
Abstract
Olefins are the backbone of the petrochemical conversion industries, like polymers, plastic, lubricating oil, surfactants, and synthetic fuels. It is a wide but challenging process to customize. Metal–organic frameworks (MOFs) are highly regarded for their potential in Fischer–Tropsch synthesis (FTS), yet they often have inadequate thermal stability. This study demonstrated the remarkable potential of the Fe-NDC MOF. It maintains its initial structure until it reaches a temperature of 500 °C (Fe@C-500), which is efficient for syngas conversion to olefin. The Fe@C-500 catalyst exceeded a twofold increase in the ratio of olefin to paraffin compared to Fe@C-600 (2 vs. 0.8). The maintained structure of Fe@C-500 enhances the transport of reactants and restricts the hydrogenation of olefins. The Fe@C-500 catalyst showed ~ 50% and 27% selectivity to total olefin and light olefin, respectively, with a Fe-time yield (FTY) for light olefins of 180 mmolCO g−1Fe h−1. In contrast, Fe@C-600 exhibits a shift in product selectivity towards paraffin (~ 70%) at a lower FTY for light olefins of 130 mmolCO g−1Fe h−1. The performance of the Fe@C-500 catalyst is particularly intriguing and warrants further investigation. Retaining the porous structure of MOF-derived catalysts might greatly enhance olefin production.
Similar content being viewed by others
Introduction
Considerable research effort has been devoted to developing sustainable alternative feedstocks for critical petrochemical products in response to rising concerns about the carbon footprint of fossil fuel consumption1,2. Designing optimized pathways for converting carbon emissions into valuable compounds and high-quality, sustainable hydrocarbon fuels is critical for establishing a carbon–neutral society3. Modern sustainable technologies, such as Power-to-X (PtX), biomass gasification to syngas, and emission-to-liquid (EtL) relying on Fischer–Tropsch Synthesis (FTS), are now cost-effective, large-scale options for fuel production, especially olefins4,5. Olefin supply and cost affect the global economy. Light olefins are the building blocks of essential materials such as polymers, plastics, and solvents6. Heavy olefins produce fuels such as gasoline by fluid catalytic cracking7. Biomass to liquid (BTL) and emissions to liquid (ETL) are receiving more attention because of the growing demand for clean and low-carbon energy and the enforcement of stricter environmental policies on the fuel industry8,9.
Olefins production relying on the FTS approach typically requires supported catalysts to foster a complex series of heterogeneous chemical reactions. The careful selection of catalyst and support contributes to keeping the Fischer–Tropsch reaction stable, active, and selective10. Carbon-supported iron nanoparticles are frequently studied for FTS activity due to the synergetic effect of iron’s high activity under broad process conditions and the mild interaction with the thermally stable carbon supports, ensuring better dispersion and reducibility11,12. Added to the abundance of cheap iron sources, various economic carbon materials have been investigated, such as biomass13, carbon fiber8,14, activated carbon15,16, graphene17,18,19, carbon nanotubes20,21, and more recently, Metal–Organic Frameworks (MOFs)22,23,24,25.
MOFs are exceptionally porous materials with a versatile crystalline structure composed of organic linkers and metal clusters26. Unlike common catalysts, MOFs avoid using post-synthetic treatments (e.g., impregnation) that decrease porosity and suppress active sites. MOF pyrolysis results in catalysts with metal dispersion at the atomic level in a carbon matrix27,28. While high metal loading normally limits dispersion and surface area (restricting at ~ 30 wt%), MOF-based catalysts overcome this barrier by achieving both high metal loading and well-dispersed small nanoparticles29,30. MOFs exhibit inferior thermal and chemical stability than typical inorganic porous materials, limiting their usage in thermocatalytic processes31. While certain MOF-based catalysts are quite resilient, their stability must be demonstrated experimentally. Additionally, the high cost of some organic linker precursors must be rationalized by the catalyst’s activity and product yield. Researchers make ongoing efforts to optimize these catalysts to increase the economic viability and efficiency of the process32. One attractive strategy is exposing catalysts to the pyrolysis process. Pyrolysis is a thermal treatment method where a material is exposed to elevated temperatures in an inert or reducing environment. Pyrolysis has shown potential in adjusting the active phase, surface area, and porosity, or incorporating promoters into the catalyst structure to enhance FTS-related challenges such as catalytic efficiency, product selectivity, and lifetime33,34,35.
Several studies applied the pyrolysis-mediated strategy to derive Fe nanoparticles on carbon support from Fe-MOF precursor with high loading and degree of dispersion7,36,37,38,39. For example, through the pyrolysis of Fe-MIL-88B and Fe-MIL-88B NH2, An et al.37 prepared active iron carbide catalysts with high catalytic activity. At various pyrolysis temperatures and in response to the addition of structural promoters, the Gascon group reported a series of catalysts fabricated from diverse MOFs, including Basolite F300, MIL-88A, MIL-127, MIL-68, MIL-101-NH2, and MIL-100, that exhibited distinct active iron carbide phases34,36,40,41,42. Their findings determined that the characteristics and structure of the initial MOFs, along with pyrolysis temperature, significantly impact the resulting catalyst in the context of catalytic efficiency and product selectivity.
Prior research on the pyrolysis of Fe-MOF for FTS aimed to fully break down the MOF structure to produce a carbon-supported catalyst with a high concentration of homogenously distributed iron. The pyrolysis temperature must be equal to or greater than the catalyst reduction and FTS reaction temperatures (up to 450 °C) to maintain the thermal stability of the catalyst throughout the process. The ideal temperature for Fe-MOF pyrolysis, based on prior studies spanning ranges from 400 to 900 °C, is 500 °C34. Maintaining the structural integrity of the MOF following pyrolysis at 500 °C without full collapse is challenging. The key is to increase the thermal stability of the MOF structure. Applying the MOF linker with multiple aromatic rings is expected to enhance the thermal stability of MOFs. The expanded aromatic system provides enhanced π-π Stacking, a stronger metal–ligand bond, and effective heat transfer, slowing thermal breakdown43,44,45. Moreover, a larger aromatic surface area limits water penetration into the framework and maintains metal–ligand bonds46. During FTS, it might help prevent catalyst deactivation by the water formed.
The 2,6-naphthalene dicarboxylic acid (NDC) is a linker used to synthesize Zr-NDC and Fe-NDC (MIL-142B) MOFs47,48. The NDC linker has two benzene rings in its structure, unlike conventional carboxylic acid linkers with one aromatic ring, such as benzene dicarboxylic (BDC) and tricarboxylic (BTC) acid. Zhang et al.48 reported the synthesis of Zr-NDC MOFs stable above 500 °C compared to Zr-BDC UiO-66(Zr) (400–500 °C)43,45,49. Higher Hydrophobicity for the NDC linker provides better resistance to hydrolysis than the BDC linker46. The prepared Fe-NDC catalyst shows superior thermal stability to other mono-benzene MOF peers, as confirmed by thermogravimetric analysis (TGA) conducted under inert and oxygen-rich environments. As far as we know, it is the first time using the partially pyrolyzed and thermally stable Fe-NDC MOF catalysts for the FTS process.
In brief, we demonstrate the solvothermal synthesis of Fe-NDC MOF, followed by pyrolysis at varying temperatures (500 °C and 600 °C) as considered by TGA. The catalysts were examined using thorough characterization methods to assess pyrolysis-induced changes. The catalysts were tested for FTS activity in a solar-powered fixed-bed reactor utilizing green hydrogen produced by electrolysis of solar desalinated water. The tests were conducted at a semi-industrial high gas hourly space velocity (GHSV) of 20,000 mL g−1cat h−1. In this study, we examine the pyrolysis temperature to tailor the properties of the resulting catalysts at a lower temperature while maintaining the original MOF structure intact. These catalysts are intended for use in a green FTS system to support sustainable and efficient hydrocarbon production.
