
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a highly lethal malignancy that urgently needs more effective therapies. Cancer-associated fibroblasts (CAFs) contribute to the aggressive and chemo-resistant nature of the disease by creating a drug-impeding fibrotic microenvironment. We developed novel compounds, the racemate CRO-05 and its active enantiomer CRO-67, which target both pancreatic tumor and CAF cells with robust anti-cancer activity. These compounds were designed using rational medicinal chemistry based on chromans, a class of anti-cancer drugs. Their therapeutic potential and efficacy were assessed in a clinically relevant patient-derived PDAC tumor explant model, which mimics the disease’s 3-dimensional complexity. CRO-67 treatment in these explants significantly reduced tumor cell and αSMA+ CAF frequency, decreased cell proliferation and increased cell death. CRO-67 also significantly decreased cell proliferation and enhanced apoptosis by inhibiting cell cycle progression through G2/M phase in PDAC cells and patient-derived CAFs in vitro. CRO-67 treatment of orthotopic PDAC tumors in mice significantly reduced tumor growth in tumors with active growth (> 150% growth at endpoint), and remodeled tumor stroma (reduced αSMA+ CAF frequency, loosened tumor fibrosis and normalized tumor vasculature). Finally, CRO-67 sensitized PDAC cells to multiple standard-of-care chemotherapeutics in vitro, paving the way for future combination therapy development and validation.
Similar content being viewed by others

Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a highly lethal malignancy with a 5-year survival rate of less than 13%1. Surgery has curative potential, but only 15–20% of patients are eligible and most still succumb to the disease2. Most patients with non-resectable PDAC depend on chemotherapy regimens such as FOLFIRINOX or gemcitabine/nab-paclitaxel (Abraxane), which provide only modest improvements in survival outcomes3. Therefore, there is an urgent need to develop new therapeutic strategies to improve the dismal survival rates of PDAC. A key driver of PDAC’s aggressive nature is its highly complex, multicellular, and heterogeneous microenvironment2. This microenvironment consists of a dense fibrotic matrix that can serve as a physical barrier to drug delivery and immune cell access4,5,6,7,8. The fibrosis in PDAC compresses tumor vasculature, creating a highly hypoxic and nutrient deprived landscape that drives selection of aggressive PDAC subclones with chemo-resistant and metastatic phenotypes9,10,11,12. Cancer-associated fibroblasts (CAFs) play a significant role in orchestrating fibrosis in PDAC9,12,13,14. They also engage in bi-directional crosstalk with PDAC cells through the secretion of cytokines, growth factors, nutrients, and immunosuppressive factors to promote tumor progression, chemoresistance, and metastasis9,13,15. However, a growing body of research on CAF heterogeneity indicates that within a single patient, CAFs can comprise a diverse mix of subpopulations, some of which promote PDAC progression, while others may exhibit anti-tumor effects16,17,18. The plasticity of CAFs further complicates this landscape, allowing them to adapt their functions in response to the tumor microenvironment. Therefore, targeting specific CAF subpopulations proves challenging19. Since CAFs and cancer cells are deeply interconnected within the PDAC tumor microenvironment, novel therapeutic strategies should aim to simultaneously target both tumor cells and the pro-tumor activity of CAFs.
To effectively translate such therapeutics to the clinic, preclinical models that accurately mimic the complex and heterogeneous nature of human PDAC are needed. Traditionally, mouse models such as the genetically engineered KPC model and orthotopic xenograft models have been the gold standard for drug testing in PDAC20. However, these animal models have limitations in recapitulating the functional complexity of the human PDAC microenvironment due to mouse and human species differences2. Patient-derived tumor organoids have recently gained traction as a rapid method to evaluate therapeutic response in the presence of interpatient heterogeneity but consist of tumor cells only and lack the presence and functionality of the PDAC stroma2,21,22,23,24. Recently, our group developed a new PDAC preclinical model using whole tissue explants (1–2 mm diameter) cultured ex vivo for 12 days25. These explants retained the 3D tumor and stromal multicellular architecture, fibrosis, and cell proliferation of patient PDAC tumors, while also maintaining the heterogeneity of the patient’s CAF and tumor cells25. Thus, this patient-derived tumor explant model, which mimics the complex tumor microenvironment and heterogeneity of PDAC, is an attractive tool for evaluating novel therapeutics that target both the tumor and stroma in PDAC2,12,25,26.
