
Abstract
The process of drug discovery is intricate, and encompasses a series of detailed phases of research, development, and testing, aimed at evaluating the safety and effectiveness of prospective therapeutic agents. Artificial Intelligence has emerged as a transformative tool in this domain, adept at analysing vast datasets to uncover intricate patterns and relationships unperceivable to humans. This study introduces a bioactivity prediction application employing the Quantitative Structure-Activity Relationship model to forecast bioactivity against Angiotensin II receptor, a major drug target in hypertension management. Angiotensin II receptor modulation holds promise for treating a spectrum of diseases, including hypertension, cardiovascular ailments, and renal disorders. Through AI-driven approaches researchers in the field of drug discovery are able to effectively identify a majority of promising drug candidates, expediting the lead optimization process while reducing costs. This paradigm shift not only accelerates therapeutic development but also minimizes the need for exhaustive in vitro or in vivo testing, thus enhancing the efficiency of drug discovery endeavours.
Similar content being viewed by others

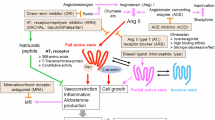

Introduction
Hypertension, defined by international guidelines as sustained blood pressure ≥ 130/80 mmHg and, a major risk factor for cardiovascular disease, stroke, and renal failure, accounting for an estimated 9.4 million deaths annually, according to the World Health Organization (WHO). Despite the availability of several antihypertensive drug classes, hypertension remains under diagnosed and poorly managed, particularly in low- and middle-income countries. This highlights the urgent need for improved diagnostic and therapeutic strategies1,2. The Renin-Angiotensin-Aldosterone System (RAAS) is a critical hormonal cascade that regulates blood pressure, fluid balance, and systemic vascular resistance. Activation of the classical RAAS begins with the release of renin from the juxtaglomerular apparatus of kidneys, which catalyzes the formation of angiotensin I from angiotensinogen. Angiotensin-converting enzyme (ACE) then converts angiotensin (Ang) I to Ang II, a potent vasoconstrictor that exerts its physiological effects primarily through the Angiotensin II Type 1 Receptor (AT1R)3. AT1Rs are widely expressed across key organs involved in cardiovascular regulation, including the vascular system, brain, kidneys, lungs, liver, and adrenal glands. Through AT1R activation, Ang II increases blood pressure by promoting vasoconstriction, stimulating aldosterone secretion, and indirectly enhancing sympathetic nervous system activity. This peptide hormone exerts a profound influence over cardiovascular homeostasis, and deciphering its structural and functional interactions with AT1R is central to advancing hypertension research and drug development4,5,6. Advances in structural biology and computer-aided drug design (CADD) provide new opportunities to develop next-generation AT1R modulators with improved efficacy and safety profiles. Therefore, sustained research into AT1R remains essential for advancing precision medicine in hypertension management and overcoming the limitations of current therapies.
CADD leverages computational tools to explore structure-activity relationships and predict compound bioactivity7,8,9,10. Quantitative Structure-Activity Relationship (QSAR) modeling, enhanced by machine learning (ML), enables improved predictive accuracy, effective feature selection, and broad applicability to diverse chemical scaffolds11,12,13. Their integration has significantly advanced virtual screening, lead optimization, and drug repurposing. While challenges such as model interpretability, overfitting, and the need for external validation persist, ML-based QSAR remains central to the rapid and cost-effective evaluation of large compound libraries14,15,16. This study utilizes bioactivity data from ChEMBL (CHEMBL227) to build ML-based QSAR models using Random Forest (RF) classifiers, used to derive molecular fingerprints and stratify compounds as active or inactive. RF algorithms were implemented to build predictive models, which were evaluated using accuracy, sensitivity, specificity, and Matthews correlation coefficient (MCC). Structural analysis identified key substructures contributing to receptor binding. Additionally, a web-based platform was developed to facilitate interactive exploration of predicted bioactivity and ligand–receptor interactions. These findings demonstrate the potential of ML-enhanced QSAR to accelerate the identification and optimization of novel antihypertensive agents.