Methods
Materials
Ferric Nitrate nonahydrate (Fe(NO3)3·9H2O) (≥ 98% purity) and n-octane (≥ 99% purity) were purchased from Fisher Scientific, UK, with laboratory reagent grade. 2,6-naphthalenedicarboxylic acid (99%), N,N-dimethylformamide (ReagentPlus®, ≥ 99%), and N-dodecane (ReagentPlus®, ≥ 99%) were provided by Sigma-Aldrich, USA. γ-Al2O3 (340 m2 g−1) was acquired from BASF. An on-demand hydrogen generating system (H-Genie®) at 100 bar pressure produced grade 4, 99.99% hydrogen gas from pure water. Post-electrolyzer purification system yields grade 5, 99.999% hydrogen gas. All other employed gases supplied or imported by Air Supply Co., Egypt, with grade 5, 99.999%.
Catalysts synthesis
Synthesis of Fe-NDC (MIL-142B) MOF: The Fe-NDC MOF was prepared according to Ibrahim et al.47 using the solvothermal method by dissolving 0.692 mmol (279.6 mg) of iron nitrate nonahydrate (Fe(NO3)3.9H2O) and 0.692 mmol (149.7 mg) of 2,6-naphthalenedicarboxylic acid in 30 mL of N,N-dimethylformamide (DMF). Subsequently, the solution was transferred into a 60-mL Teflon autoclave and heated in an oven at a temperature of 100 °C for 24 h. The resultant yellow mixture underwent centrifugation and was rinsed twice with fresh DMF to eliminate any remaining unreacted components. Eventually, the developed MOF was dried in a vacuum oven at 100 °C to eliminate residual solvent.
Preparation of Fe-NDC derived catalyst by pyrolysis: The dehydrated MOF was milled and then exposed to pyrolysis in a nitrogen stream for 4 h at two different temperatures, namely 500 and 600 °C. The temperature was held constant at 60 °C for 1 h, after which it was gradually raised to 500 or 600 °C at a rate of 5 °C per minute. Finally, the produced catalysts Fe@C-500 and Fe@C-600 were passivated by gradually exposing them to a continuous flow of 1% O2/Ar gas mixture to avoid combustion upon exposure to the atmosphere.
Preparation of Fe@Al2O3 catalyst: An iron reference catalyst supported on Alumina (with a Fe mass fraction of 30%) was synthesized using the incipient wetness impregnation technique (IWI) according to previous work7. To guarantee complete incorporation within the pore volume of alumina, a solution of Fe(NO3)3·9H2O (15.192 g, 37.6 mmol) was introduced onto the one gram alumina until it was thoroughly wetted. After drying, it was subjected to calcination in air at a temperature of 500 °C for 2 h, with a heating rate of 10 °C per minute.
Catalyst characterization
The X-ray diffraction (XRD) patterns were generated using a Shimadzu XRD-6100 instrument, utilizing Cu-Kα radiation within the 10–80° range to identify the present phases. The determination of particle size was performed using the Debye–Scherrer equation using MDI Jade software (v. 6). The chemical characteristics change before and after pyrolysis were investigated using Fourier-transform infrared spectroscopy (FTIR) spectra produced using the Bruker Vertex 70 instrument.
The iron content of the MOF-based catalyst was assessed using thermogravimetric analysis (TGA) conducted under air and atomic absorption spectroscopy (AAS). The TGA-50 instrument from Shimadzu was used, with a temperature range of 25–800 °C and a heating rate of 10 °C per minute. Iron loadings were estimated via TGA using formulae derived from Oar-Arteta et al.41, assuming Fe2O3 is the main oxidized phase. The chemical and electronic characteristics of the surface were investigated via X-ray photoelectron spectroscopy (XPS) employing K-ALPHA (Thermo Fisher Scientific, USA) equipped with monochromatic X-ray Al Kα radiation ranging from −10 to 1350 eV, a spot size of 400 µm and a pass pressure of 10–9 mbar, pass energy of 200 eV for the entire spectrum, and 50 eV for the narrow spectrum.
Scanning and transmission electron microscopy (SEM and TEM–EDX) are two techniques used for imaging and analyzing materials at the microscopic level. JEOL JSM-6010LV and JEOL JEM-2100F microscopes were used to obtain images to examine the morphological structure, particle size distribution, and dispersion. The N2 adsorption/desorption isotherms at 77 K were measured by BEL Japan (Belsorp II mini) and analyzed using the Brunauer–Emmett–Teller (BET) and BJH t-plot techniques to obtain the BET surface area, mean pore diameter, and total pore volume. Before measurement, the samples were degassed at 150 °C for a whole night. The H2-temperature-programmed reduction (H2-TPR) data were collected using a MICROTRAC BELCAT II catalyst analyzer fitted with a thermal conductivity detector. This data was used to determine the sequence and extent of reduction. Data was collected from ambient temperature to 900 °C, with a heating rate of 10 °C per minute, while a stream of 30 mL per minute containing 5% H2/Ar was used. Chemisorption measurements for CO and H2, specifically temperature-programmed desorption (TPD), were conducted using the MICROTRAC BELCAT II catalyst analyzer.
Catalytic performance evaluation
Figure 1 demonstrates the components of the applied Power-to-X system. Green hydrogen was generated from a Proton Exchange Membrane (PEM) water electrolyzer. Fresh water was supplied by a multi-effect desalination (MED) system running on solar energy from concentrated solar power (CSP). A photovoltaic (PV) system powers the electrolyzer. The choice of the fixed bed reactor material was stainless steel (316 grade), primarily based on its cost-effectiveness and high degree of corrosion resistance compared to other materials50,51,52. The reactor furnace’s temperature was calibrated based on earlier research53.

Process scheme of the Power-to-X system.
For each trial, the fixed bed reactor was filled with the catalyst (0.5 g) and an equivalent amount of SiC. A control experiment using identical reaction conditions revealed that the reactor, glass beads, glass wool, and SiC were devoid of catalytic activity. The catalyst was reduced using a flow rate of 50 ml per minute of hydrogen gas for 4 h at a temperature of 400 °C, with a heating rate of 10 °C per minute. Subsequently, the catalyst was cooled down to a temperature of 180 °C. Upon reaching the target temperature of 340 °C, syngas was introduced at a Gas Hourly Space Velocity (GHSV) of 20,000 mL g−1cat h−1, under a pressure of 20 bar, and with an H2/CO ratio of 1. A cryogenic trap containing n-octane (2 g) was used to capture a portion of liquid hydrocarbon at a temperature of 3 °C.
Permanent gas composition was monitored using a Shimadzu-GC-2014 GC/TCD instrument with a 3 m Shincarbon Restek Column. The three gases’ molar concentrations were verified using an external standard. He used a carrier gas with a flow of 20 ml min−1, heating the injection ports to 100 °C, the column to 120 °C, and the TCD detector to 180 °C. A Shimadzu-GC-2014 GC/FID system with a Rt-Alumina BOND/Na2SO4 column (30 m, 0.32 mm ID, 5 µm film) was used to identify the light hydrocarbon fraction (C1–C6). For light hydrocarbon calibration, a petroleum company supplied C1–C6 reference gas into the GC/FID system. After 6 min at 40 °C, the FID oven was heated to 100 °C (15 °C min−1) for 37 min. Samples were injected at a 150:1 split ratio with 40 cm s−1 helium linear velocity.
Using a GC/FID system (SRI-8610C-GC), the liquid fraction was fed onto an MXT-1 Restek column (60 m, 0.53 mm ID, 5 m film) with an internal standard of N-dodecane. After 3 min at 35 °C, the column’s oven temperature was increased to 240 °C (5 °C min−1), then maintained at 300 °C (10 °C min−1) for 60 min at 5 ml min−1 He. The olefin yield and selectivity of liquid samples were measured following sulfuric acid treatment. The catalyst performance was determined using previously reported calculations54. The average catalytic activity in moles for feed carbon (CO) and product carbon (residual CO, CO2, and hydrocarbons) was used to estimate the carbon balance.