Chromans are a class of anti-cancer drugs, which have been extensively studied for more than a decade, demonstrating anti-cancer activity in melanoma, ovarian and breast cancers27,28. In collaboration with Noxopharm Limited, an Australian biotechnology company, we developed a series of new drugs within the chroman family, namely CRO-05, CRO-67, and CRO-68 (Fig. 1). CRO-05 is a functionalized dihydrobenzopyran that is a racemic mix of two cis-enantiomers, CRO-67 and CRO-68. The development of these small molecules involved modifications to the chroman scaffold, such as the replacement of a phenol group with amine on the 4-phenyl ring, which markedly increased resistance to Phase II metabolism and improved half-life (outlined in the composition of matter patent [Patent #PCT/AU2023/050505]).

Synthesis of CRO-05, CRO-67, and CRO-68. Synthesis of the racemic CRO-05 1 was performed on multi-gram scale at Jubilant Biosys (Greater Noida, India), which was subsequently separated at ChemPartner Co., Ltd (Shanghai, China) via Supercritical Fluid Chromatography (SFC) to obtain enantiopure (+)-CRO-67 6 and (−)-CRO-68 7. Resorcinol 2, aryl ketone 3, O-acylated coumarin derivative 4, 2H-chromene 5. (i) 4-nitrobenzoylchloride, AlCl3, DCM, 0 to 50 °C, 16 h, 23%; (ii) 3,4-dimethylphenylacetic acid, diisopropylethylamine (DIPEA), Ac2O, 130 °C, 16 h, 31%; (iii) BH3:DMS, THF, 0 °C to room temperature, 16 h, 59%; (iv) Pd/C (cat.), H2 (g, 10 kg/cm2), EtOAc, 45 °C, 16 h, 73%; (v) Supercritical Fluid Chromatography (SFC), (+)-CRO-67, 96%, enantiomeric excess (ee) 99.52%, (−)-CRO-68, 98%, ee 100%.
In this study, we utilized an advanced PDAC tumor explant model that is clinically relevant to evaluate the therapeutic efficacy of CRO-05, CRO-67, and CRO-68. The racemate CRO-05 significantly decreased PDAC tumor and αSMA+ CAF cell frequency in patient PDAC explants, as well as decreased cell proliferation and increased cell death. However, given that racemic mixes are less desirable in medicinal drug development due to the potential for adverse effects from “inactive” enantiomers29, CRO-67 the active enantiomer, became the focus of subsequent assessments. CRO-67 treatment potently reduced tumor cell frequency, depleted αSMA+ CAFs, and decreased cell proliferation in patient PDAC tumor samples treated with CRO-67. CRO-67 also exerted significant anti-proliferative and pro-apoptotic effects in PDAC and CAF cells in vitro, through cell cycle disruption. We demonstrated that CRO-67 reduced tumor growth in orthotopic PDAC tumors with > 150% growth at endpoint (actively proliferating). CRO-67 also alleviated physical barriers to drug delivery in orthotopic PDAC tumors through stromal remodeling (i.e. reduced αSMA+ CAF frequency, loosened tumor fibrosis, and normalized tumor vasculature). Notably, CRO-67’s anti-tumor effects in vivo were less extensive when compared to those observed in patient explants. However, lower dose treatment of explants with CRO-67 demonstrated comparable results to those observed in vivo, implying combination therapy would be required to elicit more effective anti-tumor effects in vivo. Finally, we identified potential candidates for combination therapy with CRO-67 in vitro.
Collectively, our findings demonstrated that: (1) CRO-67 is a dual-cell therapy drug that targets both PDAC tumor cells and stromal-CAFs, (2) CRO-67 interferes with cell cycle progression, and (3) CRO-67 has the potential to improve sensitivity of PDAC cells to chemotherapy drugs and enhance therapeutic response. These results support further preclinical development of CRO-67 as a prospective combination therapy drug for pancreatic cancer.