Materials and methods
Dataset
The ChEMBL web service package is a powerful tool employed to retrieve essential bioactivity data pertaining to drugs targeting specific entities like Ang II. ChEMBL is extensively utilized in the fields of drug discovery and pharmaceutical research for the purpose of accessing and analyzing data pertaining to drug targets, compounds, and their associated activities17,18,19,20. In the context of the dataset identified by the ChEMBL id CHEMBL227, the target is classified as a “single protein,” and the target organism is specified as “Homo sapiens”. To enhance the specificity of the search for the target ID, a standard measurement type utilizing an IC50 value, which reflects the inhibitory concentration, is employed with efficacy. Initially, the dataset consisted of 1179 rows featuring various compounds, with each compound having a comprehensive set of 45 data columns. The initial compounds are 1179, were the 9 have missing values and removed from the initial compounds. The 1170 are the training after filtering based on the docking scores and Lipinski’s rule finally 758 compounds are filtered. These columns encompassed crucial information such as the standard upper value, assay chembl_id, canonical smiles, and other pertinent details essential in the field of drug discovery and pharmaceutical research.
Data pre-processing
Data pre-processing is essential in the initial stages of preparing/ gathering datasets for utilization in ML models. Random forest was used as the ML model as it is a collection of decision trees that work together to make predictions. Here, we used model performance of the R- value from the training and test are (Training R² = 0.72, Test R² = 0.48) and the hyperparameters of the random forest are the number of Trees (n_estimators) is 100, Maximum Depth (max_depth) is Tuned 20, Minimum Samples per Leaf optimal is 2 and Minimum Samples per Split (min_samples_split) are 4. Within the realm of drug discovery, a series of pre-processing measures have been implemented to scrub and prime the dataset for integration into a QSAR model. The initial step entailed the removal of any compounds that exhibited missing values in the canonical SMILES and standard value columns; as such omissions could lead to inaccuracies in the ML models, ultimately undermining the accuracy of the predictions. Following this, any instances of redundant canonical SMILES representations were eliminated from the dataset, a necessary action to guarantee the uniqueness of each compound and to avert any biases that might skew the performance of the ML models. Subsequently, the essential columns, namely molecule_chembl_id, standard_value, and canonical_smiles, were amalgamated to form a cohesive data frame. This consolidation was undertaken with the aim of crafting a robust dataset tailored for the training of a ML model geared towards the prediction of bioactivity. Moreover, the compounds were categorized into three distinct labels - active, intermediate, or inactive - based on their IC50 values (Fig. 1). This categorization process was instrumental in segregating the compounds according to their bioactivity levels, serving as a pivotal component in the training of a classification ML model. The initial dataset of 1179 compounds was selected based on their relevance to the Ang II receptor, obtained from the ChEMBL database. However, this was only the first stage of data preprocessing. We further refined the dataset through:
-
(1)
Feature filtering: Removal of compounds with missing or incomplete molecular descriptors.
-
(2)
Redundancy elimination: Duplicate or highly similar compounds were removed to ensure diverse chemical space coverage.
-
(3)
Docking score-based selection: Compounds were ranked based on molecular docking affinities to select the most promising candidates.
These preprocessing steps ensured that only high-quality compounds proceeded to ML-based bioactivity prediction, which improves efficiency in identifying potential drug candidates. Upon the culmination of all pre-processing procedures, the dataset boasted a total of 1170 rows and 4 columns, a dataset that was subsequently employed in training a QSAR model dedicated to predicting the bioactivity of novel compounds.

Comparison of active and inactive bioactivity class frequencies.