Results and discussion
Catalyst characterization
TGA profiles were produced for fresh Fe-NDC MOF under an inert atmosphere (Nitrogen) to investigate thermal stability and determine the suitable pyrolysis temperature. Figure 2a shows that weight loss increased as temperature rose throughout two successive stages. The first weight loss seen between room temperature and 369 °C (about 4% weight loss of Fe-NDC) is likely due to the elimination of moisture and DMF solvent molecules from the MOF’s porosity. A significant breakdown occurred between 369 and 538 °C, resulting in a weight loss of roughly 36.7 wt% of the original mass. This may be attributed to the heat decomposition of the structurally coordinated 2,6-NDC ligands. Another weight loss occurred between 538 and 736 °C, resulting in a mass loss of 16.2 wt%. The loss is likely caused by the breakdown of the iron metal clusters from the MOF framework47. The findings proved that Fe-NDC-MOF is more thermally stable than conventional Fe-MOFs previously reported 7,38. The respective weight losses resulting from the two stages are 36.7% and 16.2%. Therefore, a temperature of 500 °C was anticipated to cause partial pyrolysis of the MOF. The pyrolyzed MOF at 600 °C likely resulted in the complete disintegration of the MOF structure.

TGA and DTG of Fe-NDC (a), XRD patterns (b), and FTIR spectra (c) of Fe-NDC MOF and derived catalysts. MOF under nitrogen flow.
Figure 2b depicts the XRD patterns of Fe-NDC MOF and active catalysts. The presence of distinct diffraction peaks (2θ = 7.2°, 9.7°, 10.2°, 10.5°, and 14.7°) provides confirmation that the synthesized MOFs possess well-defined crystalline structures, matching published research47,55. Unlike Fe@C-500, the XRD pattern of Fe@C-600 is free of MOF characteristic diffraction peaks, confirming the collapse of the MOF framework, see Fig. 2b. The XRD patterns reveal that magnetite (Fe3O4) is the predominant iron phase, exhibiting a fcc structure as per the JCPDS file, No. 19-062941. Fe@C-500 pyrolyzed at a lower temperature shows pronounced peaks at 7.2°, 14.7°, and 25.1°, confirming the residual structure of the fresh MOF structure. Reduced magnetite peaks suggest a smaller particle size of Fe@C-500.
As illustrated in Supplementary Fig. S1, the differential thermogravimetric (DTG) and TGA profiles of fresh and pyrolyzed Fe-NDC were generated in the air. The first weight reduction in the TGA curves of the parent MOF, occurring before 300 °C, is likely due to the volatilization of solvents and water. Fe-NDC displayed a two-step substantial weight loss, with the highest rate of MOF breakdown seen at 355 °C and 435 °C. The first loss begins at 300 °C and concludes at 490 °C. The second loss occurs between 490 and 690 °C. The respective weight losses resulting from the two stages are 54% and 15%. Therefore, a temperature of 500 °C was anticipated to cause partial pyrolysis of the MOF. The pyrolyzed MOF at 600 °C likely resulted in the complete disintegration of the MOF structure. The XRD results indicate that the material was not completely destroyed at 500 °C since specific peaks typical of MOF were still present, Fig. 2b. TGA was used to determine the Fe loading pre- and post-pyrolysis. The remaining mass was projected to consist only of Fe2O3. The Fe loadings derived from TGA and AAS had comparable percentages, as seen in Table 1.
Other characterization techniques were needed to evaluate the different properties of the MOF and the produced catalysts. The IR spectrum of Fe-NDC in Fig. 2c is similar to those in earlier studies47,55. The FTIR data suggest that iron ions are fully coordinated with the NDC linker. The carboxylic acid vibrations related to NDC, − OH (3059–2500 cm−1), and C = O stretching (1696 and 1300 cm−1) were either minimal or undetected. The weak bands at 1658 and 1696 cm−1 correspond to traces of DMF and unbound NDC acid within the pores, respectively. On the other hand, strong coordination bands associated with the vibrations of C = O, –COO − , and out-of-plane − CH (1602, 1559, 1500, 1419, 1363, and 771 cm−1) of the aromatic NDC were detected. In addition, the two peaks at 1197 and 916 cm−1 correspond to the in-plane − CH vibrations. Furthermore, the 482 cm−1 band, characteristic of Fe–O, is shown in Fig. 2c. The findings confirm the effective coordination of Fe3+ ions with NDC to form Fe-NDC crystals. Following the pyrolysis of the produced Fe-NDC at 600 °C, a significant decrease in coordination peaks indicates successful pyrolysis. The strong band observed at 578 cm−1 and the less pronounced one at 482 cm−1 resemble the characteristics of magnetite as previously documented56, in accordance with XRD findings. The Fe-NDC MOF shows partial collapse at a lower pyrolysis temperature of 500 °C, as confirmed by the presence of most of the IR peaks of the original MOF. Strong Fe–O vibrations from the MOF structure are seen at 482 cm−1, along with a weak magnetite band at 578 cm−1.
As XRD findings provide information on the bulk phase only, XPS of Fe2p, O1s, and C1s was conducted to analyze the chemical structure and electronic characteristics at the surface, Fig. 3. Fe2p core level spectra of Fe@C-500 and Fe@C-600 are shown in Fig. 3b and e. The leading bands at 724.5 eV and 710.8 eV suggest the existence of Fe3O4. The resolved peaks at 710.8 and 713.5 eV (Fe 2p3/2) and 723.8 and 726.2 eV (Fe 2p1/2) are attributed to Fe(II) and Fe(III) correspondingly36,57. A satellite peak at 719 eV and Fe 2p1/2 shake-up of about 733.4 eV were also identified58. No metallic iron is apparent in the Fe 2p3/2 and Fe 2p1/2 spectra36, consistent with X-ray diffraction patterns.

XPS spectra of (a–c) Fe@C-500 and (d–f) Fe@C-600.
The amorphous C–C configurations are the main feature in the C1s spectra of the catalysts, appearing at binding energies between 284.1 and 284.4 eV59, refer to Fig. 3c and f. This characteristic promotes the even distribution of iron oxide, facilitating the creation of active surface iron carbide species when exposed to syngas60. At elevated heat treatment, the absence of the O–C = O peak (288.1 eV) indicates the elimination of oxygen species due to the breakdown of the coordinated NDC linker and its transformation to carbon species61, as verified by XRD and FTIR.
XPS spectra of the O1s core level for Fe@C-500 and Fe@C-600 are shown in Supplementary Fig. S2. Fe@C-500 exhibits two distinct surface oxygen species: the significant bands at 529.8–531.2 eV indicate the presence of Fe oxide phases (specifically FeO-MOF cluster and Fe3O4), whereas the bands with higher binding energies at around 532.2 eV are attributed to the surface oxygen C = O group of the preserved structure of the Fe-NDC MOF, as confirmed by XRD36. The O1s XPS spectra of Fe@C-600 exhibit a lower representation of surface oxygen groups, as presented in the survey Fig. 3a and c. The lower heat treatment effectively maintains a greater O/Fe ratio and the relative quantity of oxygen groups on the surface of the porous carbon. On Fe@C-500, the relative surface percentage of oxygen is 20.92%, but on Fe@C-600, it reduces to 8.1%.
XPS analysis indicates that the concentration of Fe atoms on the outer surface of Fe@C-500 is double that of Fe@C-600, demonstrating that a higher pyrolysis temperature results in increased encapsulation of the Fe phase inside the carbon matrix. The Fe@C-500 treatment maintained the MOF structure and Fe–O clusters, increasing Fe and O surface atomic%. The increased heat treatment (Fe@C-600) led to a more significant conversion of –OH and –COOH groups into carbon-based support, decreasing the surface atomic% of oxygen. The carbon matrix covered the Fe nanoparticles, leading to a decrease in Fe and an increase in C percentage on the surface. Refer to Table 1 for XPS atomic percentages.