Results
CRO-05 treatment significantly decreased tumor and αSMA + CAF cell frequency, reduced cell proliferation, and increased cell death in human PDAC explants
Four PDAC patient explants (Patient 1–4, Fig. 2, Supplementary Table S1 and Supplementary Fig. S1) were treated with CRO-05 every 72 h. On day 12, explants were fixed, embedded, and sectioned for analysis. Explant sections were stained for cytokeratin (cancer cells), αSMA (CAFs), BrdU (proliferation) and TUNEL (cell death). Note that within explant regions, there were areas with neither cytokeratin nor αSMA staining that were largely stromal scar tissue. Note that thresholds for detection of αSMA using Qupath software was set per patient, and did not distinguish between αSMA high versus low CAF subsets. Overall, the results demonstrated that CRO-05 treatment had significant effects on tumor cell and αSMA+ CAF frequency, proliferation and cell death (Fig. 2). The most potent and consistent effects were observed at 50 µg/mL (145 µM) CRO-05, which resulted in a significant reduction in tumor cell frequency, αSMA+ CAF frequency, and cell proliferation, and increased cell death, relative to controls (Fig. 2b–e).

CRO-05 treatment in PDAC tumor explants from patients reduced tumor and CAF cell frequency, reduced cell proliferation, and increased cell death. PDAC tumor explants from N = 4 patients were cultured for 12 days and treated with 0, 10, 20 and 50 μg/mL of CRO-05 every 72 h. (a) Representative immunohistochemistry (IHC) images for Patient 1 explants stained for tumor cells (cytokeratin), CAFs (α-smooth muscle actin [αSMA]), proliferating cells (BrdU), and cell death (TUNEL). Positive staining for IHC is brown, positive TUNEL staining is green, and nuclei are blue. Quantification of IHC stain for (b) tumor cells, (c) CAFs, (d) proliferating cells, and (e) cell death was performed using QuPath software. Inset images for BrdU staining show nuclei positive for BrdU in brown compared to negative nuclear-only staining in blue. Cytokeratin staining was not quantifiable in Patient 2 (green symbols) due to a lack of tumor cells present in all tumor sections irrespective of control or CRO-05 treatments. Symbols represent the mean of 2–4 explants per treatment from each patient. Colors indicate percentage relative to control for each patient. Patient 3 explants (blue symbols) were not treated with 10 μg/mL CRO-05 due to limited availability of tumor tissue. Bars represent mean ± SD (N < 5). Asterisks indicate statistical significance as assessed by one-way ANOVA, and Bonferroni’s multiple comparisons tests (*p ≤ 0.05, **p ≤ 0.01).
CRO-67 is the active enantiomer of the CRO-05 racemic compound
CRO-05 is a racemic mixture of two enantiomers: CRO-67 and CRO-68. We next evaluated which of the enantiomers was responsible for the effects observed with CRO-05 in the patient PDAC explants (Fig. 3). MiaPaCa-2 cells and patient-derived CAFs (N = 3) were treated with vehicle or 1.5 μM of CRO-05, CRO-67 or CRO-68. Cell viability and apoptosis were measured 24 h post-treatment. CRO-05 and CRO-67 treatments in MiaPaCa-2 cells and CAFs significantly decreased cell viability (Fig. 3a,b), and increased apoptosis relative to controls (Fig. 3c,d), while CRO-68 had no effect on cell viability nor apoptosis in either cell type (Fig. 3a–d and Supplementary Fig. S2).

CRO-67 is the active enantiomer of CRO-05 that decreased viability and increased apoptosis in PDAC and CAF cells. MiaPaCa-2 (N = 3) and primary patient-derived CAFs isolated from N = 3 PDAC patients were treated with vehicle control, CRO-05, CRO-67 and CRO-68 (1.5 μM) and analyzed 24 h post-treatment. (a, b) Cell viability was measured by counting live cells via trypan blue exclusion assay. CRO-05 and CRO-67 treated MiaPaCa-2 cells and patient-derived CAFs demonstrated a reduction in cell viability while CRO-68 treated cells showed no change. (c, d) Apoptosis was measured by flow cytometry for Annexin V/DAPI. MiaPaCa-2 (N = 3) and CAFs (N = 3) demonstrated an increase in apoptosis 24 h post-treatment with CRO-05 and CRO-67, but no change when treated with CRO-68. Bars represent mean ± SD (N = 3). Asterisks indicate significance as assessed by one-way ANOVA, and Bonferroni’s multiple comparisons tests (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001).