Calculation of descriptors
Initial step in this process involves calculating the Lipinski descriptors for each compound in the dataset. This includes a set of 4 different physicochemical parameters that are typically utilized to evaluate the drug-like properties of compounds (Fig. 2). Furthermore, the columns are merged with the pre-existing columns, resulting in a dataset consisting of 1170 rows and 8 columns. In order to ensure a more consistent distribution of IC50 data, the IC50 values are transformed into pIC50 values, essentially representing the negative logarithm of the original IC50 values. This transformation enables the conversion of IC50 values spanning across various orders of magnitude into a standardized scale, which in turn facilitates the process of data analysis and comparison. Subsequent to the execution of exploratory data analysis (EDA) utilizing Lipinski descriptors, the dataset is adjusted by eliminating values with intermediate pIC50 values. This adjustment guarantees that the dataset accurately represents both active and inactive compounds, without any bias towards compounds exhibiting intermediate levels of bioactivity.

Box plot of Lipinski’s rule-of five descriptors (A-E) and Chemical space analysis (F).
QSAR model
In the process of QSAR analysis, the input variables consist of the calculated descriptors, while the output variable corresponds to the pIC50 value (Fig. 3). The PaDEL-Descriptor software was employed to calculate the molecular fingerprints of the dataset21. Initially, the data undergoes a division into training and testing sets following 80:20 ratios. Subsequently, a random forest model is developed utilizing the training dataset, and the performance of the model is evaluated by computing the R-squared statistics. A comprehensive analysis involving around 39 distinct ML models from the lazypredict Python framework has been conducted. We conducted an extensive hyperparameter tuning process using Grid Search and Random Search techniques. The 39 models represent different parameter combinations tested to maximize predictive accuracy. These models are meticulously scrutinized using the pre-processed dataset pertaining to activity for Ang-II, with the default parameters being employed.

Work flow of proposed QSAR model.
Creation of web application
After developing a QSAR model for forecasting pIC50 values, the subsequent stage involves the creation of a web application that takes advantage of the model’s capabilities. This web application was developed using Streamlit, which is a Python library designed for the purpose of crafting interactive web applications. The primary objective of this application is to furnish users with a user-friendly platform through which they can carry out predictions of pIC50 values for novel or pre-existing drug molecules targeting the specified enzyme/protein.
Molecular Docking studies
Several studies have shown that Syzygium cumini (Jamun fruit/Indian black cherry) possesses anti-hypertensive properties22. The PIC50 values of phytocompounds of Jamun fruit was predicted using ML approach. The molecular docking study was used for validating the result obtained from the ML approach.
A molecular docking study was conducted to evaluate the binding orientations and interaction affinities between the target proteins and ligands. The top 10 phytocompounds obtained from the ML approach were docked against the target Angiotensin receptor. The 3D structure of the Angiotensin receptor (PDB ID: 4YAY) was retrieved from PDB and before the analysis, protein preparation and energy minimization was done using PyRx software. The 2D structure of top compounds from Syzygium cumini (Jamun fruit/Indian black cherry) was obtained from the PubChem database. The ligands were minimized and converted to .pdbqt format using PyRx. The grid box was generated by enclosing the entire protein with a dimension of X axis: 75.68, Y axis: 63.82 and Z axis: 90.80 (Å). The docking was performed using AutoDock Vina plugin in PyRx 0.8. The scoring function is critical in forecasting the effectiveness of ligand interactions with the target protein. The scoring function employed was AutoDock Vina. The Discovery Studio visualization tool was employed for visualizing the docked compounds23. The Pro-ToX 3.0 server is used to evaluate the pharmacokinetic properties of the top lead compounds24.