Figure 4 displays SEM and TEM images showing consistent crystalline hexagonal rods of fresh Fe-NDC MOF (Fig. 4(A-1 and A-2)). Following pyrolysis, the TEM images show evenly distributed Fe nanoparticles on the hexagonal rods for Fe@C-500 (Fig. 4(B-1 and B-2)) with sizes between 10 and 50 nm and an average size of 27 nm, as determined by particle size distribution (PSD) analysis (Fig. 4(D-1)). Particles ranging in size from 10 to 120 nm, with an average size of around 40 nm, Fig. 4(D-2), were identified for Fe@C-600 scattered on a carbon matrix, Fig. 4(C-1 and C-2). Elemental mapping of pyrolyzed Fe-NDC indicates that Fe@C-500 exhibits superior dispersion of iron with high intensity in the carbon matrix compared to Fe@C-600, as shown in Fig. 4(B-4 and C-4). In agreement with the XRD data, the HR-TEM images (Fig. 4(B-3 and C-3)) of pyrolyzed MOF reveal the interplanar spacings of 0.251 nm and 0.206 nm, which match the (311) and (400) lattice planes of the magnetite fcc structure. Based on the above results, the structural change of Fe-NDC MOF at different pyrolysis stages is suggested in Fig. 4E. After 500 °C pyrolysis, the parent Fe-NDC MOF’s retained hexagonal rods contain well-dispersed Fe nanoparticles. Following the collapse of the hexagonal rod structure at 600 °C, larger Fe nanoparticles were obtained and distributed in a carbon matrix.

SEM, TEM, HRTEM, STEM, and iron elemental mapping (red) images of (A) Fe-NDC, (B) Fe@C-500, and (C) Fe@C-600. (D) PSD of derived catalysts. (E) Schematic of the structural change at different pyrolysis temperatures.
BET measurements were used to analyze the surface characteristics of the catalyst by comparing the Fe-NDC sample before and after pyrolysis. The BET Analysis indicates that Fe-NDC MOF has a comparatively low BET surface area of 25.9 m2 g−1 and a non-microporous adsorption behavior in contrast to the reported literature55; see Table 2 and Fig. 5a. The decreased surface area and lack of microporous isotherm might originate from the tunnel-shaped pores55 in the Fe-NDC structure being obstructed by remnants of DMF solvent or free NDC acid, evidenced by the FTIR spectra. Following pyrolysis at 500 °C, the adsorption–desorption isotherm exhibited a slight increase in surface area (62.1 m2 g−1) and total pore volume with respect to the original MOF. Increasing the pyrolysis temperature from 500 to 600 °C resulted in a higher amount of N2 adsorbed, suggesting an expansion of the porous structure due to the collapse of the MOF caused by carbon gasification.

(a) Nitrogen adsorption isotherms of Fe-NDC and derived catalysts. (b) H2-TPR. (c) H2 and (d) CO chemisorption of derived catalysts.
Figure 5a shows that Fe@C-600 combines type-I and type-IV isotherms. Type-I microporosity originates from the parent MOF following the breakdown of free NDC acid, whereas mesoporosity results from the liberation of carbon due to MOF failure8. The BET surface area increases from 62.1 to 282 m2 g−1 after the temperature rises from 500 to 600 °C. The total volume increases, as well, from 0.01 to 0.18 cm3 g−1. The larger surface area and mesoporosity of the carbon support may aid in the diffusion of gases, leading to improved CO conversion8.
The TPR profiles of Fe@C-500 and Fe@C-600 in Fig. 5b indicate hydrogen utilization of 7.25 and 1.99 mmol g−1, highlighting the greater surface exposure of Fe nanoparticles in Fe@C-500, as validated by elemental mapping and Fe surface atomic% from XPS in Table 1. Fe@C-500 consumed more hydrogen due to the reduced amount of oxygen surface groups. Figure 5b illustrates two significant reduction stages for both catalysts. In the first two peaks, Fe3O4 is reduced to FeO and FeO is reduced to Fe, respectively. This aligns well with the reduction characteristics of iron oxides28,37. The higher heat treatment shifted the first reduction peaks to lower values (349 °C) for the Fe@C-600 catalyst, compared to Fe@C-500 (518 °C), as the Fe–O clusters in the maintained MOF structure of Fe@C-500 need more energy to be reduced. Beyond 600 °C, negative peaks in both catalysts originate from removing –OH and –COOH groups remaining in the pyrolyzed MOF.
The catalyst was evaluated for H2 and CO chemisorption to determine its ability to adsorb the supplied gases. The olefin selectivity is anticipated to be influenced by the variation of CO and H2 bindings58. Figure 5c demonstrates both weak and strong H2 adsorption in the H2 desorption curve. Fe@C-500 exhibits reduced hydrogen affinity at lower desorption temperatures (455 °C and 630 °C) than Fe@C-600 (567 °C and 734 °C). Fe@C-600 shows double the hydrogen uptake in relation to Fe@C-500, as seen in Table 2. The data suggest that a catalyst subjected to lower heat treatment exhibits greater olefin selectivity due to reduced availability of hydrogen for olefin hydrogenation.
The CO desorption curve in the Fig. 5d indicates that Fe@C-600 exhibits a small CO desorption peak at 152 °C, which is attributed to its limited adsorption strength. An enhanced and more distinct CO desorption peak at 611 °C was observed, suggesting the liberation of strongly adsorbed CO, as shown in Fig. 5d. Fe@C-500 exhibits a noticeable weak-desorption peak at 127 °C and a less pronounced strong-desorption peak at 515 °C. Desorbed CO is greater for Fe@C-600 compared to Fe@C-500, as indicated in Fig. 5c and d and Table 2. The findings suggest that Fe@C-600 exhibits more catalytic activity for CO conversion because of its higher CO uptake and higher desorption temperatures.
Catalytic activity results
FTS performance evaluation for prepared catalysts
The high thermal stability of the prepared Fe-NDC MOF, as shown by TGA results, provides the opportunity to pyrolyze the MOF partially. This could be done at a temperature (500 °C) higher than the FTS reaction conditions while retaining a large share of the MOF structure without a complete collapse of MOF driven by pyrolysis heat. Therefore, the FTS performance was compared between the prepared partially and fully pyrolyzed catalysts (Fe@C-500 and Fe@C-600). The catalytic performance of the catalysts and Fe/Al2O3 as a control catalyst was evaluated in the Fischer–Tropsch synthesis process under specific reaction conditions: T = 340 °C, P = 20 bar, GHSV = 20,000 mL g−1cat h−1, and H2/CO ratio of 1. Table 3 summarizes the FTS CO conversion, Fe-time yield (FTY), chain growth probability (α), carbon balance, carbon selectivity/yield of various product ranges, and the O/P ratio values after 17 h of operation.
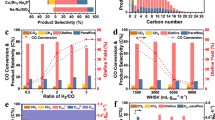
Figure 6a and b illustrates the changes in XCO and selectivity of carbon dioxide (\({\text{S}}_{{\text{CO}}_{2}}\)) and methane \(({\text{S}}_{{\text{CH}}_{4}})\) with TOS for Fe@C-500 and Fe@C-600 catalysts. Fe@C-600 has a higher XCO (89.7%) than Fe@C-500 (66.1%), but with comparable \({\text{S}}_{{\text{CO}}_{2}}\) (44%) and FTY values (660.8 vs. 709.7 mmolCOg−1Fe h−1). Figure 6c demonstrates the hydrocarbon selectivity distribution of \(({\text{S}}_{{\text{CH}}_{4}})\), C2–C4 selectivity \({(\text{S}}_{{\text{C}}_{2}-{\text{C}}_{4}})\), \({\text{S}}_{{\text{C}}_{5+}}\), and FTY for Fe@C-500 and Fe@C-600 catalysts. Fe@C-600 shows higher hydrocarbon methane selectivity, whereas \({\text{S}}_{{\text{C}}_{2}-{\text{C}}_{4}}\) remains unchanged and \({\text{S}}_{{\text{C}}_{5+}}\) is moderately increased for Fe@C-500 at the same α values (0.56–0.57). Interestingly, Fe@C-500 and Fe@C-600 catalysts produce the same amount of olefin (olefin yield of 17.5% vs. 16.5%), even though Fe@C-500 has lower XCO (66.1 vs. 89.7%) and greater olefin selectivity (48% vs. 33%).