CRO-67 treatment decreased tumor and CAF cell frequency, reduced cell proliferation, and increased cell death in human PDAC explants
CRO-67 efficacy was tested in five PDAC patient explants (Patients 1 and 5–8, Fig. 4, Supplementary Table S1 and Supplementary Fig. S3) at concentrations of 0, 10, 20 and 50 µg/mL treated every 72 h. Fixed sections were evaluated by immunohistochemistry as per CRO-05 experiments above. CRO-67 almost completely depleted tumor and αSMA+ CAFs, reduced cell proliferation, and increased cell death to comparable or greater levels (Fig. 4) as those observed with CRO-05 (Fig. 2). Notably, we observed higher potency with CRO-67 at the lowest tested dose (10 µg/mL) across all measured markers, when compared to CRO-05. To validate our in vitro results on CRO-68, we included a simultaneous comparison of efficacy using CRO-67 and CRO-68 in matched patient explants (Patient 7, Supplementary Figs. S4–S6). In contrast to CRO-67 treatment, CRO-68 treated Patient 7 explants showed no significant changes to tumor and αSMA+ CAF cell frequency, cell proliferation, or cell death at any dose, relative to the control (Supplementary Fig. S6). These results validated that CRO-67 was the active enantiomer of the pair.

CRO-67 treatment in PDAC tumor explants from patients reduced tumor and CAF cell frequency, reduced cell proliferation, and increased cell death. PDAC tumor explants from N = 5 patients were cultured for 12 days and treated with 0, 10, 20 and 50 μg/mL of CRO-67 every 72 h. (a) Representative immunohistochemistry (IHC) images for Patient 5 explants stained for tumor cells (cytokeratin), CAFs (α-smooth muscle actin [αSMA]), proliferating cells (BrdU), and cell death (TUNEL). Positive staining for IHC is brown, positive TUNEL staining is green, and nuclei are blue. Quantification of IHC stain for (b) tumor cells, (c) CAFs, (d) proliferating cells, and (e) cell death was performed using QuPath software. Inset images for BrdU staining show nuclei positive for BrdU in brown compared to BrdU negative nuclear-only staining in blue. Symbols represent the mean of 2–4 explants per treatment from each patient. Colors indicate percentage relative to control for each patient. Bars represent mean ± SEM (N = 5). Asterisks indicate statistical significance as assessed by one-way ANOVA, and Bonferroni’s multiple comparisons tests (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001).
Potent activity of CRO-67 through anti-proliferative effects and activation of apoptosis in patient-derived CAFs and PDAC cell lines
The effects of CRO-67 on PDAC cell and CAF proliferation, cell death and cell cycle progression were further evaluated in vitro. The results demonstrated that CRO-67 almost completely halted the proliferation of MiaPaCa-2, PANC-1 and CAFs, relative to controls (Fig. 5a–c). CRO-67 treatment also significantly increased apoptosis in MiaPaCa-2, PANC-1 and CAFs compared to controls (Fig. 5d–f). Furthermore, we observed that CRO-67 significantly increased the accumulation of cells in G2/M cell cycle phase from as early as 4 h post-treatment in MiaPaCa-2 and PANC-1, and more prominently at 24 h post-treatment in MiaPaCa-2, PANC-1 and CAFs (Fig. 5g–i, Supplementary Fig. S7).

CRO-67 activated apoptosis and reduced proliferation in two PDAC cell lines and patient-derived CAFs. PDAC cell lines (MiaPaCa-2 and PANC-1) and primary patient-derived CAFs isolated from N = 5 PDAC patients were treated with vehicle control or CRO-67 (1.5 μM) and analyzed 24 h post-treatment. (a–c) Phase-contrast confluency measurements were taken every hour for 48 h following CRO-67 treatment of PDAC cell lines and CAFs. MiaPaCa-2 (N = 3), PANC-1 (N = 3) PDAC cells and CAFs (N = 5 PDAC patients) demonstrated a decrease in cell proliferation, as measured by the IncuCyte S3 Live Cell Analysis system. (d–f) Apoptosis was measured by flow cytometry for Annexin V/DAPI. MiaPaCa-2 (N = 3), PANC-1 (N = 3) PDAC cells and CAFs (N = 5) demonstrated an increase in apoptosis 24 h post-treatment with CRO-67 (1.5 μM). (g–i) Flow cytometry was used to quantify the percentages of cells in the G1, S, and G2/M phases of the cell cycle at 4 and 24 h post-treatment with CRO-67 (1.5 μM). CRO-67 treatment in MiaPaCa-2 (N = 3), PANC-1 (N = 3) and CAF (N = 3) cells resulted in the accumulation of cells in the G2/M phase at 4 and 24 h post-treatment, suggesting mitotic arrest. Bars represent mean ± SD (N = 3). Asterisks indicate significance as assessed by one-way ANOVA, and Bonferroni’s multiple comparisons tests; performed on endpoint data at 48 h timepoint for cell proliferation (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001).