Results and discussion
Chemical space analysis
The dataset employed in this study comprises PubChem fingerprints, which are a set of binary structural descriptors generated by the PubChem database to represent the presence or absence of specific molecular substructures and chemical features within each compound25, which includes the SMILES identifiers for 758 compounds, along with their corresponding references. The investigation of the fundamental differences between active and inactive compounds constitutes a primary impetus for conducting chemical space analysis. In this study, the distribution of active and inactive compounds was examined by visualizing the relationship between molecular weight (MW) and the Ghose–Crippen–Viswanadhan octanol–water partition coefficient (logP). This logP value, commonly referred to as ALogP, is a computed estimate of a compound’s lipophilicity, representing the logarithm of its partition coefficient between n-octanol and water. It is widely used in drug discovery to evaluate a molecule’s membrane permeability and potential bioavailability. The calculation relies on an atomistic fragment-based approach originally developed by Ghose and Crippen and later refined by Viswanadhan, and is implemented in cheminformatics tools such as RDKit and Open Babel26. Subsequently, Lipinski’s Rule of Five (Ro5) was employed to analyze the relationship between MW and ALogP for both active and inactive compounds. Consequently, most of the compounds are situated within the MW range of 250–600 Da and exhibit an ALogP value between 0 and 6. A significant proportion of these compounds satisfy the Ro5 criteria. Additionally, the results of the statistical analysis, conducted using the Mann–Whitney U test, reveal a significant distinction between the active and inactive compounds (Table 1).
The ALogP values for inactive compounds were observed to be greater than those for active compounds. While the nHBDon values for both active and inactive compounds were similar, the nHBAcc values for active compounds were determined to be lower than those of their inactive counterparts.
Correlation between predicted bioactivity and binding affinity
The additional testing aimed to examine the correlation between the anticipated bioactivity of various compounds and their respective binding affinities to Ang-II through a comprehensive integration of protein structure extracted from the PDB database (PDB ID: 4YAY) facilitated by our bespoke web-application platform. Initially, compounds from Syzygium cumini were curated in the format of SMILES notation, accompanied by unique compound identifiers, which were subsequently inputted into our web-based tool for the purpose of predicting bioactivity levels expressed as pIC50 values (Table 2). Following this, the 3D molecular structures of the most promising compounds exhibiting elevated pIC50 values were meticulously crafted utilizing the Open Babel software and meticulously primed for molecular docking studies.
The main goal underpinning this methodology was to assess the propensity that the compounds predicted to possess elevated pIC50 values in effectively binding to the target protease with notable degrees of affinity. Following the balancing process, the dataset consisting of 478 compounds was arbitrarily partitioned into internal (80%) and exterior subsets (20%). The internal subset was utilized as the training set to develop predictive models for the external subset. It was observed that the chemical space distribution of the external set fell comfortably within the limits of the internal set. Consequently, the Applicability Domain for the proposed QSAR model appears to be adequately characterized.
In order to verify the robustness of our predictive models, we employed multiple validation techniques, including cross-validation and external validation. Cross-validation was conducted using a k-fold method, which involved dividing the internal dataset into k subsets. The model underwent training and validated k times, with each iteration utilizing a distinct subset for validation while the other k-1 subsets served as the training set. This technique allowed us to assess the model’s generalizability and reduce over fitting. The performance of our predictive models was evaluated using various statistical metrics, including Mean Squared Error (MSE), R-squared, and Root Mean Squared Error (RMSE). These metrics offered valuable information regarding the accuracy and precision of our models in predicting the bioactivity of the compounds (Fig. 4). Furthermore, molecular docking studies were conducted to validate the results obtained from ML approach. The crystallographic 3D configuration of AngII was earmarked for deployment as the receptor molecule throughout the docking experiments. The molecular docking studies were orchestrated utilizing PyRx, with a standardized set of grid box parameters meticulously encompassing the key ligand binding residues as reference points.

Comparison of predicted and experimental pIC50 values.
The docking studies revealed that all the compounds obtained from ML approach shows better binding affinity to the target protein (Table 3). The interacting residues and the pharmacokinetic properties of the top lead compounds are shown in Table 4. The compound Friedelanol shows higher bioactivity in the ML approach as well as good binding affinity among the compounds subjected for docking. The 2D interactions of all the top compounds are shown in Fig. 5.

2D interactions of top 10 compounds of Syzygium cumin.
