
Abstract
In this study, a novel series of mercapto-phenyl-1,2,4-triazole-bearing thio-quinoline moieties was designed, synthesized, and evaluated for their anti-tyrosinase activities. All compounds were tested for inhibitory activity against tyrosinase, compound 12j was found to be the most potent with IC50 = 10.49 ± 1.02 µM. Structure–activity relationship (SAR) analysis indicated that the introduction of electron-donating and electron-withdrawing groups at specific positions influenced the inhibitory efficacy. The antioxidant activity of all derivatives were also performed, and 12j showed IC50 = 102.36 ± 3.33 µM. In silico molecular docking studies showed that compound 12j had the strongest binding affinity (binding energy = − 8.04 kcal/mol) and formed stable hydrogen bonds with key active site residues (e.g., His85, His259, and His296) of tyrosinase. Molecular dynamics simulations have further exhibited the high stability and compactness of the 12j–tyrosinase complex, with minimum RMSD fluctuations and stable hydrogen bonding patterns. These results suggest the potency of these derivatives as promising tyrosinase inhibitors with useful information into their mechanism, establishing a foundation for future therapeutic applications in hyperpigmentation and related disorders.
Similar content being viewed by others
Introduction
Melanin is a natural pigment responsible for the color of skin, hair, and eyes in humans and other animals1. Melanin exists mostly in two forms: eumelanin, which produces black and brown tones, and pheomelanin, responsible for red and yellow hues. However, over-expression of melanin is associated with the development of hyperpigmentation-related complications such as melasma, freckles, age spots, and post-inflammatory hyperpigmentation2.
The production of melanin is controlled by a key enzyme called tyrosinase, which initiates and regulates the complex biochemical pathway of melanin synthesis (melanogenesis)3. Tyrosinase is a copper-containing enzyme that catalyzes the hydroxylation of tyrosine to L-DOPA and its subsequent oxidation to o-quinones, which are key precursors in melanin synthesis. This reaction leads to the formation of dopachrome, activating a cascade of biochemical events that culminate in melanogenesis4,5. Tyrosinase has diverse roles, including plant browning, skin pigmentation in humans, arthropod immune responses, and fungal spore development6,7. These observations confirm the importance of tyrosinase inhibitors which effectively reduce melanin synthesis, offering a valuable approach to managing pigmentation disorder8,9. Additionally, tyrosinase inhibitors can be used in food preservation to prevent the browning of fruits and vegetables10,11,12. Kojic acid as a tyrosinase inhibitor is widely used as a skin-whitening agent. However, kojic acid is associated with potential side effects such as dermatitis, sensitization, and erythema13. As a result, the evaluation of a wide range of tyrosinase inhibitors has yielded valuable outcomes. Numerous studies have shown that the presence of a thiol (–SH) group in the molecular structure significantly enhances the inhibitory potential of compounds against this enzyme due to interaction with two copper cofactors of tyrosinase. Considering this observation, extensive research has been conducted to identify and integrate highly potent pharmacophores containing the –SH group into their structures, aiming to develop more effective tyrosinase inhibitors14,15.
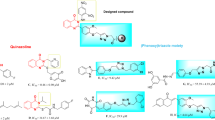
Heterocyclic compounds are highly favored by chemists due to their remarkable biological and pharmacological properties16,17,18,19. Among these, the quinoline framework stands out as a prominent heterocyclic scaffold due to its straightforward synthesis and its broad spectrum of biological properties as tyrosinase inhibitors20,21,22,23. Thio-quinazolinone derivatives conjugated with kojic acid (compound A, Fig. 1) were synthesized and assessed for their inhibitory effects on tyrosinase. Among these, the most potent derivative exhibited notable tyrosinase inhibition, showing a 68.99% reduction in melanin content at a concentration of 8 µM, demonstrating its potential for managing pigmentation disorders24. Structure–activity relationship (SAR) analysis of thioquinoline derivatives linked to thiosemicarbazide revealed that introducing an electron-withdrawing group at the para position of the aromatic ring significantly enhanced the inhibitory activity compared to other derivatives. Notably, derivative B exhibited a significant reduction in melanin content when tested against A375 cell lines, supporting its potential as a promising agent for pigmentation regulation25.

Designing strategy.
Also, thio-1,2,4-triazole has demonstrated significant tyrosinase inhibition, and compound C exhibited an IC50 of 4.52 ± 0.09 µM, outperforming kojic acid (IC50 = 30.34 ± 0.75 µM)26. Another 2024 study evaluated pyrazole-1,2,4-triazole derivatives, identifying the most potent compound as being 14 times more effective than kojic acid. Fluorescence quenching and ANS-fluorescence quenching assays revealed that compound D (Fig. 1) could bind to tyrosinase and alter its intrinsic fluorescence intensity, indicating strong enzyme interaction27. Furthermore, the effectiveness of 1,2,4-triazole-thiol derivatives was confirmed in another study, where a derivative exhibited an IC50 of 0.9 ± 0.1 µM, significantly surpassing kojic acid (IC50 = 64.1 ± 7.8 µM) in tyrosinase inhibition. These findings underscore the potential of thio-1,2,4-triazole derivatives as tyrosinase inhibitors with applications in managing pigmentation disorders and food browning28.
Apart from their pigmentation function, tyrosinase and its oxidative pathways are related to oxidative stress and reactive oxygen species (ROS) production. Of particular interest, heterocyclic frameworks of quinoline and triazole were found to have potent antioxidant capacity29,30,31. Based on the fact that oxidative damage is involved in the majority of skin diseases and the aging process, derivatives containing antioxidant and tyrosinase inhibition activities may have more potential in therapeutic efficacy applications related to cosmeceutical and dermatological applications32.
In silico approaches are essential to contemporary drug design as they provide fast, low-cost prediction of biological activity, target identification, and pharmacokinetic characteristics. These computational approaches, on one hand, allow the virtual screening of compound libraries, and the interactions between a ligand and receptor at the molecular level will be monitored33,34,35,36.
Based on these insights, we designed and synthesized a series of mercapto-phenyl-1,2,4-triazole-bearing thio-quinoline derivatives and evaluated their anti-tyrosinase activity to develop more efficient anti-browning agents. The inhibitory mechanism of these compounds against tyrosinase was investigated by molecular docking studies to elucidate their binding interactions with the enzyme’s active site. Furthermore, molecular dynamics simulations were conducted to confirm the stability and consistency of the ligand-enzyme complexes, providing deeper insights into their inhibitory potential and mechanism of action.