FTS activity of (a) Fe@C-500, (b) Fe@C-600 as a function of TOS, (c) Hydrocarbon distribution and FTY, and (d–f) olefin product distribution, at P = 20 bar, GHSV = 20,000 mL g−1cat h−1, and H2/CO ratio of 1.
Figure 6d–f shows the olefin product distribution of Fe@C-500 and Fe@C-600 catalysts. Figure 6d indicates that the Fe@C-500 catalyst has larger olefin selectivity and enhanced O/P. The overall O/P ratio of Fe@C-500 (0.92) is approximately twofold higher than Fe@C-600 (0.49), see supplementary Fig. S3b and Table 3. For Fe@C-500, the light olefin selectivity \({(\text{S}}_{{\text{C}}_{2}-{{\text{C}}_{4}}^{=}})\) = 27.2%, whereas the O/P of C2-C4 approaches 2 including 66% olefins of the C2-C4 fraction, Fig. 6e, and heavy olefins (O/P (C5+)) of 1.44, Fig. 6d. On the other hand, Fe@C-600 shows \({\text{S}}_{{\text{C}}_{2}-{{\text{C}}_{4}}^{=}}\) of 18.3%, O/P of C2–C4 of 0.8, which is less than half of the value of Fe@C-500, with only 45% of olefins in the C2–C4 fraction. In addition, a lower O/P (C5+) of 1.03 is observed for Fe@C-600. Fe@C-500 outperforms Fe@C-600, showing larger FTY for light olefins, Fig. 6f, \({(\text{FTY}}_{{\text{C}}_{2}-{{\text{C}}_{4}}^{=}})\) of 179.5 mmolCO g−1Fe h−1 compared to 129.6 mmolCOg−1Fe h−1. It can be concluded that the O/P of C2–C4 and C5+ increase as the pyrolysis temperature of Fe-NDC decreases. Unlike the trend of olefin selectivity, the total olefin yield of C2-C4 and C5+ is relatively constant for both catalysts, as presented in supplementary Fig. S3b–d.
Figure 7a summarizes the FTS activity and product distribution after 17 h TOS for Fe@C-500 and Fe@C-600 catalysts. Furthermore, Fig. 7b and c show the Anderson–Shulz–Flory (ASF) plot of the prepared catalysts. The catalysts’ product spectrum adheres strictly to the Anderson-Schulz-Flory distribution, with the chain growth probability marginally shifting from 0.56 (for Fe@C-600) to 0.57 (for Fe@C-500). Both catalysts show high catalytic activity and olefin yield. However, the Fe@C-500 catalyst shows relatively lower carbon monoxide conversion while having high olefin/paraffin selectivity. In contrast, the Fe@C-600 catalyst has higher CO conversion and a lower olefin/paraffin selectivity. For example, Fe@C-600 has an ethene-to-ethane ratio of 0.16, whereas for Fe@C-500, the ratio increases to 0.6. Similarly, higher ratios were obtained with higher carbon numbers; the O/P C3 ratio increased from 1.42 to 3.62, and the O/P C4 ratio from 2.12 to 3.76, respectively.

(a) Carbon number product distribution and ASF distribution of (b) Fe@C-500 and (c) Fe@C-600 at T = 340 °C, P = 20 bar, GHSV of 20,000 mL g−1cat h−1, and H2/CO of 1.
Unexpectedly, despite the lower XCO of Fe@C-500, it shows comparable FTY and olefin yield with the more catalytically active Fe@C-600, as displayed in Fig. 6 and Table 3. The considerable FTY value of Fe@C-500 (660.8 mmolCOg−1Fe h−1) could be attributed to several factors: the relatively lower Fe loading as depicted from AAS, the higher surface concentration of Fe as observed from XPS, and the better dispersion of Fe phase as seen from elemental mapping images. The second aspect is that olefin production potential (Selectivity and O/P) increases with a decrease in the pyrolysis temperature. This trend is reported by Luo et al. and Valero-Romero et al.8,30. This could be accounted for the higher surface oxygen content, as shown from XPS, that might mitigate the secondary hydrogenation of hydrocarbon products. This is verified by hydrogen chemisorption experiments showing the lower capability of Fe@C-500 for hydrogen adsorption.
Particle and pore size are other factors to consider. The impact of surface area and pore structure on product dispersion in FTS is well recognized62,63,64. The increased pore capacity of the catalysts allows for greater exposure of active metals during the FTS reaction and facilitates gas diffusion. The presence of bigger pores increases the likelihood of chain elongation and allows for better control of hydrogenation, resulting in higher levels of \({\text{S}}_{{\text{C}}_{5+}}\) and greater selectivity towards olefins62,65. For example, Fe@C-600 has a lower pore size (2.5 nm) than Fe@C-500 (6.3 nm), which may result in lower olefin selectivity and \({\text{S}}_{{\text{C}}_{5+}}\). Moreover, the Fe@C-500 catalyst combines the unique porous properties of MOF with the mesoporous structure of the pyrolyzed part, which may provide easier transport and lower secondary olefin hydrogenation8.
Evaluation of the FTS catalytic stability of the prepared Fe-BTC catalyst
To check the stability of the catalyst, longer catalytic tests were performed for 70 h at T = 340 °C, P = 20 bar, GHSV = 20,000 mL g−1cat h−1, and H2/CO ratio of 1, Fig. 8a. The catalysts show satisfactory stability for continuous TOS up to 70 h at a constant temperature of 340 °C and high GHSV of 20,000 mL g−1cat h−1, Fig. 8a. Fe@C-500 exhibits a smaller decline in activity after 70 h (about 6%) in comparison to Fe@C-600 (approximately 10%). However, the activity could be restored to its starting value by catalyst reactivation under H2 flow at 400 °C for 2 h 7. The Fe@C-500 catalyst was reactivated after 70 h on stream, where the activity was back to its original value. The catalyst shows satisfactory stability for continuous TOS up to 140 h at a constant temperature of 340 °C and GHSV of 20,000 mL g−1cat h−1. The noticed reduced stability, particularly common in systems with Fe particles exceeding 6 nm, is typically linked to ongoing chemical and structural alteration in the iron phases, plus the deposition of carbon on the catalyst surface36,38.

(a) Catalytic performance stability of catalysts at T = 340 °C, P = 20 bar, GHSV = 20,000 mL g−1cat h−1, and H2/CO ratio of 1. (b) XRD of spent catalysts.
Figure 8b displays the XRD patterns for the spent catalysts. The primary phases present are Fe3O4 and χ-Fe5C2 (JCPDS file No. 19-0629 and No. 36-1248, respectively), along with SiC, which is used to dilute the catalyst. Weak peaks of iron carbide phase χ-Fe5C2 are shown with the presence of a substantial graphite peak at 25° in the XRD pattern of the spent Fe@C-600. Figure 8b provides evidence of observed less stability and deterioration of catalytic activity resulting from the deposition of carbon when compared with the more stable Fe@C-500. This is confirmed by the stronger typical peaks of the active iron carbide phase χ-Fe5C2 43.4° and 44.1° of Fe@C-500.
Effect of operating conditions
Table 3 and Fig. 9 show how temperature and gas hourly space velocity (GHSV) affect olefin quality, product selectivity, and FTS activity. By raising the temperature from 300 to 340 °C, the CO conversion increased slightly (68.56–71.86%) at a constant GHSV of 4200 mL g−1cat h−1. However, the methane (CH₄) selectivity increased (13.45–17.02%) and the C5 + selectivity decreased (51.45–45.05%), suggesting that cracking reactions are enhanced at higher temperatures. The findings show that heavier hydrocarbons are suppressed, whereas light olefin generation is favoured by increased temperature and gas hourly space velocity (GHSV). At a constant GHSV (4200 mL g−1cat h−1), raising the temperature from 300 to 340 °C improved the selectivity of C2–C4 olefins (O/P ratio strengthened from 2.00 to 2.53) while decreasing C₅ + olefins (O/P dropped from 2.16 to 1.84). This implies that light olefin production is stimulated by higher temperatures because of improved cracking and dehydrogenation processes.