CRO-67 treatment significantly decreased αSMA + CAF frequency and increased the fraction of opened blood vessels and low-density collagen fibrils in mouse tumors
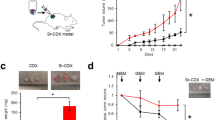
To further evaluate the efficacy of CRO-67, we assessed its therapeutic potential in an orthotopic PDAC mouse model (tumor cells + CAF co-injection; Fig. 6a). 4 weeks post-surgery, the mice were randomized into treatment groups based on ultrasound measurements of tumor size (Fig. 6b), then treatments performed as shown in Fig. 6a. The treatment was well tolerated and had no significant effect on animal weight (Supplementary Fig. S8a). Morphological changes observed in the liver in response to treatment may be indicative of slight toxicity relative to controls, but overall no adverse effects were observed in mice and all other organs presented normal histology (Supplementary Fig. S8b). Overall, tumor growth and metastatic spread was not significantly affected by CRO-67 treatment (Fig. 6c, Supplementary Fig. S8c), but we did observe that in a sub-cohort analysis restricted to tumors with > 150% growth at endpoint [active tumor cell proliferation (Supplementary Fig. S8d–f)], CRO-67 treatment did significantly reduce both tumor growth and metastatic spread (Fig. 6d), consistent with its proposed function as a cell cycle disruptor. The sub-cohort analysis was performed based on the predicted mechanism of action of CRO-67 (anti-mitotic effects in vitro) where the drug’s potency could be dependent on cell proliferation. Accordingly, this sub-cohort analysis excludes five PDAC tumors that had slower (< 150% from start) growth at endpoint (Supplementary Fig. S9). Note, these tumors were still included in the full cohort analysis as they sat within the average tumor size for their respective groups at 4 weeks post-surgery, and some demonstrated metastatic spread at endpoint, both of which imply viable tumors at the start of treatment (Fig. 6b–d, Supplementary Fig. S9). Importantly, CRO-67 treatment significantly decreased intratumoral αSMA+ CAFs and increased intratumoral open blood vessels in the full cohort (i.e. regardless of effects on tumor growth; Fig. 6e-f and Supplementary Fig. S8g). These effects were likely a consequence of loosening of CAF-deposited collagen (Fig. 6g–l; increased low birefringence collagen), even though overall tumor collagen content was unchanged (Fig. 6i). These findings suggest that CRO-67 could alleviate drug delivery barriers in PDAC tumors in vivo.

CRO-67 treatment significantly decreased αSMA+ CAF frequency, and increased the fraction of opened blood vessels and low-density collagen fibrils in PDAC orthotopic mouse tumors. (a) Schematic representation of orthotopic PDAC mouse model made with Biorender. 7 weeks old, female Balb/c nude mice were injected with luciferase-expressing MiaPaCa-2 cells and immortalized human primary CAFs at a 1:1 ratio of cells (106 each) in the pancreas. 4 weeks post-surgery, mice were randomized into the vehicle control or CRO-67 treatment group (10 mice/group), based on (b) mouse tumor volume via ultrasound measurements. Mice were treated with Vehicle Control (90% saline + 10% DMSO) or CRO-67 (60 mg/kg), twice daily via intraperitoneal injection (i.p.), for 5 weeks. At endpoint, mouse pancreatic tumors were harvested, fixed in 4% paraformaldehyde, for tissue processing and subsequent immunohistochemistry analyses. Ex vivo organ luminescence for metastasis detection was performed via bioluminescence imaging using IVIS SpectrumCT. Individual tumor volume was measured by ultrasound as a percentage of start and number of metastatic sites per mouse with the percentage of mice presenting any metastasis were quantified for (c) all tumors and for (d) tumors with > 150% growth at endpoint compared to start. (e) Representative images and quantification of α-smooth muscle actin (αSMA) positive cells. (f) Representative images and quantification of CD31 positive cells. (g) Representative bright field images showing picrosirius red stained tumor sections and (h) representative images of picrosirius red stained imaged using polarized light. Quantification of total picrosirius red coverage (i) and low–high birefrigent collagen (j–l) are shown in the graphs. Quantification of each stain was performed using MATLAB programming. Symbols show quantification of individual mice. Bars represent mean ± SEM. Asterisks indicate statistical significance as assessed by t-test (*p ≤ 0.05, **p ≤ 0.01).