Results and discussion
Chemistry
The synthesis of these compounds involves multiple chemical reactions (Fig. 2). Initially, potassium hydroxide and carbon disulfide were mixed with benzohydrazide (compound 1) in ethanol, followed by the addition of dry ether to precipitate (compound 3), which was then reacted with hydrazine hydrate and water under reflux to synthesize 4-amino-5-phenyl-4H-1,2,4-triazole-3-thiol (compound 4). Next, 2-chloroquinoline-3-carbaldehyde (compound 6) was synthesized using a Meth–Cohn reaction involving acetanilide and Vilsmeier–Haack reagent, yielding a 95% product after cooling and filtering. The compound 6 was then reacted with sodium sulfide in DMF, and the product was extracted with ethyl acetate to synthesize 2-mercaptoquinoline-3-carbaldehyde (compound 7). Additionally, different aniline (8a–j) was reacted with chloroacetyl chloride in DMF, followed by filtration after cooling to synthesize 10a–j. A mixture of 2-mercaptoquinoline-3-carbaldehyde (compound 7) and substituted phenyl anilide (10a–j) was stirred with potassium carbonate, and the product was filtered to synthesize 11a–j intermediate. Lastly, an aldehyde (11a–j) was combined with 4-amino-5-phenyl-4H-1,2,4-triazole-3-thiol (compound 4) in ethanol with glacial acetic acid, refluxed, cooled, and the precipitate was filtered and washed to obtain final products (12a–j).

Synthesis of 12a–j. Reaction condition: (First step) 1 (10 mmol), 2 (15 mmol), KOH (15 mmol), EtOH (125 mL), Diethyl ether (250 mL), r.t. 3 (10 mmol), N2H4 (3 mL), water (2 mL), 3 h under reflux condition. (Second step) 5 (1 mmol), POCl3 (7 eq), DMF (3 eq), 8 h under reflux conditions. (Third step) 6 (0.71 mmol), Na2S (1.07 mmol) in DMF for 5 h at r.t. (fourth step) 8a–j (0.05 mol), 9 (0.06 mol), DMF (40 mL), r.t. (fifth step) 7 (1 mmol), 10a–j (1 mmol), acetone (15 mL), K2CO3 (3 mmol), r.t. (final step) 11a–j (6 mmol), 4 (6 mmol), Acetic acid (5 mL), Ethanol (10 mL), 6 h at 78 °C.
To provide a comprehensive explanation, the detailed characterization of compound 12a was thoroughly examined. The FT-IR spectrum features a stretching bond at 3330 cm−1, corresponding to the NH amide. Aromatic and aliphatic CH groups appear at 3025 cm−1 and 2960 cm−1, respectively. Additionally, a distinct peak associated with C=O is detected at 1667 cm−1. The 1H NMR spectrum of this compound exhibits a singlet for the amide proton at 14.37 and three other singlets for the thiol proton, proton of carbon number 4 of quinoline, and imine proton at 10.51, 10.26, and 8.81, respectively. Aromatic protons appear at 8.10 (d, J = 8.1 Hz, 1H), 8.00 (dd, J = 6.7, 3.0 Hz, 2H), 7.93–7.81 (m, 2H), 7.65 (dd, J = 9.1, 5.0 Hz, 2H), 7.61–7.50 (m, 5H), 7.16 (t, J = 8.9 Hz, 2H). Additionally, a singlet peak at 4.24 is attributed to two protons of an aliphatic carbon. In the 13C-NMR spectrum, the carbonyl peak appears at 167.12. The aromatic and olefinic peaks are found at 162.72, 157.94, 149.25, 148.38, 139.57, 136.11, 136.10, 133.08, 131.26, 129.79, 129.18, 129.04, 127.62, 126.99, 125.75, 125.20, 124.07, 121.30, 121.22, and 115.96. Meanwhile, the aliphatic peak is observed at 35.91.
Biological evaluation against tyrosinase
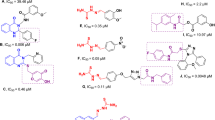
The SAR analysis for the synthesized derivatives highlights the impact of various substituents on the aromatic ring in modulating tyrosinase inhibition (Table 1).
The unsubstituted phenyl derivative (compound 12a) demonstrates moderate tyrosinase inhibition (% inhibition = 32.25% at 50 µM), serving as a baseline for comparison. Substituting the 2-position of the phenyl ring with chlorine (compound 12b) exhibited a higher inhibition (55.72% at 50 µM) with an IC50 value of 31.03 µM. The chlorine at the 3-position in compound 12c also improved the activity, achieving an IC50 of 30.98 µM. The 4-chloro substitution in compound 12d enhanced inhibition, with 65.98% inhibition at 50 µM and an IC50 of 28.30 µM, suggesting that the position of chlorine on the aromatic ring influences activity. This trend might be explained by the electron-withdrawing nature of the mono-chlorine substituent, which affects the electronic distribution of aromatic ring. Although the differences among the chlorine-substituted derivatives are not significant, the para-chlorine substitution appears slightly more favorable. Interestingly, the 2,4-dichloro substitution in compound 12e resulted in reduced inhibition (48.34% at 50 µM), indicating that the presence of two chlorine atoms reduced the overall activity. This reduction may be due to increased steric hindrance and altered physicochemical properties, such as decreased solubility or changes in the molecular shape that would lessen the ability of the compound to efficiently gain access to the enzyme active site.
The methoxy group at the para position in compound 12f slightly improves tyrosinase inhibition compared to the unsubstituted phenyl derivative (40.87 at 50 µM). Methyl groups, which are small electron-donating groups with lipophilic characteristics, were also evaluated. Compound 12g (4-methylphenyl) showed moderate inhibition (51.44% at 50 µM) with an IC50 of 25.89 µM. In contrast, 2-methylphenyl (compound 12h) and 2,3-dimethylphenyl (compound 12i) exhibited similar inhibition values (45.98% and 43.81%, respectively), but these modifications did not significantly improve the potency compared to compound 12a.
Methoxy is a strong electron-donating group through resonance, which increases the electron density in the aromatic ring. This affects lipophilicity and probably causes a slight decrease in activity. Conversely, methyl is a weak electron-donating group; its major effect is to increase hydrophobicity. The para-methyl derivative (12g) showed enhancement activity probably due to hydrophobic character and minimal steric hindrance due to its size. However, ortho-substitution in 12h and 12i probably introduces steric hindrance that may interfere with molecular planarity or conformational flexibility, thereby counterbalancing the beneficial effects coming from electron donation and hydrophobicity.
Finally, the unsubstituted compound 12j, which lacks any modifications, demonstrated the best inhibition (86.59% at 50 µM) and a low IC50 of 10.49 µM, indicating that increasing the linker length is more favorable. This could be due to the small active site of tyrosinase, where increasing the linker length may offer more flexibility, aiding in proper orientation and binding within the enzyme’s active site.