Model deployment as the web-app for bioactivity analysis and evaluation
To facilitate the application of the prediction model by biologists and chemists lacking a background in computer science, we have created and deployed a publicly accessible web application known as the Bioactivity web-app, which can be accessed at Briefly (Fig. 6). A text file (.txt) must be generated that contains the SMILES ID of the selected compounds, with each identifier separated by spaces. SMILES ID for various small compounds can be obtained from multiple databases, such as PubChem, ChemSpider, and/ or Drugbank. Additionally, custom compounds can be represented using the ChemDraw, ChemSketch, and/or JSME structure editor to generate the SMILES notation for unknown compounds. To access the web application, the specified URL should be entered into any web browser. The generated text file can then be uploaded to the web application by selecting the “Browse files” button. The prediction process can be initiated by clicking the “Predict!” button. The results will be displayed in a designated area below the “Prediction results” heading, where users can select the model they have developed, such as the Ang-II model that has been developed by our team. Typically, the web application requires only a few seconds to complete the processing task. Furthermore, users have the option to download the predicted results in CSV format by clicking the “Download Predictions” button.

Homepage of the deployed web app.
Descriptor calculation
Specifically, there were approximately 667 instances classified as active, 350 instances classified as inactive, and 153 instances classified as intermediate, contributing to the distribution of bioactivity classes as illustrated in Fig. 1. Additionally, a total of 881 descriptors are computed utilizing PaDEL Descriptor software, which is capable of generating an extensive array of molecular descriptors capturing diverse facets of molecular properties including structural, topological, and electronic characteristics. Nevertheless, not all these descriptors prove to be effective in predicting bioactivity, thereby necessitating the removal of descriptors with low variance. Following the process of balancing and elimination of low variance descriptors, the dataset is comprised of 1169 rows and 175 columns. This pre-processed dataset is now ready to be utilized for training a ML model aimed at predicting the bioactivity of compounds. The heatmap (Fig. 7) provided depicts a correlation matrix illustrating the relationship between various molecular properties.

Correlation heatmap of molecular properties in drug discovery.
Text labels on the heatmap represent abbreviated chemical properties as follows: MW: mass of a molecule, LogP: ratio of a molecule’s concentration between two solvents, typically octanol and water, NumHDonors: Number of hydrogen bond donor atoms in the molecule, NumHAcceptors: Number of hydrogen bond acceptor atoms in the molecule and pIC50: Half-maximal inhibitory concentration (IC50) at a physiological pH.The colour intensity within each cell indicates the strength of correlation between the two properties represented by the row and column labels. For instance, a strong negative correlation is observed in the bottom right corner between pIC50 and nHBAcc, indicating that an increase in nHBAcc is associated with a decrease in the pIC50 value.
It is noted that in this particular scenario, the random forest model has demonstrated a commendable R-squared value of 0.89 for the training set and 0.48 for the test set, respectively. Based on the performance metrics (Training R² = 0.72, Test R² = 0.48), the Decision Tree Regressor emerges as the top-performer among the various models, showcasing an impressive R-squared value of 0.92 and a RMSE value of 0.42. These outcomes can be graphically represented to facilitate a deeper comprehension of the findings.
Conclusion
The development of the bioactivity predictor application for Ang-II involved using QSAR modeling techniques in order to forecast the bioactivity levels of newly synthesized compounds. Initially, the molecular descriptors for the chemical compounds under consideration were computed through the application of the PaDEL software. Subsequently, a rigorous process was implemented to eliminate descriptors with low variance and maximum informativeness from the dataset. The tailored bioactivity predictor application, specifically tailored for Ang-II, showcased an impressive level of accuracy, boasting an 85% success rate in aligning its prognostications with the authentic experimental PIC50 values associated with the compounds that were subjected to analysis. In order to delve deeper into the assessment of the reliability and overall performance metrics of the bioactivity predictor application, a visual representation in the form of a scatter plot was generated. This scatter plot was instrumental in illustrating the relationship between the standardized residuals (i.e. the deviations between the predicted bioactivity values and the actual experimental results, standardized for comparison) and the experimental pIC50 values. A thorough analysis of the scatter plot revealed that the data points were distributed across the plot in a manner that appeared to be random, with some points positioned higher and others lower than the zero thresholds on the normalized residual axis. This particular distribution pattern is often interpreted as a positive sign, indicating the absence of any discernible systematic errors in the predictive capabilities of the bioactivity predictor application.
