
Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is a genetic disorder characterized by cyst formation in the kidneys, and is associated with an elevated risk of intracranial aneurysms (IAs). Although a family history is a recognized risk factor for IAs in patients with ADPKD, emerging research suggests that gut microbiome composition may influence IA development. We investigated the relationship between the gut microbiome and the development of IA in patients with ADPKD. We recruited patients with ADPKD with (IA group) and without (non-IA group) IA from Osaka University between October 2021 and December 2023. Fecal samples were analyzed using 16S rRNA sequencing. Data were processed using the QIIME 2 pipeline to determine microbial diversity and composition. We included 60 patients: 26 in the IA and 34 in the non-IA groups. There were significant differences in microbial beta diversity between the groups. The IA group had higher abundances of Eubacterium siraeum group, Oscillibacter, Fournierella, Negativibacillus, Colidextribacter, and Adlercreutzia. The non-IA group had higher abundances of Bifidobacterium, Megamonas, Acidaminococcus, Megasphaera, and Merdibacter. There was a significant association between the gut microbiome composition and the presence of IAs in patients with ADPKD. Specific bacterial taxa were differentially abundant between patients with ADPKD with and without IAs, suggesting a potential role of the gut microbiome in the pathogenesis of IAs in this genetically predisposed population.
Similar content being viewed by others


Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is a genetic disorder that causes cystic changes in the kidneys and is associated with a high risk of intracranial aneurysms (IAs). Patients with ADPKD have a four times higher prevalence of IAs than the general population, with rates ranging from 9 to 12%1,2. Additionally, subarachnoid hemorrhage (SAH) in this population occurs at median age of 43 years, approximately ten years earlier than that in the general population3. A family history of IAs is a widely known risk factor for ADPKD aneurysms, and some reports have suggested an association with total kidney volume (TKV) and kidney unction4,5,6. However, no association with environmental factors has been identified, and there is no means of preventing the development of IAs in this population.
Recently, several reports have investigated the relationship between the gut microbiome and the occurrence of IAs or SAH7,8,9,10,11,12,13. The gut microbiome is a potential intervening environmental factor that could lead to the establishment of preventive treatments for the development and rupture of IAs. However, there have been no reports on the association between the gut microbiome and IAs specifically in patients with ADPKD.
We hypothesized that the gut microbiome may influence the development of IAs even in patients with ADPKD, who are genetically predisposed to develop IAs. In addition, we also hypothesized that studies in populations at high risk of developing IAs are best suited to identify the types of gut microbiome that are associated with IAs, which could lead to the discovery of future therapies, including those in the general population. Therefore, we aimed to determine the impact of the gut microbiome on the development of IAs in patients with ADPKD.
Results
Patients’ characteristics
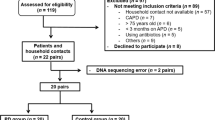
A total of 60 patients were recruited between October 2021 and December 2023. Consequently, 26 patients in the IA group and 34 in the non-IA group were included in this study (Fig. 1). Three patients, two in the IA group and one in the non-IA group, were from the same family. The characteristics of the patients and aneurysms are summarized in Table 1 and Supplementary Table S1. Patient characteristics, including age, sex, dyslipidemia, diabetes status, renal function (estimated glomerular filtration rate, blood urea nitrogen, chronic kidney disease stage, and TKV), ischemic heart disease, smoking, alcohol consumption, body mass index, and modified Rankin Scale (mRS), did not differ between patients with IAs and those without IAs. However, patients with a family history of IAs were significantly more common in the IA group (p = 0.0032), as were patients with hypertension (HT, P = 0.0044). The 26 patients in the IA group had 39 aneurysms. The average aneurysm size was 3.82 mm, and the most common location was the middle cerebral artery. Four patients had a history of SAH. Of the 39 aneurysms, eight underwent clipping, four underwent coil embolization, and the remaining 27 were untreated and followed up. Regarding medication history, the use of calcium-channel blockers (P = 0.0299) and spironolactone (P = 0.0076) was significantly higher in the IA group (Supplementary Table S2).