Our results show that compound 12j, with ethyl-substituted phenyl, displayed a strong tyrosinase inhibitory activity with an IC50 value of 10.49 ± 1.02 µM, which highlights the importance of substitution towards inhibitory potency. Our SAR shows that electron-donating groups, especially ethyl substitution, are a contributing factor towards enhanced activity. This aligns with the result of Saeed et al.26, which also emphasized that electron-donating groups are important in the design of tyrosinase inhibitors but mentioned methyl substituents to be superior to ethyl group. In contrast, Noori et al.25 observed that electron-withdrawing groups, particularly at the para position, enhanced tyrosinase inhibition of thioquinoline derivatives. This is also seen in our study that para-chloroquine group was also categorized as one of the potent analogs. In another investigation, the meta or para positions of methoxy and hydroxyl substitutions enhanced tyrosinase inhibitory activity; however, in the current study, the methyl substitution was better than methoxy37. Gultekin et al.38 found that the introduction of electron-donating methyl and methoxy groups in their 1,2,4-triazole–(thio)semicarbazide hybrids enhanced inhibition, while their best compound possessed an IC50 significantly lower than that of compound 12j.
Antioxidant activity
The antioxidant activity of the synthesized derivatives (12a–j) was evaluated, with their IC50 values compared to quercetin as a standard (Table 2). Overall, the compounds exhibited weak to moderate antioxidant activity compared to quercetin (IC50 = 18.56 ± 2.19 µM), demonstrated low radical scavenging potential.
The unsubstituted phenyl derivative (12a) exhibited IC50 = 107.18 ± 3.49 µM, categorized as a base compound for evaluating the effect of substitutions. Among halogen-substituted derivatives, 2-chlorophenyl (12b) and 3-chlorophenyl (12c) showed improved activity (IC50 = 105.93 ± 1.72 µM and 94.46 ± 4.61 µM, respectively), predicting the positive effect of chloro, particularly at the meta position. However, 4-chlorophenyl (12d) exhibited reduced activity (IC50 = 116.15 ± 1.89 µM). The 2,4-dichlorophenyl derivative (12e) further decreased activity (IC50 = 123.74 ± 1.01 µM). Among electron-donating substituents, the 4-methoxyphenyl derivative (12f) demonstrated improved activity (IC50 = 98.91 ± 4.83 µM) versus 12a, possibly due to its dual electronic effects and moderate lipophilicity. In contrast, the 4-methylphenyl derivative (12g) and 2-methylphenyl derivative (12h) showed reduced activity, highlighting the limited effect of alkyl groups. The 2,3-dimethylphenyl derivative (12i) exhibited further diminished activity (IC50 = 142.25 ± 1.39 µM). The ethylphenyl derivative (12j) exhibited moderate activity (IC50 = 102.36 ± 3.33 µM), categorized as the third potent group.
In summary, SAR analysis suggests that electron-donating groups (e.g. methoxy) and specific halogenations, particularly meta-chlorine, slightly enhance antioxidant activity relative to the unsubstituted analog. However, bulky or multiple substituents tend to reduce efficacy, likely due to steric or unfavorable electronic effects.
Kinetic study
The mechanism and type of inhibition were investigated through comprehensive kinetic studies. To determine the mode of enzyme inhibition, we conducted a Lineweaver–Burk plot analysis using the most effective derivative, compound 12j (Fig. 3). The Lineweaver–Burk plot, which graphs 1/V against 1/[S], was employed to assess the inhibition of tyrosinase by compound 12j at various concentrations of the substrate (L-DOPA). In this analysis, Km represents the Michaelis–Menten constant, while Vmax corresponds to the maximum reaction velocity.

Kinetic study of compound 12j against tyrosinase. (a) The Lineweaver–Burk plot in the presence of different concentrations of compound 12j.
The data analysis revealed that the Vmax of the enzyme remained unchanged, while the Km values increased with higher concentrations of compound 12j. This pattern indicates that compound 12j functions as a competitive inhibitor. The plot of the slope of the lines against varying concentrations of 12j allowed us to estimate the inhibition constant (Ki), which was determined to be 9.06 µM (Fig. 4).

The secondary plot of the slope of lines and various concentrations of compound 12j.
Molecular docking study
Molecular docking studies were performed to elucidate the molecular interactions between 12a–j and the tyrosinase enzyme. Before docking, the protocol was validated by redocking tropolone, the crystallographic ligand, into the active site of tyrosinase (PDB ID: 2Y9X). The root mean square deviation (RMSD) value was less than 2 Å, confirming the accuracy and reliability of the docking protocol. The docking results for 12j in the active site of tyrosinase showed binding energy of − 8.04 kcal/mol. Compound 12j, bearing an ethyl–phenyl substituent, showed the highest inhibition (86.59 ± 6.13%) due to a comprehensive network of interactions within the tyrosinase active site. The compound engages in π-π stacking with His259, His263, and Phe264, stabilizing its binding orientation. The mercapto triazole moiety also forms a critical coordination bond with Cu ions, essential for activity, while additional H-bonding with His85 and π-π stacking with His244 enhance the compound’s binding strength (Fig. 5).

Compound 12j in the active site of tyrosinase.
Molecular dynamic simulation
To further investigate the stability and interactions of compound 12j with the tyrosinase enzyme, molecular dynamics simulations were performed over a 100-ns to evaluate the dynamic behavior and binding stability of the enzyme-12j complex within the active site. As illustrated in Fig. 6, the RMSD analysis demonstrated that the enzyme alone exhibited a gradual increase in RMSD after 40 ns, reaching a peak of approximately 4.2 Å, followed by several fluctuations before stabilizing around 60 ns with an RMSD value of approximately 3.5 Å. In contrast, the enzyme-12j complex displayed only a slight increase in RMSD after 40 ns, followed by minimal fluctuations and stabilization at an RMSD value of approximately 2.5 Å. According to the Schrodinger software report, an RMSD value below 3.0 Å is considered indicative of a stable complex. These results confirm that compound 12j is well-fitted within the enzyme’s active site, effectively stabilizing the enzyme-inhibitor complex and underscoring its potential as a promising tyrosinase inhibitor.

RMSD of tyrosinase versus tyrosinase-12j.
To gain a deeper understanding of the tyrosinase-inhibitor complex’s behavior, we analyzed the root mean square fluctuation (RMSF) values for both the enzyme-12j complex and the enzyme alone. As shown in Fig. 7, the complex exhibited lower RMSF values across most regions, particularly in critical regions, including residues 68–84, 178–208, and 240–295. However, an unexpected increase in RMSF was observed between residues 312–335, suggesting this region does not interact directly with the 12j and may contribute to a slight instability. To further explore these observations, we examined the timeline representation of interactions and noted key residues involved. This interaction data aligns with the RMSD and RMSF values. The residues His264, His259, and His296 displayed consistent interactions with the thio-triazole functionality of the inhibitor, which contributed to the reduced RMSF values. Additionally, His85, His94, and His61 maintained interactions more than 50% of the time during the MD simulation (Fig. 8a).

