vmax) 3335 (NH), 3040 (CH Aromatic), 2950 (CH Aliphatic), 1676 (C = O) Cm−1; 1H NMR (301 MHz, DMSO) δ 10.08 (s, 1H), 8.00 (s, 1H), 7.74 (d, J = 7.9 Hz, 1H), 7.53 (d, J = 8.1 Hz, 3 H), 7.39 (d, J = 8.1 Hz, 2 H), 7.35–7.20 (m, 5 H), 7.11 (d, J = 8.0 Hz, 2 H), 5.39 (s, 2 H), 4.92 (s, 2 H), 2.39 (s, 3 H); 13C NMR (76 MHz, DMSO) δ 165.27, 144.12, 142.96, 139.28, 135.48, 134.57, 130.21, 130.09, 129.39, 128.86, 128.29, 128.03, 127.83, 127.24, 126.78, 126.33, 126.18, 52.38, 46.47, 21.50. Anal. Calcd for C24H22ClN5O3S: C, 58.12; H, 4.47; N, 14.12, Found: C, 58.23; H, 4.58; N, 14.02.
N-(4-chlorophenyl)−2-(4-(((4-methyl-N-phenylphenyl)sulfonamido)methyl)−1 H-1,2,3-triazol-1-yl)acetamide (11 L).
Yield: 82%; Light yellow solid; mp: 228–231 °C; IR (KBr, vmax) 3325(NH), 3035(CH Aromatic), 2960 (CH Aliphatic) 1668 (C = O) Cm−1; 1H NMR (301 MHz, DMSO) δ 10.60 (s, 1H), 7.99 (s, 1H), 7.62 (d, J = 8.6 Hz, 2 H), 7.53 (d, J = 8.0 Hz, 2 H), 7.45–7.37 (m, 4 H), 7.34–7.27 (m, 3 H), 7.16–7.08 (m, 2 H), 5.29 (s, 2 H), 4.92 (s, 2 H), 2.41 (s, 3 H); 13C NMR (76 MHz, DMSO) δ 164.78, 144.05, 142.92, 139.35, 137.84, 135.54, 130.20, 129.37, 129.32, 128.86, 128.24, 127.85, 126.13, 121.26, 52.61, 46.47, 21.52. Anal. Calcd for C24H22ClN5O3S: C, 58.12; H, 4.47; N, 14.12, Found: C, 58.31; H, 4.56; N, 14.01.
N-(4-bromophenyl)−2-(4-(((4-methyl-N-phenylphenyl)sulfonamido)methyl)−1 H-1,2,3-triazol-1-yl)acetamide (11 m).
Yield: 82%; Yellow solid; mp: 233–236 °C; IR (KBr, vmax) 3320 (NH), 3030 (CH Aromatic), 2860 (CH Aliphatic), 1668 (C = O) Cm−1; 1H NMR (301 MHz, DMSO) δ 10.60 (s, 1H), 7.99 (s, 1H), 7.62–7.49 (m, 6 H), 7.40 (d, J = 8.0 Hz, 2 H), 7.34–7.26 (m, 3 H), 7.16–7.07 (m, 2 H), 5.28 (s, 2 H), 4.92 (s, 2 H), 2.41 (s, 3 H); 13C NMR (76 MHz, DMSO) δ 164.81, 144.05, 142.93, 139.35, 138.26, 135.54, 132.23, 130.20, 129.37, 128.86, 128.24, 127.85, 126.14, 121.63, 115.88, 52.64, 46.47, 21.53. Anal. Calcd for C24H22BrN5O3S: C, 53.34; H, 4.10; N, 12.96, Found: C, 53.59; H, 4.23; N, 12.78.
N-benzyl-2-(4-(((4-methyl-N-phenylphenyl)sulfonamido)methyl)−1 H-1,2,3-triazol-1-yl)acetamide (11n).
Yield: 80%; Light yellow solid; mp: 171–174 °C; IR (KBr, vmax) 3280(NH), 2990 (CH Aromatic), 2935 (CH Aliphatic), 1662 (C = O) Cm−1; 1H NMR (301 MHz, DMSO) δ 8.82 (t, J = 6.0 Hz, 1H), 7.95 (s, 1H), 7.54 (d, J = 6.4 Hz, 2 H), 7.40 (d, J = 7.2 Hz, 2 H), 7.35 (d, J = 7.7 Hz, 2 H), 7.32–7.24 (m, 6 H), 7.12 (d, J = 6.8 Hz, 2 H), 5.13 (s, 2 H), 4.92 (s, 2 H), 4.35 (d, J = 5.8 Hz, 2 H), 2.41 (s, 3 H); 13C NMR (76 MHz, DMSO) δ 165.82, 144.04, 142.82, 139.38, 139.18, 135.58, 130.20, 129.36, 128.85, 128.22, 127.86, 127.49, 125.99, 52.06, 46.49, 42.86, 21.52. Anal. Calcd for C25H25N5O3S: C, 63.14; H, 5.30; N, 14.73, Found: C, 63.27; H, 5.42; N, 14.51.