Lower doses of CRO-67 treatment significantly decreased tumor and αSMA + CAF cell frequency and reduced cell proliferation in human PDAC explants
Given the dose of CRO-67 administered in vivo was 15-fold higher than the highest dose tested in explants, the reduced anti-tumor potency of CRO-67 in vivo suggested that the dose of CRO-67 reaching the tumor site may be less than what was tested in our initial explant screens. To investigate this further, we treated PDAC explants from an additional five patients (Patients 11–15) with CRO-67 at 1–2 μg/mL (5–tenfold lower concentrations than lowest CRO-67 doses in Fig. 4 explant experiments; Fig. 7, Supplementary Table S1, and Supplementary Fig. S10). We observed that at 1 µg/mL, CRO-67 reduced tumor and αSMA+ CAF cell frequency in patient explants, but on average these effects were closer to the effects observed in vivo than the near depletion of cells observed at higher CRO-67 concentrations (Fig. 7b,c versus Fig. 4b,c). Notably, the reductions in tumor cells at 1 µg/mL were mixed across patients (Fig. 7b; ≤ 10% reductions in 2/5 patient explants, 50% reduction in 1/5, and ≥ 75% in 2/5). Moreover, cell-death induction was no longer significant at lower concentrations of CRO-67 (Fig. 7e) compared to the significant increase in the initial explant screens (Fig. 4e). Our results suggested that we likely require an ideal combination therapy to take advantage of CRO-67-induced stromal remodeling in vivo, to enhance anti-tumor cell effects.

Low doses of CRO-67 treatment in PDAC tumor explants from patients reduced tumor and CAF cell frequency, and reduced cell proliferation. PDAC tumor explants from N = 5 patients were cultured for 12 days and treated with 0, 1, and 2 μg/mL of CRO-67 every 72 h. (a) Representative immunohistochemistry (IHC) images for Patient 12 explants stained for tumor cells (cytokeratin), CAFs (α-smooth muscle actin [αSMA]), proliferating cells (BrdU), and cell death (TUNEL). Positive staining for IHC is brown, positive TUNEL staining is red, and nuclei are blue. Please note that the red stain present in the control representative image was predominantly non-nuclear background staining, and was not included in quantification. Quantification of IHC stain for (b) tumor cell, (c) CAFs, (d) proliferating cells, and (e) cell death was performed using QuPath software. Inset images for BrdU staining show nuclei positive for BrdU in brown compared to BrdU negative nuclear-only staining in blue. TUNEL was not quantifiable in Patient 11 explants (Red symbols) due to dead areas not presenting nuclei. Patient 12 explants (light blue symbols) were not treated with 2 μg/mL CRO-67 due to limited availability of tumor tissue. Symbols represent the mean of 2–4 explants per treatment from each patient. Colors indicate percentage relative to control for each patient. Bars represent mean ± SEM (N = 5). Asterisks indicate statistical significance as assessed by one-way ANOVA, and Bonferroni’s multiple comparisons tests (**p ≤ 0.01, ***p ≤ 0.001).