RMSF of tyrosinase versus tyrosinase-12j complex.

(a) 2D ligand interactions with the protein residue (b) Protein interaction types with the 12j throughout the simulation.
A detailed analysis of 12j-protein interactions revealed significant contacts. Interactions over 20% of the simulation time were evaluated. These results support our design strategy: the thio-triazole moiety showed critical interactions with the copper cofactor, connecting to His61, Cys83, His85, His94, Asn220, His263, and His296. Additionally, the phenyl linker formed π–π interactions with Phe264, and the quinoline moiety displayed π–π interactions with His85. The side linker also formed hydrogen bonds with Tyr82, reinforcing the complex’s stability (Fig. 8b).
Prime/MM-GBSA analysis
In addition to the interaction analysis, the Prime/MM-GBSA module was utilized to quantitatively assess the interaction strengths of the 12j-tyrosinase complex. The computed ΔGbind values for the complex formed between compound 12j and tyrosinase were determined to be − 89.42 kcal/mol, which supports the high affinity and stability of the compound 12j-tyrosinase complex.
In silico drug-likeness, ADME, and toxicity studies
The drug-likeness and ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties of the most potent compound, 12j were evaluated using computational methods to estimate its suitability as orally administered agents. The results, summarized in Table 3 and Fig. 9, revealed that compound 12j possesses properties within the desirable range for an ideal oral therapeutic agent. This includes favorable bioavailability, chemical stability, and non-toxicity, aligning with drug-like characteristics.

Drug-likeness prediction for 12j.
Conclusion
In this study, we designed, synthesized, and evaluated a series of mercapto-phenyl-1,2,4-triazole-bearing thio-quinoline derivatives for their anti-tyrosinase activity. Among the synthesized derivatives, compound 12j exhibited the highest inhibitory potency, with an IC50 of 10.49 µM, surpassing the activity of the standard inhibitor kojic acid. Kinetic studies revealed that compound 12j acts as a competitive inhibitor, with a Ki value of 9.06 µM, suggesting its ability to bind effectively at the active site of tyrosinase. Molecular docking and dynamics simulations further confirmed the stable interactions of these derivatives with the enzyme’s active site, providing insight into their mechanism of inhibition. Furthermore, the SAR analysis and molecular docking studies suggest that key interactions, such as metal coordination with Cu ions and π–π stacking with critical residues, are essential for achieving high inhibitory potency. In addition, the antioxidant activity of the synthesized compounds was evaluated with DPPH assays, with several derivatives, particularly compound 12c, demonstrating moderate radical scavenging effects. This work highlights the potential of mercapto-phenyl-1,2,4-triazole-bearing thio-quinoline derivatives as promising candidates for managing hyperpigmentation disorders and preventing enzymatic browning in food products. These findings contribute to the ongoing development of safer and more effective tyrosinase inhibitors, paving the way for future research and application in medical, cosmetic, and industrial fields.
Materials and methods
General information
All chemicals and solvents were sourced from commercial suppliers, including Merck and Sigma-Aldrich, and were used without further purification. Thin-layer chromatography (TLC) was performed using Merck silica gel 60 F254 plates (0.25 mm thickness) and visualized at 254 nm under ultraviolet light. Nuclear magnetic resonance (NMR) spectra for 1H and 13C were obtained with Bruker Ultra Shield spectrometer at frequencies of 400 MHz (1H) and 101 MHz (13C), with data reported in terms of chemical shifts, multiplicity, coupling constants (Hz), and integration. Infrared spectra were taken using a Bruker–Equinox 55 spectrophotometer in KBr. Melting points were determined using a Kofler hot-stage apparatus. Mass spectra were obtained using a Varian Mat CH-7 spectrometer operating at 70 eV.
General procedure for the synthesis of 12a–j
Benzoic acid hydrazide 1 (10 mmol, 1.36 g) was added to a mixture of potassium hydroxide (15 mmol, 8.5 g) and carbon disulfide 2 (15 mmol, 11.2 g) in ethanol (125 mL) and stirred. Afterward, dry ether (250 mL) was introduced, and the solution was further stirred in room temperature, leading to the formation of a yellow precipitate, which was subsequently filtered. Without purification, the obtained compound 3 (10 mmol) was combined with hydrazine hydrate (3 mL) and water (2 mL) and heated under reflux conditions for 3 h. The reaction progress was tracked using thin-layer chromatography (TLC) until it reached completion. Upon completion of the reaction, the green solution was cooled and acidified with concentrated hydrochloric acid, resulting in the formation of a white precipitate 4, which was then filtered39.
A solution of phosphoryl chloride (7 equivalents) was gradually added to ice-chilled N,N-dimethylformamide (3 equivalents) while continuously stirring. After 30 min of stirring, acetanilide 5 (1 mmol) was introduced, and the mixture was briefly stirred at room temperature. The reaction was then refluxed for 8 h. Once completed, the cooled reaction mixture was poured into cold water and crushed ice, leading to the precipitation of the final product, 2-chloroquinoline-3-carbaldehyde 6, which was collected by filtration with a yield of 95%40.
2-Chloroquinoline-3-carbaldehyde 6 (0.71 mmol) and sodium sulfide (0.08 g, 1.07 mmol) were stirred in N,N-dimethylformamide at room temperature for 5 h. The mixture was then poured into water, and the product 7 was extracted using ethyl acetate41.
A mixture of aniline 8a–j (0.050 mol) and N,N-dimethylformamide (40 mL) was stirred, then chloroacetyl chloride 9 (6.78 g, 0.06 mol), dissolved in DMF (15 mL), was added while stirring in an ice bath. The mixture was stirred for 2 h at room temperature. After the reaction was completed, the mixture was poured into ice water and then filtered42.
A mixture of 2-mercaptoquinoline-3-carbaldehyde 7 (1 mmol) and the appropriate 2-chloro-N-(substituted phenyl) anilide 10a–j (1 mmol) in acetone (15 mL), along with anhydrous potassium carbonate (3 mmol, 415 mg), was stirred at room temperature for 10–12 h. The product 11a–j was then filtered43.
Glacial acetic acid (5 mL) was added to a mixture of aldehyde 11a–j (6 mmol) and 4-amino-5-phenyl-4H-1,2,4-triazole-3-thiol 4 (6 mmol) in ethanol (10 mL). The mixture was then refluxed for 6 h, cooled, and the resulting precipitate 12a–j was filtered and washed with ethanol44.