Flow diagram for the screening of patients with ADPKD. IA; intracranial aneurysm, UIA; unruptured intracranial aneurysm, ADPKD; autosomal dominant polycystic kidney disease.
Differences in gut microbiome between the IA and non-IA groups
The datasets for the IA and non-IA groups comprised 4882 features that classified 266 genera, 91 families, 48 orders, 23 classes, and 12 phyla. No difference was observed in alpha diversity between the IA and non-IA groups, including the observed species, Shannon index, evenness, and phylogenetic diversity of the microbial community (Fig. 2A). However, a significant difference in beta diversity was observed between the weighted (p = 0.033) and unweighted UniFrac distances (P = 0.012) (Fig. 2B).

Comparison of microbial diversity analysis between IA and non-IA groups. (A) No significant differences were found in alpha diversity based on the observed Shannon, evenness, and faith indices (p > 0.05). (B) Principal coordinate analysis illustrating the grouping patterns of IA and non-IA groups based on weighted and unweighted UniFrac distances. Red points; IA group, and green points; non-IA group. There were significant differences in beta diversity between the weighted (p = 0.033) and unweighted (p = 0.012) UniFrac distances.
The dominant phyla in all the patients were Firmicutes and Bacteroidetes (Table 2, Fig. 3A,B). Although significant differences in the relative abundance of Bacteroidota and Actinobacteriota were observed at the phylum level in univariate analysis, these differences did not remain significant in multivariate analysis or after correction for false discovery rate.

Comparison of microbiome composition at the phylum level between IA and non-IA groups. (A, B) Distribution of the relative abundance of bacteria at the phylum level. The relative abundance of Firmicutes, Bacteroidota and Actinobacteriota did not differ significantly between the groups.
Via linear discriminant analysis (LDA) and effect size (LEfSe), we identified two bacterial phyla, three classes, five orders, five families, and 16 genera that had significantly different relative abundances between the IA and non-IA groups (Fig. 4). Among these, the bacteria that could be classified at the genus level were Eubacterium siraeum group, Oscillibacter, Fournierella, Negativibacillus, Colidextribacter, and Adlercreutzia in the IA group, and Bifidobacterium, Megamonas, Acidaminococcus, Megasphaera, and Merdibacter in the non-IA group.

Discriminative taxa between IA and non-IA groups. (A) Discriminative taxa between the IA and non-IA groups were determined using linear discriminant analysis effect size (LEfSe). LEfSe revealed that two bacterial phyla, classes, orders, families, and genera were significantly different between the IA and non-IA groups. The green bar represents the bacteria that were more abundant in the non-IA group, and the red bar represents the bacteria that were more abundant in the IA group. (B) The cladograms report the taxa showing different abundance values according to LEfSe.
Discussion
In this study, we compared the gut microbiome of patients with ADPKD classified into the IA group and non-IA group and found significant differences at the genus level. The IA group had higher levels of E. siraeum, Oscillibacter, Fournierella, Negativibacillus, Colidextribacter, and Adlercreutzia, whereas the non-IA group had higher levels of Bifidobacterium, Megamonas, Acidaminococcus, Megasphaera, Merdibacter Megasphaera, Merdibacter, and Bifidobacterium. There are no reports showing the relationship between IAs and the gut microbiome in patients with ADPKD; thus, this study is the first.
Only one study has examined the gut microbiome of patients with ADPKD14; however, its association with IAs remains uninvestigated. In our study, the most common bacteria in the IA group were E. siraeum, Oscillibacter, Fournierella, Negativibacillus, Colidextribacter, and Adlercreutzia. Of these, Fournierella and Adlercreutzia have been reported to be associated with IAs in the general population10,12. Fournierella has been reported to be more common in symptomatic unruptured IAs presenting with oculomotor nerve palsy or headache10, and Adlercreutzia is positively associated with the risk of unruptured IAs12. However, there are no reports suggesting an association between E. siraeum, Oscillibacter, Negativibacillus, and Colidextribacter and IAs. These bacteria may also be involved in the development of IAs. Other studies that focused on IAs in non-ADPKD patients reported that Campylobacter is associated with rupture of IAs7 or that Hungatella hathewayi is associated with the development of IAs8, but these microorganisms were not identified in our study.