CRO-67 sensitized PDAC cells to chemotherapy drugs in vitro by exerting enhanced anti-proliferative effects in combination treatments
We evaluated the anti-proliferative effect of CRO-67 in combination with PDAC standard-of-care chemotherapy drugs in vitro using MiaPaCa-2 and AsPC-1 PDAC cell lines. Concentrations of all drugs for evaluating CRO-67 sensitization with chemotherapeutics were selected based on a ≤ 50% drop in proliferation, allowing sufficient room to observe further anti-proliferative effects when used in combination. It is important to note that AsPC-1 is a PDAC metastatic line that is less sensitive to CRO-67 treatment compared to MiaPaCa-2. Hence, CRO-67 treatment was at a concentration of 1.2 μM for AsPC-1 compared to 0.5 μM for MiaPaCa-2 to achieve comparable anti-proliferative effects. At endpoint, CRO-67 treatments significantly reduced both MiaPaCa-2 and AsPC-1 cell proliferation relative to controls, confirming the anti-proliferative effect of CRO-67 as previously observed (Fig. 8a–h). The treatment of MiaPaCa-2 cells with CRO-67 significantly increased its sensitivity to gemcitabine, 5-fluorouracil, and oxaliplatin, as shown by the significant reduction in cell proliferation in combination treatments compared to individual drug treatments (Fig. 8a,c,e). In contrast, AsPC-1 cells treated with CRO-67 only exhibited increased sensitivity to oxaliplatin (Fig. 8f). The treatment of both MiaPaCa-2 and AsPC-1 cells with CRO-67 in combination with irinotecan had no effect on cell proliferation compared to individual drug treatment alone (Fig. 8g, h).

CRO-67 sensitized PDAC cell lines to gemcitabine and oxaliplatin, partial sensitization to 5-fluorouracil, and no sensitization with irinotecan in vitro. PDAC cell lines (MiaPaCa-2 and AsPC-1) were treated with vehicle DMSO control, CRO-67 (0.5 µM for MiaPaCa-2 and 1.2 µM for AsPC-1) alone or in combination with chemotherapy drugs: (a, b) gemcitabine (GEM; 0.02 µM for MiaPaCa-2 and 0.05 µM for AsPC-1), (c, d) 5-fluorouracil (5-FU; 5 µM), (e, f) oxaliplatin (OXAL; 2.5 µM), or (g, h) irinotecan (IRINO; 2.5 µM), and phase-contrast confluency measurements were taken every 4 h for 72 h with the IncuCyte S3 Live Cell Analysis System. CRO-67 treatment alone and chemotherapy drug treatment alone significantly decreased proliferation of both MiaPaCa-2 (N = 3) and AsPC-1 (N = 3) cells relative to controls. CRO-67 in combination with GEM, 5-FU, or OXAL demonstrated a further decrease in MiaPaCa-2 cell proliferation relative to single drug treatments. In AsPC-1 (N = 3) PDAC cells, only CRO-67 in combination with OXAL further decreased cell proliferation relative to single drug treatments. Bars represent mean ± SD (N = 3). Asterisks indicate significance as assessed by one-way ANOVA, and Bonferroni’s multiple comparisons tests, performed on endpoint data at 72 h timepoint (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001).
Discussion
The majority of PDAC patients have unresectable disease at diagnosis, resulting in reliance on standard of care chemotherapy for treatment, which offers patients little hope of long-term survival. Traditional chemotherapy approaches for PDAC have primarily focused on targeting PDAC cells exclusively, neglecting the pro-tumor influence of stromal CAFs12,30. CAFs play a major role in shaping the characteristic stromal barrier of the PDAC microenvironment, promoting tumor growth, and limiting drug penetrance, all which contribute to the aggressive nature of the disease and chemoresistance12,30. Herein, the novel compound CRO-05 and its enantiomers CRO-67 and CRO-68, were developed in partnership with Noxopharm Limited as a potential dual-cell therapy for PDAC. Our unique compounds were rationally designed from a novel class of anti-cancer drug called chromans, details of which are outlined in the composition of matter patent (Patent #PCT/AU2023/050505). We demonstrated in clinically relevant patient-derived pancreatic cancer models, that CRO-05 and its active enantiomer CRO-67, are potent drugs that target both PDAC cells and αSMA+ CAFs. However, racemic mixes such as CRO-05 are less desirable in medicinal chemistry due to potential side effects or toxicity associated with inactive enantiomers29. As such, for the preclinical validation experiments detailed in this study, CRO-67 was directly synthesized from a precursor of CRO-05 via hydrogenation in the presence of a chiral catalyst (iridium, rhodium or ruthenium based)31, which produces a high yield of the compound while retaining purity and efficacy.