Product distribution comparison of Fe@C-500 at different temperatures and GHSV at P = 20 bar and H2/CO = 1.
CO conversion reduced significantly (66.08%) when GHSV was boosted to 20,000 mL g−1cat h−1 at 340 °C. At the same time, CH₄ and C₂–C₄ selectivity increased further (23.32% and 41.37%, respectively), while C₅ + selectivity declined substantially (35.30%). Shorter residence duration at greater GHSV promoted shorter hydrocarbon chains. The Fe-time yield (FTY) increased dramatically (660.8 mmolCO g−1Fe h−1) despite greater GHSV, at the expense of heavier hydrocarbon (C5+) and CO conversion, potentially because of improved mass transfer. C₂–C₄ olefin selectivity maintained its value (O/P = 1.95) when GHSV was raised to 20,000 mL g−1cat h⁻1 at 340 °C, while C₅ + olefins further decreased (O/P = 1.44). Interestingly, the FTY for light hydrocarbons increased (from 48.27–179.5 mmolCO g−1Fe h−1) while the yield of C₂–C₄ olefins stayed constant (9.25–9.90 mmolCO g−1Fe h−1). This suggests that higher GHSV increases light hydrocarbon productivity without compromising light olefin selectivity. These trends imply that higher temperature and GHSV cause the product distribution to shift towards lighter olefins, most likely as a result of shorter residence times and enhanced cracking.
Comparison with prepared reference catalyst
The IWI-synthesized Alumina supported catalyst (Fe/Al2O3) prepared in our previous study7 was tested as a reference catalyst at the same conditions, T = 340 °C, P = 20 bar, GHSV = 20,000 mL g−1cat h−1, and H2/CO ratio of 1. Figure 10 highlights the diverse FTS behavior between the reference and prepared Fe-NDC-derived catalysts. The promoter-free Fe/Al2O3 catalyst has equivalent carbon monoxide conversion to that of Fe@C-500. However, Fe/Al2O3 has the highest methane (42.3%), carbon dioxide selectivity (50.5%), and light hydrocarbons (C1–C4 = 92.4%), Fig. 10(a and b) and Table 3. Furthermore, it has a smaller C5+ of 7.6% with α = 0.39. Besides, Fe/Al2O3 has the lowest total olefin selectivity/yield, O/P, FTY, and \({\text{FTY}}_{{\text{C}}_{2}-{{\text{C}}_{4}}^{=}}\), supplementary Fig. S4. However, it has a comparable light olefin selectivity (19.3%) and total paraffin selectivity (75.5%) to that of Fe@C-600. In contrast to Fe/Al2O3, promoter-free Fe-NDC catalysts exhibit enhanced results. The mismatch proves the beneficial impact of the carbon support of the Fe-NDC catalyst. The higher C5+ (29.3–35.3%) and gasoline-range hydrocarbons (C5–C12), ~ 30% for both catalysts, could be attributed to the porous carbon support.

(a) Product distribution comparison and (b) olefin selectivity and FTY of Fe@C-500, Fe@C-600, and Fe/Al2O3, at T = 340 °C, P = 20 bar, GHSV = 20,000 mL g−1cat h−1, and H2/CO = 1.
Comparison with previous work
Supporting Table S1 summarizes comparisons with other Fe-MOF-generated catalysts, references, and commercial catalysts published in the literature. Using various promoters and operating scenarios, such as gas GHSV and pressure, complicates the evaluation task. However, according to the table, the XCO value for the prepared catalysts (66.1–89.7%) is similar to or greater than the reported Fe-MOF-derived catalysts. The overall Fe-time yield (660.8–709.7 mmolCOg−1Fe h−1) and \({\text{FTY}}_{{\text{C}}_{2}-{{\text{C}}_{4}}^{=}}\) (129.6–179.5 mmolCOg-1Fe h-1) are higher than most past studies, except for those that used higher GHSV values than 20,000 mL g−1cat h−1.
The catalyst’s exceptional performance is highlighted when compared to the recognized Ruhrchemie industrial catalyst (Fe-Cu-K-SiO2)66,67, Table S1. The comparison shows that the Fe@C-500 catalyst, which was not promoted, has a lower XCO but still maintains the same level of olefin selectivity/yield as the promoted Ruhrchemie catalyst. Additionally, the Fe@C-500 catalyst has higher total and light olefin FTY, as well as higher \({\text{S}}_{{\text{C}}_{5+}}\) values.
Fe@C-500 exhibits notable performance in terms of total olefin selectivity (~ 50%) and light olefin yield and selectivity (17.5% and 27%, respectively). These values are either greater or equivalent to the majority of research, as shown in Table S1. Furthermore, it is equivalent to our previously documented Fe-BTC/C catalyst under the same conditions7, as shown in the Fig. 11. Furthermore, all experimental outcomes were achieved using catalysts without promoters, which boost the catalytic activity and olefin production.

Olefin indicators produced in this work compared to state-of-the-art Fe-MOF-based catalysts.
Eventually, the Fe@C-500 catalyst described here demonstrates a superior combination of C2-C4 olefin selectivity, yield, and FTY, considering the current state of the art (see supplementary Table S1), exceeds a wide range of the existing literature7,23,28,34,36,37,38,41,61,66,67,68,69,70 (see Fig. 11). For example, the material significantly surpasses two of the recently published Fe-MOF catalysts (first and third row in Table S1), showing a C2–C4 olefins FTY ~ 27 and 6 times higher than the promoter-free Fe-MIL-88B and Fe-MIL-100-derived catalyst reported by Wang et al. and Qin et al., respectively68,69.
While this study reveals Fe@C-500’s better performance in selective olefin synthesis, various limitations should be noted. The investigation focused on only two pyrolysis temperatures (500 °C and 600 °C). The best temperature for balancing structural retention and activity was 500 °C. Lower temperatures (< 500 °C) were not examined due to potential negative effects such as excessive preservation of the MOF framework, which might mask active Fe sites and drastically reduce syngas conversion. Pure CO and green H2 were employed in experiments. However, real-world syngas from coal/biomass has variable H2/CO ratios while hosting other gases and impurities (e.g., H₂S) that might poison the catalyst, which was not explored in this work. Testing under authentic feedstock scenarios is required for evaluating catalytic performance. The catalyst’s performance was examined under controlled, short-term operating conditions (70 h). Continuous operation (> 100 h) can result in deactivation pathways (e.g., coking, sintering), as indicated by XRD of spent catalysts for Fe@C-600. The significant role of residual MOF porosity in suppressing olefin hydrogenation, although noticeable from selectivity trends, needs additional confirmation using in-situ spectroscopy or computational modelling.
Conclusions
In summary, the Fe@C-500 catalyst produced from Fe-NDC doubles the olefin/paraffin ratio while retaining MOF-like porosity at 500 °C. The sustained structure of the catalyst allowed reactant movement and limited olefin hydrogenation, leading to an improved olefin/paraffin ratio. However, its untested stability for a prolonged time and limited for pure syngas warrant further optimization. Despite these limitations, the promoter-free Fe@C-500 catalyst’s light olefin selectivity and yield are more significant or equivalent to state-of-the-art Fe-MOF-derived catalysts. Additionally, it shows comparable olefin selectivity and yield, increased Fe-time yield for light olefins, and higher C5+ values compared to the promoted Ruhrchemie industrial catalyst. Going forward with practical applications, further research should examine long-term stability under a continuous flow of industrial syngas. These results establish Fe-NDC as a potential precursor for structure-controlled catalysts for converting syngas into valuable chemicals.
Data availability
All relevant data are available from the corresponding author at reasonable request.
References
Eldesouki, M. H., Rashed, A. E. & El-Moneim, A. A. A comprehensive overview of carbon dioxide, including emission sources, capture technologies, and the conversion into value-added products. Clean Technol. Environ. Policy 25, 3131–3148 (2023).
Choi, Y. H. et al. Carbon dioxide Fischer–Tropsch synthesis: A new path to carbon-neutral fuels. Appl. Catal. B 202, 605–610 (2017).