This study demonstrates the successful integration of ML-based QSAR modeling with molecular docking to identify and evaluate potential Ang II receptor inhibitors. By leveraging a curated dataset from ChEMBL and employing algorithms, we developed a predictive model capable of estimating compound bioactivity with reasonable accuracy (Training R² = 0.72, Test R² = 0.48). The model effectively stratified compounds based on their predicted pIC₅₀ values, enabling the identification of high-affinity candidates for further investigation. To validate these predictions, molecular docking studies were conducted using AutoDock Vina via PyRx. The docking results confirmed that several phytochemicals from Syzygium cuminiexhibited favorable binding affinities with the Ang II receptor. Notably, compounds such as Friedelanol and Myricetin 3-O-glucoside displayed both high predicted pIC₅₀ values and strong docking scores, suggesting their potential as lead molecules for antihypertensive therapy. This combined computational approach not only enhances the efficiency of early-stage drug discovery but also offers a robust framework for identifying bioactive compounds with therapeutic relevance. Future studies incorporating molecular dynamics simulations and in vitro validation would further substantiate the findings and support the development of novel antihypertensive agents targeting the Ang II receptor.
Data availability
All the data is provided within the manuscript.
References
Laurent, S. & Boutouyrie, P. Arterial stiffness and hypertension in the elderly. Front. Cardiovasc. Med. 7, 544302. https://doi.org/10.3389/fcvm.2020.544302 (2020).
Flack, J. M. & Adekola, B. Blood pressure and the new ACC/AHA hypertension guidelines. Trends Cardiovasc. Med. 30 (3), 160–164 (2020).
Su, C., Xue, J., Ye, C. & Chen, A. Role of the central renin-angiotensin system in hypertension. International J. Mol. Medicine 47(6), (2021).
Paul, M., Poyan Mehr, A. & Kreutz, R. Physiology of local renin-angiotensin systems. Physiol. Rev. 86 (3), 747–803. https://doi.org/10.1152/physrev.00036.2005 (2006). https://journals.physiology.org/doi/full/
Guo, D. F., Uno, S., Ishihata, A., Nakamura, N. & Inagami, T. Identification of a cis-acting glucocorticoid responsive element in the rat angiotensin II type 1A promoter. Circul. Res. 77 (2), 249–257. https://doi.org/10.1161/01.RES.77.2.249 (1995).
Dasgupta, C. & Zhang, L. Angiotensin II receptors and drug discovery in cardiovascular disease. Drug Discovery Today. 16 (1–2), 22–34. https://doi.org/10.1016/j.drudis.2010.11.016 (2011).
Prachayasittikul, V. et al. Computer-aided drug design of bioactive natural products. Curr. Top. Med. Chem. 15 (18), 1780–1800 (2015).
Nantasenamat, C., Isarankura-Na-Ayudhya, C. & Prachayasittikul, V. Advances in computational methods to predict the biological activity of compounds. Expert Opin. Drug Discov. 5 (7), 633–654. https://doi.org/10.1517/17460441.2010.492827 (2010).
Suvannang, N. et al. Probing the origin of Estrogen receptor alpha Inhibition via large-scale QSAR study. RSC Adv. 8 (21), 11344–11356. https://doi.org/10.1039/C7RA10979B (2018).