Chemistry
2-((3-(((3-mercapto-5-phenyl-4H-1,2,4-triazol-4-yl)imino)methyl)quinolin-2-yl)thio)-N-phenylacetamide (12a)
Brown solid; Yield: 94%; MP = 189-191 °C; IR (KBr, vmax) 3330 (NH), 3025 (CH Aromatic), 2960 (CH Aliphatic) 1667 (C=O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 14.37 (s, 1H), 10.51 (s, 1H), 10.26 (s, 1H), 8.81 (s, 1H), 8.10 (d, J = 8.1 Hz, 1H), 8.00 (dd, J = 6.7, 3.0 Hz, 2H), 7.93–7.81 (m, 2H), 7.65 (dd, J = 9.1, 5.0 Hz, 2H), 7.61–7.50 (m, 5H), 7.16 (t, J = 8.9 Hz, 2H), 4.24 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 167.12, 162.72, 157.94, 149.25, 148.38, 139.57, 136.12, 136.10, 133.08, 131.26, 129.79, 129.18, 129.04, 127.62, 126.99, 125.75, 125.20, 124.07, 121.30, 121.22, 115.96, 35.91 Anal. Calcd for C26H20N6OS2: C, 62.88; H, 4.06; N, 16.92; Found: C, 63.4; H, 4.28; N, 17.11.
N-(2-chlorophenyl)-2-((3-(((3-mercapto-5-phenyl-4H-1,2,4-triazol-4-yl)imino)methyl)quinolin-2-yl)thio)acetamide (12b)
Brown solid; Yield: 93%; MP = 218–220 °C IR (KBr, vmax) 3330 (NH), 3040 (CH Aromatic), 2975 (CH Aliphatic) 1664 (C=O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 14.37 (s, 1H), 10.28 (s, 1H), 9.87 (s, 1H), 8.83 (s, 1H), 8.11 (d, J = 8.0 Hz, 1H), 8.02–7.94 (m, 3H), 7.86 (t, J = 7.7 Hz, 1H), 7.81 (d, J = 8.1 Hz, 1H), 7.60 (t, J = 7.5 Hz, 1H), 7.55 (dd, J = 5.1, 1.9 Hz, 3H), 7.47 (d, J = 7.9 Hz, 1H), 7.31 (t, J = 7.9 Hz, 1H), 7.16 (t, J = 7.7 Hz, 1H), 4.34 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.74, 162.73, 157.60, 149.26, 148.40, 139.71, 135.38, 133.04, 131.27, 129.91, 129.76, 129.18, 129.05, 127.97, 127.79, 127.08, 126.46, 125.91, 125.75, 125.49, 125.29, 124.15, 35.22; Anal. Calcd: C26H19ClN6OS2; C, 58.81; H, 3.61; N, 15.83; Found C, 58.98; H, 3.79; N, 16.02.
N-(3-chlorophenyl)-2-((3-(((3-mercapto-5-phenyl-4H-1,2,4-triazol-4-yl)imino)methyl)quinolin-2-yl)thio)acetamide (12c)
Brown solid; Yield: 89%; MP = 204–206 °C IR (KBr, vmax) 3340 (NH), 3050 (CH Aromatic), 2880 (CH Aliphatic), 1662 (C=O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 14.37 (s, 1H), 10.28 (s, 1H), 9.87 (s, 1H), 8.83 (s, 1H), 8.10 (d, J = 8.1 Hz, 1H), 8.04–7.93 (m, 3H), 7.85 (t, J = 7.5 Hz, 1H), 7.81 (d, J = 7.9 Hz, 1H), 7.59 (t, J = 7.9 Hz, 1H), 7.57–7.52 (m, 3H), 7.47 (d, J = 8.0 Hz, 1H), 7.30 (t, J = 7.8 Hz, 1H), 7.15 (t, J = 7.7 Hz, 1H), 4.34 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.74, 162.74, 157.60, 149.26, 148.40, 139.70, 135.38, 133.03, 131.26, 129.91, 129.76, 129.17, 129.05, 127.97, 127.79, 127.07, 126.45, 125.90, 125.75, 125.47, 125.29, 124.15, 35.22: Anal. Calcd C26H19ClN6OS2; C, 58.81; H, 3.61; N, 15.83; Found; C, 59.03; H, 3.82; N, 15.98.
N-(4-chlorophenyl)-2-((3-(((3-mercapto-5-phenyl-4H-1,2,4-triazol-4-yl)imino)methyl)quinolin-2-yl)thio)acetamide (12d)
Brown solid; Yield: 89%; MP = 208–210 °C IR (KBr, vmax) 3350 (NH), 3030 (CH Aromatic), 2870 (CH Aliphatic), 1666 (C=O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 14.34 (s, 1H), 10.26 (s, 1H), 9.86 (s, 1H), 8.84 (s, 1H), 8.05 (d, J = 8.1 Hz, 1H), 8.00–7.95 (m, 2H), 7.85 (d, J = 9.2 Hz, 1H), 7.80 (t, J = 8.1 Hz, 1H), 7.61–7.52 (m, 2H), 7.49–7.44 (m, 4H), 7.32 (t, J = 8.4 Hz, 1H), 7.15 (t, J = 8.2 Hz, 1H), 4.23 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 167.89, 162.79, 157.71, 149.43, 148.45, 139.63, 135.68, 133.23, 131.42, 130.20, 129.72, 129.30, 128.92, 128.11, 127.66, 127.22, 125.97, 125.70, 125.58, 125.18, 124.06, 35.00: Anal. Calcd C26H19ClN6OS2; C, 58.81; H, 3.61; N, 15.83; Found; C, 59.07; H, 3.86; N, 16.01.
N-(2,4-dichlorophenyl)-2-((3-(((3-mercapto-5-phenyl-4H-1,2,4-triazol-4-yl)imino)methyl)quinolin-2-yl)thio)acetamide (12e)
Brown solid; Yield: 93%; MP = 210–212 °C; IR (KBr, vmax) 3360 (NH), 3065 (CH Aromatic), 2960 (CH Aliphatic), 1669 (C=O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 14.37 (s, 1H), 10.27 (s, 1H), 9.96 (s, 1H), 8.83 (s, 1H), 8.11 (d, J = 8.0 Hz, 1H), 8.05–7.96 (m, 2H), 7.94 (d, J = 8.4 Hz, 1H), 7.87 (d, J = 7.6 Hz, 1H), 7.82 (d, J = 8.8 Hz, 1H), 7.69–7.63 (m, 1H), 7.60 (t, J = 7.4 Hz, 1H), 7.59–7.52 (m, 3H), 7.40 (dd, J = 8.9, 2.5 Hz, 1H), 4.34 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.94, 162.75, 157.58, 149.26, 148.37, 139.77, 134.60, 133.07, 131.26, 129.77, 129.46, 129.36, 129.18, 129.04, 128.11, 127.74, 127.09, 126.77, 126.48, 125.76, 125.29, 124.14, 35.22; Anal. Calcd: C26H18Cl2N6OS2; C, 55.22; H, 3.21; N, 14.86; Found C, 55.39; H, 3.44; N, 15.07.