Braide, D., Panaritis, C., Patience, G. & Boffito, D. C. Gas to liquids (GTL) microrefinery technologies: A review and perspective on socio-economic implications. Fuel 375, 125169 (2024).
Dieterich, V., Buttler, A., Hanel, A., Spliethoff, H. & Fendt, S. Power-to-liquid via synthesis of methanol, DME or Fischer–Tropsch-fuels: a review. Energy Environ. Sci. 13, 3207–3252 (2020).
Gao, Y. et al. Syngas production from biomass gasification: Influences of feedstock properties, reactor type, and reaction parameters. ACS Omega 8, 31620–31631 (2023).
Wang, L. et al. Sustainable light olefins synthesis via CO2 hydrogenation: Comparative exergetic, exergoeconomic, and exergoenvironmental analyses. J. Environ. Chem. Eng. 12, 113113 (2024).
Rashed, A. E., Elkady, M. F., Matsushita, Y., Nasser, A. & Abd El-Moneim, A. Syngas to FCC-like gasoline range hydrocarbons with upgraded light olefin selectivity catalyzed by readily synthesized Fe-MOF. Chem. Eng. J. 473, 145125 (2023).
Valero-Romero, M. J., García-Mateos, F. J., Kapteijn, F., Rodríguez-Mirasol, J. & Cordero, T. Fischer-Tropsch synthesis over lignin-derived cobalt-containing porous carbon fiber catalysts. Appl. Catal. B 321, 122078 (2023).
Eldesouki, M. H., Mohamed, M. M. & Abdelmoniem, A. Targeting hydrogen-rich gasoline production from CO₂ via a sustainable Fe-Cu tandem catalyst. J. Environ. Chem. Eng. 13, 116328 (2025).
Shafer, W. D. et al. Fischer–Tropsch: Product selectivity-the fingerprint of synthetic fuels. Catalysts 9, 259 (2019).
Sun, B., Xu, K., Nguyen, L., Qiao, M. & Tao, F. F. Preparation and catalysis of carbon-supported iron catalysts for Fischer-Tropsch synthesis. ChemCatChem 4, 1498–1511 (2012).
Luk, H. T. et al. Role of carbonaceous supports and potassium promoter on higher alcohols synthesis over copper-iron catalysts. ACS Catal 8, 9604–9618 (2018).
Have, I. C. T., van den Brink, R. Y., Marie-Rose, S. C., Meirer, F. & Weckhuysen, B. M. Using biomass gasification mineral residue as catalyst to produce light olefins from CO, CO2, and H2 mixtures. Chemsuschem 15, e202200436 (2022).
El-Deen, A. G. et al. High-performance asymmetric supercapacitor based hierarchical NiCo2O4@ carbon nanofibers//Activated multichannel carbon nanofibers. Nanotechnology 31, 365404 (2020).
Ravindran, M. X. Y. et al. A review of carbon-based catalyst for production of renewable hydrocarbon rich fuel. J Environ Chem Eng 12, 112330 (2024).
Okur, O. & Şakoğlu, P. Development of a green catalytic route to light olefins by Fischer-Tropsch synthesis with renewable hydrogen: Investigation of boron doped activated carbon supported iron catalyst. Int. J. Hydrogen. Energy 55, 1102–1108 (2024).
Nasser, A. H. et al. Mn-Fe nanoparticles on a reduced graphene oxide catalyst for enhanced olefin production from syngas in a slurry reactor. RSC Adv. 8, 14854–14863 (2018).
Hamed, A., Hessein, A. & Abd El-Moneim, A. Towards high performance flexible planar supercapacitors: In-situ laser scribing doping and reduction of graphene oxide films. Appl. Surf. Sci. 551, 149457 (2021).
Belal, M. A. et al. Layered structure design of inkjet-printed graphene/Co3O4for high-performance flexible microsupercapacitors. J Energy Storage 101, (2024).
Shi, X. et al. Synergistic effect of nitrogen-doped carbon-nanotube-supported Cu–Fe catalyst for the synthesis of higher alcohols from syngas. Fuel 210, 241–248 (2017).
Shaker, A., Hassanin, A. H., Shaalan, N. M., Hassan, M. A. & El-Moneim, A. A. Micropatterned flexible strain gauge sensor based on wet electrospun polyurethane/PEDOT: PSS nanofibers. Smart Mater. Struct. 28, 75029 (2019).
Rashed, A. E. et al. Highly Active Fischer-Tropsch Synthesis Fe-BDC MOF-Derived Catalyst Prepared by Modified Solvothermal Method. Key Eng Mater 891, 56–61 (2021).
Zhao, Q. et al. Highly active and controllable MOF-derived carbon nanosheets supported iron catalysts for Fischer-Tropsch synthesis. Carbon 173, 364–375 (2021).
Wang, A. et al. MOF-derived porous carbon-supported bimetallic Fischer-Tropsch synthesis catalysts. Ind. Eng. Chem. Res. 61, 3941–3951 (2022).
Kabir, L. M., Albolkany, M. K., Mohamed, M. M. & El-Moneim, A. A. Porous carbon-supported cobalt catalyst for CO hydrogenation to gasoline range hydrocarbons. Catal. Letters 154, 3372–3387 (2024).
Cui, W. G., Zhang, G. Y., Hu, T. L. & Bu, X. H. Metal-organic framework-based heterogeneous catalysts for the conversion of C1 chemistry: CO, CO2 and CH4. Coord. Chem. Rev. 387, 79–120 (2019).
Linares, N., Silvestre-Albero, A. M., Serrano, E., Silvestre-Albero, J. & García-Martínez, J. Mesoporous materials for clean energy technologies. Chem Soc Rev 43, 7681–7717 (2014).
Oschatz, M. et al. Influence of precursor porosity on sodium and sulfur promoted iron/carbon Fischer-Tropsch catalysts derived from metal-organic frameworks. Chem. Commun. 53, 10204–10207 (2017).
Chen, Y. et al. Carbon-based catalysts for Fischer-Tropsch synthesis. Chem. Soc. Rev. 50, 2337–2366 (2021).
Luo, Q. X. et al. Cobalt nanoparticles confined in carbon matrix for probing the size dependence in Fischer-Tropsch synthesis. J. Catal. 369, 143–156 (2019).
Oar-Arteta, L., Wezendonk, T., Sun, X., Kapteijn, F. & Gascon, J. Metal organic frameworks as precursors for the manufacture of advanced catalytic materials. Mater. Chem. Front 1, 1709–1745 (2017).
Zafari, R., Abdouss, M. & Zamani, Y. Application of response surface methodology for the optimization of light olefins production from CO hydrogenation using an efficient catalyst. Fuel 237, 1262–1273 (2019).
Munir, S., Amin, M., Iqbal, N., Iqbal, A. & Ghfar, A. A. Effect of Pyrolysis on iron-metal organic frameworks (MOFs) to Fe3C @ Fe5C2 for diesel production in Fischer-Tropsch Synthesis. Front Chem 11, 1–9 (2023).
Wezendonk, T. A. et al. Elucidating the nature of Fe species during pyrolysis of the Fe-BTC MOF into highly active and stable Fischer-Tropsch catalysts. ACS Catal. 6, 3236–3247 (2016).
Galallah, A. G. et al. Optimizing Mo2C-based catalytic system for efficient CO2 conversion and CO selectivity through carbon-nitrogen supporting and potassium promotion. J. Environ. Chem. Eng. 12, 113380 (2024).
Santos, V. P. et al. Metal organic framework-mediated synthesis of highly active and stable Fischer-Tropsch catalysts. Nat. Commun. 6, 6451 (2015).
An, B., Cheng, K., Wang, C., Wang, Y. & Lin, W. Pyrolysis of Metal-Organic Frameworks to Fe3O4@Fe5C2 Core-Shell Nanoparticles for Fischer-Tropsch Synthesis. ACS Catal 6, 3610–3618 (2016).
Rashed, A. E., Nasser, A., Elkady, M. F., Matsushita, Y. & El-Moneim, A. A. Fe nanoparticle size control of the Fe-MOF-derived catalyst using a solvothermal method: Effect on FTS activity and olefin production. ACS Omega 7, 8403–8419 (2022).