Bifidobacteria, Megamonas, Acidaminococcus, Megasphaera, and Merdibacter were commonly found in the non-IA group. Bifidobacterium is a major component of the gut microbiome in humans, which exhibits anti-inflammatory activity and plays a role in immune regulation15. In vascular lesions, Bifidobacterium is significantly less abundant in patients with abdominal aortic aneurysms16. The other bacteria common to the non-IA group belonged to the phylum Firmicutes. The phylum Firmicutes has also been reported to possess anti-inflammatory properties17. In summary, many bacteria reported to be associated with IAs in the general population were identified in the IA group. However, some bacteria were newly identified. In the non-IA group of patients with ADPKD, many bacteria were reported to be involved in anti-inflammatory processes. In ADPKD, a reduction in anti-inflammatory bacteria has been shown to be involved in the formation of IAs, which may also be applicable to aneurysms in the general population.
Increased expression of NFκB in vascular endothelial cells from ADPKD patients has been reported to be significantly higher than that in those from non-ADPKD patients, suggesting the involvement of inflammation in vascular lesions18. Furthermore, studies have indicated a link among ADPKD, immunity, and inflammation19,20. The gut microbiome has been shown to regulate systemic inflammation21,22,23, and its relationship with NFκB has been reported24,25. In this study, some bacteria detected in high abundance in the IA group may alter inflammation in the blood vessels of ADPKD patients; it is not clear whether these bacteria are solely involved in inflammation. Additionally, many bacterial species with anti-inflammatory properties were detected in the non-IA group, suggesting that probiotics promoting the growth of these bacteria may reduce the incidence of IA in patients with ADPKD. This could also lead to a reduction in the incidence of IAs in the general population.
ADPKD is caused by mutations in the PKD1 (16p13.3) and PKD2 (4q21) genes, and lesions of the vascular system have been attributed to decreased protein levels of polycystin 1 or 2 in vascular endothelial cells and vascular smooth muscle cells2,26. The most well-known association between the incidence of IAs and ADPKD is a family history of IAs1. Other risk factors for IAs in patients with ADPKD include female sex and HT, as well as in the general population4,5. More recently, worsening renal function and increased TKV were associated with the presence of IAs4,5,6, suggesting that ADPKD disease status may be related to the development of IAs. Although our study had a small sample size, family history of IAs and HT were correlated with the incidence of IA, but renal function and TKV were not (Table 1).
Medications administered at the time patients’ stool samples were collected were also investigated (Supplementary Table 2). This is because drugs other than antibiotics are known to alter the composition of the gut microbiome27. Calcium-channel blockers and spironolactone were more frequently administered in the IA group, possibly because of the significantly higher number of patients with HT in this IA. However, there are no reports of calcium-channel blockers and spironolactone altering the composition of the gut microbiome, and no significant differences were observed for other drugs that affect the gut microbiome, as previously described27. In addition, tolvaptan, the only approved medication for slowing the progression of rapidly progressive ADPKD, showed no significant difference between the groups in this study (Supplementary Table S2). However, this mechanism of action involves inhibiting the abnormal elevation of intracellular cyclic adenosine monophosphate levels in cystic epithelial cells28, which may contribute to inflammation suppression and could potentially influence the incidence of intracranial aneurysms in the future.
This study has certain limitations. First, the IA group included a mix of patients with unruptured aneurysms, ruptured aneurysms except those in the acute phase, and patients who underwent various therapeutic interventions. Thus, the differences in the gut microbiome revealed in this study are clearly related to the presence of IAs; however, a relationship with IA rupture cannot be demonstrated. Second, we were unable to assess the dietary habits that are thought to have the greatest impact on the gut microbiome. Third, we did not examine metabolites in this study, nor did we analyze bacteria at the species level. Further studies using metagenomic analysis or other methods are warranted. Additionally, no genetic analysis was performed in this study. As there was a significant difference in the family history of IAs between the IA and non-IA groups, the possibility that differences in genetic background may have influenced the results cannot be ruled out. Genetic variants are associated with IA formation29. However, the composition of the gut microbiome is similar in families30, and factors thought to be genetic may actually be influenced by the gut microbiome.