Abdullahi, M., Adeniji, S. E., Arthur, D. E. & Musa, S. Quantitative structure-activity relationship (QSAR) modelling study of some novel Carboxamide series as new anti-tubercular agents. Bull. Natl. Res. Centre. 44 (1), 136. https://doi.org/10.1186/s42269-020-00389-7 (2020). https://link.springer.com/article/
Prachayasittikul, V. et al. Synthesis, anticancer activity and QSAR study of 1, 4-naphthoquinone derivatives. Eur. J. Med. Chem. 84, 247–263. https://doi.org/10.1016/j.ejmech.2014.07.024 (2014).
FF da Cunha, E., Matos, S., Ramalho, C. & K., & QSAR and Docking studies of HCV NS3 Serine protease inhibitors. Med. Chem. 9 (6), 774–805 (2013).
Worachartcheewan, A., Prachayasittikul, V., Toropova, A. P., Toropov, A. A. & Nantasenamat, C. Large-scale structure-activity relationship study of hepatitis C virus NS5B polymerase Inhibition using SMILES-based descriptors. Mol. Diversity. 19, 955–964. https://doi.org/10.1007/s11030-015-9614-2 (2015).
Baskin, I. I. Machine learning methods in computational toxicology. Computational toxicology: Methods and Protocols. 119 – 39, (2018).
Muratov, E. N. et al. QSAR without borders. Chemical Soc. Reviews 49(11), (2020).
Lenselink, E. B. et al. Beyond the hype: deep neural networks outperform established methods using a chembl bioactivity benchmark set. J. Cheminform. 9, 1–4 (2017).
Bento, A. P. et al. The chembl bioactivity database: an update. Nucleic Acids Res. 42 (D1), D1083–D1090. https://doi.org/10.1093/nar/gkt1031 (2014).
Gaulton, A. et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40 (D1), D1100–D1107. https://doi.org/10.1093/nar/gkr777 (2012).
Gaulton, A. et al. The ChEMBL database in 2017. Nucleic acids research. 45(D1), D945-D954. (2017). https://doi.org/10.1093/nar/gkw1074
Mendez, D. et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 47 (D1), D930–D940. https://doi.org/10.1093/nar/gky1075 (2019).
Yap, C. W. PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 32 (7), 1466–1474. https://doi.org/10.1002/jcc.21707 (2011).
Rizvi, M. K. et al. Astounding health benefits of Jamun (Syzygium cumini) toward metabolic syndrome. Molecules 27 (21), 7184 (2022).
Tahir, M., Baharuddin, M. & Najib, A. In Silico screening of Brotowali (Tinospora Crispa L.) chemical compounds as α-glucosidase inhibitor using the pyrx program. AIP Publishing. 2595 (1). https://doi.org/10.1063/5.0123693 (2023).
Banerjee, P., Kemmler, E., Dunkel, M. & Preissner, R. ProTox 3.0: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 52(W1), W513–W520 (2024).
Kim, S. et al. PubChem substance and compound databases. Nucleic Acids Res. 44 (D1), D1202–D1213 (2016).
Viswanadhan, V. N., Ghose, A. K., Revankar, G. R. & Robins, R. K. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships. 4. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certain naturally occurring nucleoside antibiotics. J. Chem. Inf. Comput. Sci. 29 (3), 163–172 (1989).
Acknowledgements
The authors JS, VR, and KM thankfully acknowledge the DBT-Bioinformatics and Computational Biology Centre (BIC)—No.BT/PR40154/BTIS/137/34/2021 for the infrastructure facilities. The authors, BBK and AHA, extend their appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work through the Small Research Project under grant number RGP 1/75/46.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Bioinformatics investigation, Methodology, Validation and manuscript writing—Bioinformatics were performed by JS. Methodology, and data curation— ML approach was performed by VR. Writing review and editing were performed by JS and KM. Supervision and investigation were done by BBK, AHA, LSW and KM. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sankar, J., Rajendran, V., Kuriakose, B.B. et al. ML enhanced bioactivity prediction for angiotensin II receptor: A potential anti-hypertensive drug target. Sci Rep 15, 25367 (2025). https://doi.org/10.1038/s41598-025-08653-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-08653-4