2-((3-(((3-Mercapto-5-phenyl-4H-1,2,4-triazol-4-yl)imino)methyl)quinolin-2-yl)thio)-N-(4-methoxyphenyl)acetamide (12f)
Brown solid; Yield: 89%; MP = 198–200 °C IR (KBr, vmax) 3325 (NH), 3025 (CH Aromatic), 2875 (CH Aliphatic), 1655 (C=O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 14.37 (s, 1H), 10.31 (s, 1H), 10.27 (s, 1H), 8.80 (s, 1H), 8.10 (d, J = 8.1 Hz, 1H), 8.03–7.96 (m, 2H), 7.91 (d, J = 8.4 Hz, 1H), 7.84 (t, J = 7.6 Hz, 1H), 7.63–7.46 (m, 6H), 6.89 (d, J = 9.1 Hz, 2H), 4.23 (s, 2H), 3.71 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.60, 162.73, 158.03, 155.69, 149.26, 148.41, 139.41, 133.05, 132.85, 131.25, 129.77, 129.18, 129.04, 127.66, 126.96, 125.75, 125.20, 124.09, 121.08, 114.37, 55.61, 35.86. MS-EI (m/z) = 526.5 (M+). Anal. Calcd C27H22N6O2S2; C, 64.98; H, 3.92; N, 9.47; Found; C, 65.04; H, 4.08; N, 9.68.
2-((3-(((3-Mercapto-5-phenyl-4H-1,2,4-triazol-4-yl)imino)methyl)quinolin-2-yl)thio)-N-(p-tolyl)acetamide (12g)
Brown solid; Yield: 94%; MP = 193–195 °C; IR (KBr, vmax) 3325 (NH), 3030 (CH Aromatic), 2975 (CH Aliphatic) 1659 (C=O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 14.36 (s, 1H), 10.36 (s, 1H), 10.27 (s, 1H), 8.81 (s, 1H), 8.10 (d, J = 8.1 Hz, 1H), 7.99 (dd, J = 6.8, 2.7 Hz, 2H), 7.89 (d, J = 8.4 Hz, 1H), 7.84 (t, J = 7.5 Hz, 1H), 7.62–7.49 (m, 6H), 7.12 (d, J = 8.1 Hz, 2H), 4.24 (s, 2H), 2.25 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.88, 162.72, 158.01, 149.26, 148.39, 139.45, 137.21, 133.05, 132.64, 131.25, 129.77, 129.64, 129.18, 129.04, 127.65, 126.97, 125.76, 125.20, 124.09, 119.53, 35.95, 20.92; Anal. Calcd for C27H22N6OS2: C, 63.51; H, 4.34; N, 16.46; Found: C, 63.72; H, 4.52; N, 16.67.
2-((3-(((3-Mercapto-5-phenyl-4H-1,2,4-triazol-4-yl)imino)methyl)quinolin-2-yl)thio)-N-(o-tolyl)acetamide (12h)
Brown solid; Yield: 94%; MP = 194–196 °C; IR (KBr, vmax) 3330 (NH), 3020 (CH Aromatic), 2955 (CH Aliphatic) 1659 (C=O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 14.38 (s, 1H), 10.38 (s, 1H), 10.29 (s, 1H), 8.88 (s, 1H), 8.09 (d, J = 8.0 Hz, 1H), 8.01–8.00 (m, 1H), 7.96 (d, J = 8.4 Hz, 1H), 7.85 (t, J = 8.2 Hz, 1H), 7.61 (t, J = 7.5 Hz, 1H), 7.54–7.48 (m, 1H), 7.39 (d, J = 7.7 Hz, 2H), 7.22–7.01 (m, 5H), 4.31 (s, 2H), 2.15 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 166.83, 162.76, 157.93, 149.51, 148.24, 139.95, 137.49, 133.34, 132.88, 131.53, 130.02, 128.99, 127.91, 127.04, 125.89, 125.38, 124.26, 119.74, 36.25, 21.48; Anal. Calcd for C27H22N6OS2: C, 63.51; H, 4.34; N, 16.46; Found: C, 63.69; H, 4.57; N, 16.62.
N-(2,3-dimethylphenyl)-2-((3-(((3-mercapto-5-phenyl-4H-1,2,4-triazol-4-yl)imino)methyl)quinolin-2-yl)thio)acetamide (12i)
Brown solid; Yield: 89%; MP = 209–211 °C; IR (KBr, vmax) 3310 (NH), 3025 (CH Aromatic), 2930 (CH Aliphatic), 1660 (C=O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 14.37 (s, 1H), 10.31 (s, 1H), 9.80 (s, 1H), 8.82 (s, 1H), 8.11 (d, J = 8.1 Hz, 1H), 8.06–7.93 (m, 3H), 7.87 (t, J = 7.6 Hz, 1H), 7.67–7.48 (m, 4H), 7.14 (d, J = 7.5 Hz, 1H), 7.09–6.97 (m, 2H), 4.30 (s, 2H), 2.21 (s, 3H), 2.04 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.12, 162.74, 162.55, 158.04, 149.28, 148.49, 139.21, 137.43, 136.56, 133.00, 131.69, 131.25, 129.80, 129.18, 129.05, 127.75, 127.44, 126.97, 125.77, 125.65, 125.27, 124.19, 123.83, 35.16, 20.59, 14.52. MS-EI (m/z) = 524.65 (M+). Anal. Calcd for C28H24N6OS2: C, 64.10; H, 4.61; N, 16.02; Found; C, 64.32; H, 4.82; N, 16.19.
2-((3-(((3-Mercapto-5-phenyl-4H-1,2,4-triazol-4-yl)imino)methyl)quinolin-2-yl)thio)-N-phenethylacetamide (12j)
Cream solid; Yield: 73%; MP = 195–197 °C; IR (KBr, vmax) 3335 (NH), 3015 (CH Aromatic), 2940 (CHAliphatic), 1662 (C=O) cm−1; 1H NMR (400 MHz, DMSO-d6) δ 14.37 (s, 1H), 10.27 (s, 1H), 8.80 (s, 1H), 8.31 (t, J = 5.7 Hz, 1H), 8.10 (d, J = 8.1 Hz, 1H), 7.99 (dd, J = 7.4, 2.5 Hz, 2H), 7.93–7.81 (m, 2H), 7.75–7.51 (m, 4H), 7.26–7.11 (m, 5H), 4.04 (s, 2H), 3.36–3.23 (m, 2H), 2.71 (t, J = 7.3 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.05, 162.71, 162.53, 157.92, 149.26, 148.44, 139.76, 139.12, 132.99, 131.26, 129.75, 129.19, 129.03, 129.00, 128.71, 127.76, 126.96, 126.49, 125.76, 125.24, 124.16, 41.13, 35.51, 34.55; Anal. Calcd: C28H24N6OS2; C, 64.10; H, 4.61; N, 16.02; Found C, 64.32; H, 4.83; N, 16.14.