In conclusion, even in patients with ADPKD, which is thought to have a strong genetic component, the gut microbiome—an environmental factor—is related to the presence of IAs. This result may also be applicable to aneurysms in the general population. Further research may allow therapeutic intervention for the gut microbiome to reduce the incidence of IAs in patients with ADPKD.
Methods
Standard protocol approvals, registrations, and patient consent
The investigators obtained approval from the Ethical Review Board of Osaka University Hospital (no. 22310) before initiating the case–control study. Each patient was fully informed of the study and provided written informed consent prior to participation. All methods were performed in accordance with the relevant guidelines and regulations.
Study population
Participants with IA (IA group) and those without IA (non-IA group) were prospectively recruited between October 2021 and December 2023 from Osaka University.
In principle, all patients with ADPKD at our institution underwent head MRI examination, and the inclusion criteria for each group were as follows: patients meeting the diagnostic criteria for ADPKD31,32,33 with or without an IA confirmed by head magnetic resonance imaging (MRI) in 2017 or later. We excluded patients aged < 20 years, those with any other associated genetic predisposition known to contribute to IA formation (e.g., Ehlers–Danlos syndrome Type 4), those who developed SAH within 1 year, or those who used antibiotics within 1 month prior to fecal sampling. The IA group included patients with unruptured IAs and a history of SAH. The non-IA group included patients who underwent head MRI in 2017 or later and there was no detection of an IA.
Fecal sample collection
Fecal samples were collected at home and packed into frozen gel packs in insulated containers. Within 24 h, the sample collection kits were returned and stored at ‒80 °C until processing.
Bacterial DNA extraction and 16S rRNA sequencing
DNA was extracted from the fecal samples using an automated DNA extraction machine (GENE PREP STAR PI-480, Kurabo Industries, Osaka, Japan) following the manufacturer’s protocol. The V1–V2 region of the 16S rRNA gene was amplified using the forward primer (16S_27Fmod: TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG AGR GTT TGA TYM TGG CTC AG) and the reverse primer (16S_338R: GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GTG CTG CCT CCC GTA GGA GT) using the KAPA HiFi Hot Start Ready Mix (Kapa Biosystems, Wilmington, MA)34.
To sequence the 16S amplicons using the Illumina MiSeq platform (Illumina, San Diego, CA), dual-index adapters were attached using the Nextera XT Index kit (Illumina). Libraries were prepared according to the Illumina 16S library preparation protocol. Libraries were sequenced using the MiSeq Reagent Kit v2 (500 cycles) and 250 bp paired-end reads.
Microbiome bioinformatics
The generated FASTQ files were imported, demultiplexed, and processed using the Quantitative Insights into Microbial Ecology 2 (QIIME 2) pipeline35. First, partial 16S rRNA bacterial sequences were qualitatively trimmed, and the reads were truncated into operational taxonomic units (OTUs) using the software package Divisive Amplicon Denoising Algorithm 2 in QIIME2 v2023.2. Taxonomy was assigned based on the Silva 138 database release at the 99% OTU level. A phylogenetic tree was created to generate phylogenetic diversity measures using the q2-phylogeny plugin in QIIME2.
To detect the alpha diversity, including the observed species, Shannon index, evenness, and phylogenetic diversity between groups, we processed the output files generated in the previous steps using GraphPad Prism version 10.2.0 (GraphPad Software, San Diego, CA). Beta diversity was calculated using a phylogenetic tree in weighted and unweighted UniFrac software. Permutational multivariate analysis of variance was used in GraphPad Prism to assess the statistical differences in beta diversity metrics between groups.
LEfSe was performed to identify significantly different taxa between groups36. The LDA score was then used to estimate the effect size of each differentially abundant feature. The threshold of the LDA score was defined as ± 2.0 when comparing the relative taxa abundances between the two groups.