2-(4-(((4-methyl-N-phenylphenyl)sulfonamido)methyl)−1 H-1,2,3-triazol-1-yl)-N-phenethylacetamide (11o).
Yield: 78%; Light yellow solid; mp: 176–179 °C; IR (KBr, vmax) 3285(NH), 3005 (CH Aromatic), 2925 (CH Aliphatic), 1663 (C = O) Cm−1; 1H NMR (301 MHz, DMSO) δ 8.41 (t, J = 5.7 Hz, 1H), 7.89 (s, 1H), 7.54 (d, J = 8.0 Hz, 2 H), 7.40 (d, J = 8.0 Hz, 2 H), 7.34–7.27 (m, 5 H), 7.27–7.19 (m, 3 H), 7.12 (d, J = 6.6 Hz, 2 H), 5.02 (s, 2 H), 4.91 (s, 2 H), 3.35 (q, J = 7.0 Hz, 2 H), 2.75 (t, J = 7.4 Hz, 2 H), 2.41 (s, 3 H); 13C NMR (76 MHz, DMSO) δ 165.67, 144.04, 142.80, 139.64, 139.35, 135.53, 130.20, 129.36, 129.12, 128.85, 128.22, 127.86, 126.67, 125.88, 52.07, 46.46, 40.88, 35.39, 21.52. Anal. Calcd for C26H27N5O3S: C, 63.78; H, 5.56; N, 14.30, Found: C, 63.91; H, 5.69; N, 14.11.
Urease inhibitory assay
Anti-urease activity of the new synthetic compounds 11a-o was evaluated against JBU exactly according to our pervious paper20.
Docking study
In the present work, docking study on the most potent compounds and the standard inhibitor was performed by SwissDock as an online web server23,24. For these purpose, 4H9M that is a crystallographic structure of JBU was selected of PDB. Water molecules and the original inhibitors were removed from this protein structure and the final pdb file was used as the input file for SwissDock. The selected ligands were depicted by ChemDraw professional 15.1 and were copied in SMILES form. Original inhibitor of this enzyme was AHA and based on position of this inhibitor, grid box parameters were determined by BIOVIA Discovery Studio v.3.5. In this regards, the center of the this box was placed at x = 18.7825, y = −57.8095, and z = −24.1515 Å and the dimensions of the active site box were set at 24 × 24 × 24 Å. Finally, docking process was performed by SwissDock and the obtained data were Visualized by BIOVIA Discovery Studio v.3.5.
Prediction of druglikness, pharmacokinetics, and toxicity
In silico study on druglikness and pharmacokinetics of the most potent compound 11b and standard inhibitor was performed by SwissADME online web server21. Toxicity of these agents was predicted by PreADMET online server22.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Santos, M. L. C. et al. Helicobacter pylori infection: beyond gastric manifestations. World J. Gastroenterol. 26, 4076–4093 (2020).
Hassan, M. N. et al. Global prevalence of Helicobacter pylori and its effect on human health. Pure Appl. Biol. 9, 936–948 (2020).
Safavi, M., Sabourian, R. & Foroumadi, A. Treatment of Helicobacter pylori infection: current and future insights. World J. Clin. Cases. 4, 5 (2016).
Benninga, M. A. et al. Drugs in focus: proton pump inhibitors. J. Pediatr. Gastroenterol. Nutr. 72, 645–653 (2021).
Roszczenko-Jasińska, P., Wojtyś, M. I. & Jagusztyn-Krynicka, E. K. Helicobacter pylori treatment in the post-antibiotics era—searching for new drug targets. Appl. Microbiol. Biotechnol. 104, 9891–9905 (2020).
Ansari, S. & Yamaoka, Y. Survival of Helicobacter pylori in gastric acidic territory. Helicobacter 22, e12386 (2017).
Ghobadi, E., Ghanbarimasir, Z. & Emami, S. A review on the structures and biological activities of anti-Helicobacter pylori agents. Eur. J. Med. Chem. 223, 113669 (2021).
Hashem, O. et al. Design and discovery of urease and Helicobacter pylori inhibitors based on benzofuran/benzothiophene-sulfonate and sulfamate scaffolds for the treatment of ureolytic bacterial infections. Int. J. Biol. Macromol. 271, 132502 (2024).