Tyrosinase inhibitory assay
The tyrosinase inhibitory activities of derivatives were performed according to the previously reported procedures. 10 µL of test samples dissolved in DMSO were added to 160 µL of phosphate buffer (pH = 6.8), and then 10 µL tyrosinase was added. After the mixture was pre-incubated at 28 °C for 20 min, 20 µL of L-DOPA solution was added. DMSO without test compounds was used as the control, and kojic acid was used as a positive control. After 8 min incubation, the absorbance of samples was measured at 490 nm. Each assay was conducted as three separate replicates16.
Enzyme kinetic studies
The kinetic study for tyrosinase inhibition by 12j as the most potent analog was carried out using four different concentrations of inhibitor (3, 6, 10, 20, and 30 µM) against tyrosinase with different concentrations of L-DOPA (0.25, 0.5, 0.75, and 1 mM) as the substrate. The Lineweaver–Burk reciprocal plot was provided by plotting 1/V against 1/[S] at variable concentrations of the L-DOPA.
Molecular docking
The molecular docking studies were performed according to our previous study using the Maestro molecular modeling platform (version 10.5) by Schrödinger, LLC (Maestro, Schrödinger, LLC, New York, NY, 2021). The 3D crystal structure of tyrosinase was retrieved from the Protein Data Bank (PDB code: 2Y9X)45,46.
Molecular dynamics simulation
Docked tyrosinase and enzyme complexed with 12j were used for the molecular dynamics simulation according to a previous study using the Schoringer package47,48.
Prime MM-GBSA
The ligand-binding energies (ΔGBind) were calculated using molecular mechanics/generalized born surface area (MM‑GBSA) modules (Schrödinger LLC 2018).
Prediction of pharmacokinetic properties of 12j
The physicochemical and biological absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of the selected compound were performed. These predictions were generated using reliable tools, including PKCSM (https://biosig.lab.uq.edu.au/pkcsm/) and ADMETmesh (https://admetmesh.scbdd.com/) (accessed on: 12-9-2023).
Data availability
The datasets generated and/or analyzed during the current study are available in the Worldwide ProteinData Bank with the PDB ID of the 2Y9X repository.
References
Slominski, R. M. et al. Melanoma, melanin, and melanogenesis: The Yin and Yang relationship. Front. Oncol. 12, 842496 (2022).
Lee, A. Y. Skin pigmentation abnormalities and their possible relationship with skin aging. Int. J. Mol. Sci. 22(7), 3727 (2021).
Fernandes, B., Cavaco-Paulo, A. & Matamá, T. A comprehensive review of mammalian pigmentation: Paving the way for innovative hair colour-changing cosmetics. Biology 12(2), 290 (2023).
Iraji, A. et al. Design, synthesis, spectroscopic characterization, in vitro tyrosinase inhibition, antioxidant evaluation, in silico and kinetic studies of substituted indole-carbohydrazides. Bioorg. Chem. 129, 106140 (2022).
Zhao, W. et al. Potential application of natural bioactive compounds as skin-whitening agents: A review. J. Cosmet. Dermatol. 21(12), 6669–6687 (2022).
Devi Mari, S., Sheeja, L. & Aishwariyaa Lakshmi, V. Screening and evaluation of melanogenic enzyme tyrosinase producing bacterium from different crop soil. Int. J. Recent Adv. Biotechnol. Nanotechnol. 3, 18–33 (2020).
Iraji, A. et al. Synthesis, biological evaluation and molecular docking analysis of vaniline–benzylidenehydrazine hybrids as potent tyrosinase inhibitors. BMC Chem. 14, 1–11 (2020).
Karimian, S. et al. Design, synthesis, and biological evaluation of symmetrical azine derivatives as novel tyrosinase inhibitors. BMC Chem. 15(1), 54 (2021).
Alizadeh, N. et al. Evaluating the effects of disubstituted 3-hydroxy-1H-pyrrol-2(5H)-one analog as novel tyrosinase inhibitors. Bioorg. Chem. 126, 105876 (2022).
Najafi, Z. et al. Design, synthesis, and molecular dynamics simulation studies of some novel kojic acid fused 2-amino-3-cyano-4H-pyran derivatives as tyrosinase inhibitors. BMC Chem. 18(1), 41 (2024).
Chang, T. S. An updated review of tyrosinase inhibitors. Int. J. Mol. Sci. 10(6), 2440–2475 (2009).
Çakmak, R. et al. Synthesis, characterization and biological evaluation of ester derivatives of 4-(diethylamino) salicylaldehyde as cholinesterase, and tyrosinase inhibitors. Middle East J. Sci. 7(2), 137–144 (2021).
Deri, B. et al. The unravelling of the complex pattern of tyrosinase inhibition. Sci. Rep. 6(1), 34993 (2016).
Demir, E. A. et al. Investigation of tyrosinase inhibition by some 1,2,4 triazole derivative compounds: In vitro and in silico mechanisms. Turk. J. Biochem. 44(4), 473–481 (2019).
Carradori, S. et al. Chapter 8.2—Tyrosinase enzyme and its inhibitors: An update of the literature. In Metalloenzymes (eds Supuran, C. T. & Donald, W. A.) 533–546 (Academic Press, 2024).
Bagheri, A. et al. Structure-based development of 3,5-dihydroxybenzoyl-hydrazineylidene as tyrosinase inhibitor; in vitro and in silico study. Sci. Rep. 14(1), 1540 (2024).
Çakmak, R. et al. Synthesis of new fluorinated sulfonates and their Schiff bases as anti-Alzheimer drug candidates: An in vitro-in silico study. J. Mol. Struct. 1329, 141474 (2025).
Kamalı, A., Çakmak, R. & Boğa, M. Anticholinesterase and antioxidant activities of novel heterocyclic Schiff base derivatives containing an aryl sulfonate moiety. J. Chin. Chem. Soc. 69(4), 731–743 (2022).
Reşit, Ç. et al. Schiff base derivatives of 4-aminoantipyrine as promising molecules: Synthesis, structural characterization, and biological activities. Russ. J. Bioorg. Chem. 48(2), 334–344 (2022).
Huang, Y. et al. Newly designed quinazolinone derivatives as novel tyrosinase inhibitor: Synthesis, inhibitory activity, and mechanism. Molecules 27(17), 5558 (2022).
Mol Lima, R. et al. Quinoline derivatives as novel depigmenting and antioxidant agents. Lett. Drug Des. Discov. 14(4), 374–379 (2017).
Gardelly, M. et al. Anti-tyrosinase and anti-butyrylcholinesterase quinolines-based coumarin derivatives: Synthesis and insights from molecular docking studies. Chem. Afr. 4, 491–501 (2021).
Sultana, N. & Khan, T. H. Tyrosinase inhibitor fatty ester and a quinoline alkaloid from Skimmia laureola. Z. Naturforschung B 60(11), 1186–1191 (2005).