Statistical analysis
The distribution of each continuous variable was assessed using the Shapiro–Wilk W test. Normally distributed parametric data were analyzed using unpaired Student’s t-tests, and the non-normally distributed nonparametric data were analyzed using the Wilcoxon test. Categorical variables were analyzed using the chi-squared test or Fisher’s exact test as appropriate. False discovery rate correction using the Benjamin-Hochberg procedure was performed when multiple comparisons were made to evaluate differences between 2 groups. Multivariate analysis for relative abundance at the phylum level was performed by logistic regression analysis. Values of P < 0.05 or false discovery rate q < 0.05 were considered significant. Statistical analyses were performed using JMP software, version 17.1.0 (SAS Institute, Cary, NC).
Data availability
The sequencing data have been deposited in the DDBJ BioProject database under accession number PRJDB20223.
References
Xu, H.-W., Yu, S.-Q., Mei, C.-L. & Li, M.-H. Screening for intracranial aneurysm in 355 patients with autosomal-dominant polycystic kidney disease. Stroke 42, 204–206 (2011).
Cornec-Le Gall, E., Alam, A. & Perrone, R. D. Autosomal dominant polycystic kidney disease. Lancet 393, 919–935 (2019).
Nurmonen, H. J. et al. Polycystic kidney disease among 4436 intracranial aneurysm patients from a defined population. Neurology 89, 1852–1859 (2017).
Ushio, Y. et al. Factors associated with early-onset intracranial aneurysms in patients with autosomal dominant polycystic kidney disease. J. Nephrol. 37, 983–992 (2024).
Kataoka, H. et al. Impact of kidney function and kidney volume on intracranial aneurysms in patients with autosomal dominant polycystic kidney disease. Sci. Rep. 12, 18056. https://doi.org/10.1038/s41598-022-22884-9 (2022).
Yoshida, H. et al. Relationship between intracranial aneurysms and the severity of autosomal dominant polycystic kidney disease. Acta Neurochir. (Wien) 159, 2325–2330 (2017).
Kawabata, S. et al. Dysbiosis of gut microbiome is associated with rupture of cerebral aneurysms. Stroke 53, 895–903 (2022).
Li, H. et al. Alterations of gut microbiota contribute to the progression of unruptured intracranial aneurysms. Nat. Commun. 11, 3218. https://doi.org/10.1038/s41467-020-16990-3 (2020).
Shikata, F. et al. Potential influences of gut microbiota on the formation of intracranial aneurysm. Hypertension 73, 491–496 (2019).
Sun, K. et al. Altered gut microbiomes are associated with the symptomatic status of unruptured intracranial aneurysms. Front. Neurosci. 16, 1056785. https://doi.org/10.3389/fnins.2022.1056785 (2022).
Kawabata, S. et al. Association of gut microbiome with early brain injury after subarachnoid hemorrhage: An experimental study. Transl. Stroke Res. 15, 87–100 (2024).
Ma, C. et al. Association of gut microbiome with risk of intracranial aneurysm: A Mendelian randomization study. BMC Neurol. 23, 269. https://doi.org/10.1186/s12883-023-03288-2 (2023).
He, M. et al. Genetic causal association between the gut microbiome and intracranial aneurysm and subarachnoid hemorrhage: A two-sample Mendelian randomization study. Neurol. Ther. 12, 1695–1707 (2023).
Yacoub, R. et al. Fecal microbiota analysis of polycystic kidney disease patients according to renal function: A pilot study. Exp. Biol. Med. (Maywood) 244, 505–513 (2019).
Gavzy, S. J. et al. Bifidobacterium mechanisms of immune modulation and tolerance. Gut Microbes 15, 2291164. https://doi.org/10.1080/19490976.2023.2291164 (2023).
Ito, E. et al. Impact of Bifidobacterium adolescentis in patients with abdominal aortic aneurysm: A cross-sectional study. Biosci. Microbiota Food Health 42, 81–86 (2023).
Bolte, L. A. et al. Long-term dietary patterns are associated with pro-inflammatory and anti-inflammatory features of the gut microbiome. Gut 70, 1287–1298 (2021).
Nowak, K. L. et al. Vascular dysfunction, oxidative stress, and inflammation in autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 13, 1493–1501 (2018).
Zheng, D. et al. Urinary excretion of monocyte chemoattractant protein-1 in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 14, 2588–2595 (2003).