Ahmed, H. E., Rashed, A. E., El-Khouly, M. E., Albolkany, M. K. & El-Moneim, A. A. Green approach for sustainable production of paraffin fuel from CO2 hydrogenation on Fe-MOF catalyst. J. Environ. Chem. Eng. 11, 2 (2023).
Wezendonk, T. A. et al. Controlled formation of iron carbides and their performance in Fischer-Tropsch synthesis. J Catal 362, 106–117 (2018).
Oar-Arteta, L., Valero-Romero, M. J., Wezendonk, T., Kapteijn, F. & Gascon, J. Formulation and catalytic performance of MOF-derived Fe@C/Al composites for high temperature Fischer-Tropsch synthesis. Catal. Sci. Technol. 8, 210–220 (2018).
Wezendonk, T. A. et al. Structural and elemental influence from various MOFs on the performance of Fe@C catalysts for Fischer-Tropsch synthesis. Faraday Discuss 197, 225–242 (2017).
Healy, C. et al. The thermal stability of metal-organic frameworks. Coord. Chem. Rev. 419, 213388 (2020).
Escobar-Hernandez, H. U. et al. Thermal stability of metal-organic frameworks (MOFs): Concept, determination, and model prediction using computational chemistry and machine learning. Ind. Eng. Chem. Res. 61, 5853–5862 (2022).
Kandiah, M. et al. Synthesis and stability of tagged UiO-66 Zr-MOFs. Chem. Mater. 22, 6632–6640 (2010).
Xiao, C., Tian, J., Chen, Q. & Hong, M. Water-stable metal–organic frameworks (MOFs): rational construction and carbon dioxide capture. Chem. Sci. 15, 1570–1610 (2024).
Ibrahim, M., Sabouni, R. & Husseini, G. A. Synthesis of metal-organic framework from iron nitrate and 2,6-naphthalenedicarboxylic acid and its application as drug carrier. J. Nanosci. Nanotechnol. 18, 5266–5273 (2018).
Zhang, W. et al. A new metal–organic framework with high stability based on zirconium for sensing small molecules. Microporous Mesoporous Mater. 171, 118–124 (2013).
Howarth, A. J. et al. Chemical, thermal and mechanical stabilities of metal–organic frameworks. Nat. Rev. Mater. 1, 15018 (2016).
El-Khatib, K. M., Abou Helal, M. O., El-Moneim, A. A. & Tawfik, H. Corrosion stability of SUS316L HVOF sprayed coatings as lightweight bipolar plate materials in PEM fuel cells. Anti-Corrosion Methods Mater. 51, 136–142 (2004).
Hassan, S., Suzuki, M. & El-moneim, A. A. Capacitive behavior of manganese dioxide / stainless steel electrodes at different deposition currents. Am. J. Mater. Sci. 2, 11–14 (2012).
El-Moneim, A. A., Akiyama, E., Ismail, K. M. & Hashimoto, K. Corrosion behaviour of sputter-deposited Mg–Zr alloys in a borate buffer solution. Corros. Sci. 53, 2988–2993 (2011).
Rashed, A. E., Nasser, A., Elkady, M. F., Matsushita, Y. & Abd El-Moneim, A. Temperature calibration effect on FTS activity and product selectivity using Fe-MOF catalyst. Case Stud. Chem. Environ. Eng. 7, 100300 (2023).
Nasser, A. L. H., El-Naggar, H., El-Bery, H., Basha, I. & Abdelmoneim, A. Correction: Utilizing FBR to produce olefins from CO reduction using Fe-Mn nanoparticles on reduced graphene oxide catalysts and comparing the performance with SBR. RSC Adv. 9, 10937 (2019).
Chevreau, H. et al. Mixed-linker hybrid superpolyhedra for the production of a series of large-pore iron(III) carboxylate metal-organic frameworks. Angew. Chem. Int. Ed 52, 5056–5060 (2013).
Lou, X. et al. Capacity control of ferric coordination polymers by zinc nitrate for lithium-ion batteries. RSC Adv. 6, 86126–86130 (2016).
Chen, J. et al. Benchmarking promoters of Fe/activated carbon catalyst for stable hydrogenation of CO2 to liquid hydrocarbons. Appl. Catal. B 325, 122370 (2023).
Ramirez, A., Gevers, L., Bavykina, A., Ould-Chikh, S. & Gascon, J. Metal organic framework-derived iron catalysts for the direct hydrogenation of CO2 to short chain olefins. ACS Catal. 8, 9174–9182 (2018).
Liu, J. et al. Fe-MOF-derived highly active catalysts for carbon dioxide hydrogenation to valuable hydrocarbons. J. CO2 Util. 21, 100–107 (2017).
Sun, J., Wang, P. & Chen, J. Fe2O3 hollow microspheres as highly selective catalysts for the production of α-olefins. New J. Chem. 42, 17923–17930 (2018).
Wu, Y. et al. Facile synthesis of uniform metal carbide nanoparticles from metal-organic frameworks by laser metallurgy. ACS Appl. Mater. Interfaces 11, 44573–44581 (2019).
Tu, J., Yuan, J., Kang, S., Xu, Y. & Wang, T. One-pot synthesis of carbon-coated Fe3O4 nanoparticles with tunable size for production of gasoline fuels. New J. Chem. 42, 10861–10867 (2018).
Kim, J. et al. Unravelling the effects of short pore on Fischer-Tropsch synthesis and its role as selectivity controller. Chem. Eng. J. 475, 11–14 (2023).
Gorimbo, J. et al. Effect of support particle size in Fischer-Tropsch synthesis: The use of natural clinoptilolite as support. Ind. Eng. Chem. Res. 63, 3018–3029 (2024).
Janani, H., Mirzaei, A. A. & Rezvani, A. Correlation of metal–organic framework structures and catalytic performance in Fischer-Tropsch synthesis process. React. Kinet. Mech. Catal. 128, 205–215 (2019).
Torres Galvis, H. M. et al. Supported iron nanoparticles as catalysts for sustainable production of lower olefins. Science 335, 835–838 (2012).
Ma, W., Ding, Y., Vázquez, V. H. C. & Bukur, D. B. Study on catalytic performance and attrition strength of the Ruhrchemie catalyst for the Fischer-Tropsch synthesis in a stirred tank slurry reactor. Appl. Catal. A Gen. 268, 99–106 (2004).
Qin, H. et al. Metal organic framework (MOF)/wood derived multi-cylinders high-power 3D reactor. ACS Appl. Mater. Interfaces 13, 5460–5468 (2021).
Wang, A. et al. Effect of Na, Cu and Ru on metal-organic framework-derived porous carbon supported iron catalyst for Fischer-Tropsch synthesis. Mol. Catal. 509, 111601 (2021).
Yang, X. et al. Synthesis and catalytic properties of iron based Fischer-Tropsch catalyst mediated by MOFs Fe-MIL-100. Acta Chimi. Sin. 75, 360–366 (2017).
Acknowledgements
This study was completed as part of the Academy of Scientific Research and Technology (ASRT) supported research project "Green Integrated Solar Fuel Production System: Two Steps and Direct FT Synthesis Routs" (ID: 7825). The Graphene Center of Excellence for Electronic and Energy Applications (GCEE) at EJUST offered facilities, analysis, and materials for this research.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
A.E.R. was engaged in all phases, including the research idea, experimental design, interpretation of data, and manuscript review. Catalytic System was designed by A.E.R. and M.N. Material synthesis and catalytic experiments were performed by A.E.R. Material characterization and Gas Chromatography analyses were conducted by A.E.R. and M.N. A.A.E. was responsible for funding acquisition and Project administration. A.E.R. wrote the main manuscript, and all authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rashed, A.E., Nofal, M.S. & El-Moneim, A.A. Thermally stable metal–organic framework based iron 2,6-naphthalenedicarboxylic catalyst (Fe-NDC) for syngas conversion to olefin. Sci Rep 15, 26526 (2025). https://doi.org/10.1038/s41598-025-09332-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-09332-0