Sepehri, N. et al. The natural-based optimization of kojic acid conjugated to different thio-quinazolinones as potential anti-melanogenesis agents with tyrosinase inhibitory activity. Bioorg. Med. Chem. 36, 116044 (2021).
Noori, M. et al. Thioquinoline derivatives conjugated to thiosemicarbazide as potent tyrosinase inhibitors with anti-melanogenesis properties. Sci. Rep. 13(1), 2578 (2023).
Saeed, S. et al. Discovery of novel 1,2,4-triazole tethered β-hydroxy sulfides as bacterial tyrosinase inhibitors: Synthesis and biophysical evaluation through in vitro and in silico approaches. RSC Adv. 14(22), 15419–15430 (2024).
Zhang, J. et al. Development of novel pyrazole-1,2,4-triazole derivatives as tyrosinase inhibitors: Design, preparation, mechanism of action and anti-browning application. Food Chem. 460, 140722 (2024).
Abd El-Lateef, H. M. et al. Fe(III), Ni(II), and Cu(II)-moxifloxacin-tri-substituted imidazole mixed ligand complexes: Synthesis, structural, DFT, biological, and protein-binding analysis. Inorg. Chem. Commun. 158, 111486 (2023).
Çınar, E. et al. Heterocyclic Schiff base derivatives containing pyrazolone moiety: Synthesis, characterization, and in vitro biological studies. J. Chin. Chem. Soc. 68(12), 2355–2367 (2021).
Hernández-Ayala, L. F., Guzmán-López, E. G. & Galano, A. Quinoline derivatives: Promising antioxidants with neuroprotective potential. Antioxidants (Basel) 12(10), 1853 (2023).
Pachuta-Stec, A. Antioxidant activity of 1,2,4-triazole and its derivatives: A mini-review. Mini. Rev. Med. Chem. 22(7), 1081–1094 (2022).
Hussen, N. H. A. et al. Role of antioxidants in skin aging and the molecular mechanism of ROS: A comprehensive review. Asp. Mol. Med. 5, 100063 (2025).
Hacıosmanoğlu-Aldoğan, E. et al. Synthesis, characterization, and in vitro and in silico studies of substituted-vanillin hydrazones of anthranilic acid and their quinazolin-4(3H)-One analogues as anti breast cancer agents. ChemistrySelect 10(3), e202404806 (2025).
Tokalı, F. S. et al. Novel acetic acid derivatives containing quinazolin-4(3H)-one ring: Synthesis, in vitro, and in silico evaluation of potent aldose reductase inhibitors. Drug Dev. Res. 84(2), 275–295 (2023).
Şenol, H. et al. New anthranilic acid hydrazones as fenamate isosteres: Synthesis, characterization, molecular docking, dynamics and in silico ADME, in vitro anti-inflammatory and anticancer activity studies. Chem. Biodivers. 20(8), e202300773 (2023).
Tokalı, F. S. et al. Exploring highly selective polymethoxy fenamate isosteres as novel anti-prostate cancer agents: Synthesis, biological activity, molecular docking, molecular dynamics, and ADME studies. J. Mol. Struct. 1319, 139519 (2025).
Chabhadiya, B. K. et al. Design, synthesis, and evaluation of quinoline-1,2,3-triazole hybrids as CYP51 inhibitors: In silico study and in vitro antimicrobial assessment. ChemistrySelect 9(36), e202402221 (2024).
Gultekin, E. et al. Synthesis of new 1,2,4-triazole–(thio)semicarbazide hybrid molecules: Their tyrosinase inhibitor activities and molecular docking analysis. Arch. Pharm. 354(8), 2100058 (2021).
Jubie, S. et al. Synthesis, antidepressant and antimicrobial activities of some novel stearic acid analogues. Eur. J. Med. Chem. 54, 931–935 (2012).
Bazine, I. et al. Synthesis, antioxidant and anticholinesterase activities of novel quinoline-aminophosphonate derivatives. J. Heterocycl. Chem. 57(5), 2139–2149 (2020).
Velmurugan, K. et al. Imidazoloquinoline bearing thiol probe as fluorescent electrochemical sensing of Ag and relay recognition of proline. J. Photochem. Photobiol. A 333, 130–141 (2017).
Guo, X. et al. Design and bio-evaluation of indole derivatives as potent Kv1.5 inhibitors. Bioorg. Med. Chem. 21(21), 6466–6476 (2013).
Alaa, A.-M. et al. Design, synthesis of 2,3-disubstitued 4 (3H)-quinazolinone derivatives as anti-inflammatory and analgesic agents: COX-1/2 inhibitory activities and molecular docking studies. Bioorg. Med. Chem. 24(16), 3818–3828 (2016).
Wang, B.-L. et al. Synthesis, biological activities and SAR studies of new 3-substitutedphenyl-4-substitutedbenzylideneamino-1,2,4-triazole Mannich bases and bis-Mannich bases as ketol-acid reductoisomerase inhibitors. Bioorg. Med. Chem. Lett. 27(24), 5457–5462 (2017).
MoghadamFarid, S. et al. Synthesis, biological evaluations, and in silico assessments of phenylamino quinazolinones as tyrosinase inhibitors. Sci. Rep. 15(1), 846 (2025).
MoghadamFarid, S. et al. Synthesis and structure–activity relationship studies of benzimidazole-thioquinoline derivatives as α-glucosidase inhibitors. Sci. Rep. 13(1), 4392 (2023).
Azimi, M. et al. Synthesis and biological assessment of novel 4H-chromene-3-carbonitrile derivatives as tyrosinase inhibitors. BMC Chem. 18(1), 187 (2024).
ShokouhiAsl, A. S. et al. Cinnamic acid conjugated with triazole acetamides as anti-Alzheimer and anti-melanogenesis candidates: an in vitro and in silico study. Sci. Rep. 15(1), 655 (2025).
Acknowledgements
The Aida Iraji would like to thank the Vice-Chancellor for Research of Shiraz University of Medical Sciences for the financial support of this research (Grant Number: IR.SUMS.REC.1403.388). Support of the research council of University of Tehran is acknowledged.
Author information
Authors and Affiliations
Contributions
N.M.S., M.N., N.D., M.H.S., and M.M. synthesized compounds and contributed to the characterization of compounds. N.O., and B.L. performed biological activities. A.I. supervised the in silico synthetic part of the study. P.R.R. supervised the synthetic part of the study. All authors read and approved the final version of the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Motamedi Shakib, N., Sayahi, M.H., Oliyaei, N. et al. Development of mercapto-phenyl-1,2,4-triazole bearing thio-quinoline as a novel class of tyrosinase inhibitors: an in vitro and in silico study. Sci Rep 15, 25382 (2025). https://doi.org/10.1038/s41598-025-09072-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-09072-1