Lee, Y. et al. Semaphorin 7A in circulating regulatory T cells is increased in autosomal-dominant polycystic kidney disease and decreases with tolvaptan treatment. Clin. Exp. Nephrol. 22, 906–916 (2018).
Agirman, G., Yu, K. B. & Hsiao, E. Y. Signaling inflammation across the gut-brain axis. Science 374, 1087–1092 (2021).
Johnson, K.V.-A. & Foster, K. R. Why does the microbiome affect behaviour?. Nat. Rev. Microbiol. 16, 647–655 (2018).
Tang, W. H. W., Kitai, T. & Hazen, S. L. Gut microbiota in cardiovascular health and disease. Circ. Res. 120, 1183–1196 (2017).
Zhang, S., Paul, S. & Kundu, P. NF-κB regulation by gut microbiota decides homeostasis or disease outcome during ageing. Front. Cell. Dev. Biol. 10, 874940. https://doi.org/10.3389/fcell.2022.874940 (2022).
Chalouhi, N. et al. Biology of intracranial aneurysms: Role of inflammation. J. Cereb. Blood Flow Metab. 32, 1659–1676 (2012).
Vasileva, V. Y., Sultanova, R. F., Sudarikova, A. V. & Ilatovskaya, D. V. Insights into the molecular mechanisms of polycystic kidney diseases. Front. Physiol. 12, 693130. https://doi.org/10.3389/fphys.2021.693130 (2021).
Maier, L. et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 555, 623–628 (2018).
Jdiaa, S., Mustafa, R. & Yu, A. Treatment of autosomal-dominant polycystic kidney disease. Am. J. Kidney Dis. S0272–6386(24), 01032–01041. https://doi.org/10.1053/j.ajkd.2024.08.008 (2024).
Kataoka, H. et al. Mutation type and intracranial aneurysm formation in autosomal dominant polycystic kidney disease. Stroke Vasc. Interv. Neurol. https://doi.org/10.1161/SVIN.121.000203 (2022).
Valles-Colomer, M. et al. The person-to-person transmission landscape of the gut and oral microbiomes. Nature 614, 125–135 (2023).
Iliuta, I.-A. et al. Polycystic kidney disease without an apparent family history. J. Am. Soc. Nephrol. 28, 2768–2776 (2017).
Pei, Y. Diagnostic approach in autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 1, 1108–1114 (2006).
Barua, M. & Pei, Y. Diagnosis of autosomal-dominant polycystic kidney disease: An integrated approach. Semin. Nephrol. 30, 356–365 (2010).
Kim, S.-W. et al. Robustness of gut microbiota of healthy adults in response to probiotic intervention revealed by high-throughput pyrosequencing. DNA Res. 20, 241–253 (2013).
Guerrini, C. J., Botkin, J. R. & McGuire, A. L. Clarify the HIPAA right of access to individuals’ research data. Nat. Biotechnol. 37, 850–852 (2019).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. https://doi.org/10.1186/gb-2011-12-6-r60 (2011).
Acknowledgements
We thank Cykinso Inc. (Tokyo, Japan) for sequencing the 16S rRNA gene. This work was supported by JSPS KAKENHI, Grant Numbers 21K09072 and 22K09282.
Author information
Authors and Affiliations
Contributions
Conceptualization,TF and MT; Data curation, TF, RN, TM, KT, HY and MT; Formal analysis, TF, MT, DM, SN and SK; Funding acquisition, MT and RN; Investigation; TF, MT and JK; Methodology, TF, MT, DM and SN; Project administration, TF, MT, JK, YI and HK; Resources, MT, JK, HN, TO, YI and HK; Supervision, YI and HK; Visualization, TF; Writing-original draft preparation, TF; Writing-review and editing, MT, JK, DM, SN, SK, HN, TO, RN, TM, KT, HY, YI and HK; All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fukuda, T., Takagaki, M., Kaimori, J. et al. Differences in gut microbiome between autosomal dominant polycystic kidney disease with and without intracranial aneurysms. Sci Rep 15, 24204 (2025). https://doi.org/10.1038/s41598-025-08942-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-08942-y