Kataria, R. & Khatkar, A. In-silico design, synthesis, ADMET studies and biological evaluation of novel derivatives of chlorogenic acid against urease protein and H. Pylori bacterium. BMC Chem. 13, 1–17 (2019).
Ibrar, A., Khan, I. & Abbas, N. Structurally diversified heterocycles and related privileged scaffolds as potential urease inhibitors: a brief overview. Arch. Pharm. 346, 423–446 (2013).
Ghomashi, R., Ghomashi, S., Aghaei, H. & Massah, A. R. Recent advances in biological active sulfonamide based hybrid compounds part A: Two-component sulfonamide hybrids. Curr. Med. Chem. 30, 407–480 (2023).
Ahmad, S. et al. New acetamide-sulfonamide-containing scaffolds: antiurease activity screening, structure-activity relationship, kinetics mechanism, molecular docking, and Md simulation studies. Molecules 28, 5389 (2023).
Khair-ul-Bariyah, S. et al. Synthesis of 2-aminothiazole sulfonamides as potent biological agents: synthesis, structural investigations and Docking studies. Heliyon 10, e34980 (2024).
Hamad, A. et al. Bio-oriented synthesis of new sulphadiazine derivatives for urease Inhibition and their Pharmacokinetic analysis. Sci. Rep. 11, 18973 (2021).
Begum, F. et al. Synthesis and urease inhibitory potential of benzophenone sulfonamide hybrid in vitro and in Silico. Bioorg. Med. Chem. 27, 1009–1022 (2019).
Hosseinzadeh, N. et al. Synthesis, Docking Simulation, and ADMET Prediction of Novel Barbituric‐hydrazine‐phenoxy‐1, 2, 3‐triazole‐acetamide Derivatives as Potent Urease Inhibitors. ChemistrySelect 8, e202203297 (2023).
Hussain, T., Khan, M. & Siddiqui, H. Synthesis of 1, 2, 3-triazole functionalized derivatives of metronidazole as potent inhibitors of urease: synthesis via click chemistry, in vitro urease inhibition, kinetics and molecular Docking studies. J. Mol. Struct. 1319, 139475 (2025).
Hou, J., Liu, X., Shen, J., Zhao, G. & Wang, P. G. The impact of click chemistry in medicinal chemistry. Expert Opin. Drug Discov. 7, 489–501 (2012).
Kalan, R. D., Amiri, K., Rominger, F., Balalaie, S. & Bijanzadeh, H. R. Regio-and diastereoselective transition metal-free hydroalkylation of N-allenyl sulfonamides by push–pull 2-alkynylquinolines. Org. Biomol. Chem. 20, 8269–8272 (2022).
Emadi, M. et al. Design, synthesis, in vitro anti-α-glucosidase evaluations, and computational studies of new phthalimide-phenoxy-1, 2, 3-triazole-N-phenyl (or benzyl) acetamides as potential anti-diabetic agents. Sci. Rep. 13, 10030 (2023).
Dulsat, J., López-Nieto, B., Estrada-Tejedor, R. & Borrell, J. I. Evaluation of free online ADMET tools for academic or small biotech environments. Molecules 28, 776 (2023).
Daina, A., Michielin, O. & Zoete, V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717 (2017).
Bugnon, M. et al. SwissDock 2024: major enhancements for small-molecule Docking with attracting cavities and AutoDock Vina. Nucleic Acids Res. 52, W324–W332 (2024).
Eberhardt, J., Santos-Martins, D., Tillack, A. F., Forli, S. & AutoDock Vina 1.2. 0: new Docking methods, expanded force field, and python bindings. J. Chem. Inf. Model. 61, 3891–3898 (2021).
Acknowledgements
We thankfully acknowledge the support of National Institute for Medical Research Development (NIMAD) (Project code: 4030394).
Funding
NIMAD; Project code: 4030394.
Author information
Authors and Affiliations
Contributions
M.M-K., M.M., M.H., M.E., M.A., and B.L. designed the research work, performed the docking study, and wrote the manuscript. M.H.S., A.M.T, N.D., S.S., and R.Y. synthesized and purified the compounds, and carried out analysis of spectra. M.A. supervised the biological tests. A.S.S.A., M.T. and S.B.V. performed the biological tests. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Varzaneh, S.B., Shokouhi Asl, A., Sayahi, M.H. et al. Design, synthesis, and anti-urease evaluations of new sulfonamide-1,2,3-triazole-acetamide derivatives. Sci Rep 15, 22565 (2025). https://doi.org/10.1038/s41598-025-07553-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-07553-x