We assessed the effectiveness of CRO-67 as a dual-cell therapy drug in a clinically relevant human PDAC explant model. This model developed by our laboratory accurately maintains the three-dimensional architecture, fibrosis, and heterogeneity of human PDAC explants (obtained from surgical resections) for 12 days, which can test the therapeutic potential of novel agents in targeting both stromal-CAF cells and PDAC tumor cells12,25. Our studies showed that CRO-67 significantly reduced both tumor cell and αSMA+ CAF cell frequencies in patient explants, and within the remaining cells, significantly reduced cell proliferation and induced cell death. Previous studies have shown the anti-cancer cell activity of related chromans Cantrixil and Trilexium against multiple cancer types in vitro and in 3D tumor culture27,28. However, PDAC cell lines exhibited a wide range of response to Cantrixil (IC50 values between 0.2 and 3.7 µM) compared to 16 other cancer cell types in vitro, with ovarian, prostate, and lung cancers being more responsive (IC50 values ≤ 0.1 µM)27. In a different study that performed an in vitro screen of 240 cell lines, PDAC cells were considered borderline insensitive to Trilexium compared to other cancer cell lines, suggesting that Trilexium is not suitable for clinical translation for PDAC28. Importantly, these studies did not examine the direct effect that Cantrixil or Trilexium may have on CAFs, nor did they consider how CAFs and PDAC cells would respond to these drugs together. In contrast to these studies, we considered the complexity of the tumor microenvironment in our evaluation of CRO-67’s therapeutic potential by assessing its impact on both cell types in intact PDAC tumor explants.
Regarding observed reductions in CAF frequency within explants, it should be noted that detection thresholds for αSMA in our tissue sections were set to detect any level of αSMA expression (i.e. αSMA high and low CAF subsets). αSMA is a robust CAF marker due to its frequent expression in most CAFs and its association with pancreatic cancer pathology8,32, but its expression can vary between major CAF subtypes: myofibroblast CAFs [myCAFs; αSMAhigh interleukin-6 (IL-6)low] and inflammatory CAFs (iCAFs; αSMAlow IL-6high)16,17. In addition, these CAF subtypes are plastic and can transition between these states15,16,17. Many studies have attempted to target specific CAF subtypes, but this approach proves challenging and ineffective due to CAF heterogeneity, with different subtypes playing conflicting roles in tumor growth17,33,34,35,36. Importantly, previous studies have suggested against the targeted depletion of αSMA+ CAFs, as it had resulted in a more aggressive phenotype in mouse models and spheroids13,33,35,36. In contrast, our in vivo findings showed that CRO-67 treatment reduced, but did not deplete, αSMA+ CAFs in PDAC tumors. The stromal remodeling effects of CRO-67 were also linked to anti-tumor and anti-metastatic effects when restricting analyses to rapidly growing PDAC tumors (> 150% growth at endpoint). This suggested that the reduction of αSMA+ CAFs did not worsen disease progression, and instead, CRO-67’s more consistent stromal remodeling effect (loosening of tumor fibrosis, normalization of tumor vasculature) indicate its potential to improve drug penetrance to the tumor in combination therapy settings. Future studies to delineate the effects of CRO-67 on specific CAF subsets, using spatial transcriptomic approaches, would be vital to understanding how it reprograms the stroma, thereby influence therapeutic response.
Findings from our in vitro studies complemented our observations in patient explants. CRO-67 significantly reduced the viability and proliferation of both PDAC cells and CAFs, and increased apoptosis in both cell types in vitro. A potential mechanism driving these effects was cell cycle arrest in G2/M phase of the cell cycle, which we observed in both PDAC cells and CAFs in response to CRO-67 treatment in vitro. Prolonged arrest of PDAC cells at the G2/M phase checkpoint results in mitotic failure and induced apoptosis37,38,39. The medicinal chemistry relationship between CRO-67 and chromans, stemming from their shared benzopyran structure, suggests that CRO-67 may exhibit a similar mechanism of action as other chromans in inducing cell cycle arrest and apoptosis. For instance, the most clinically advanced chroman, Cantrixil, exerts anti-tumor effects by inhibiting tubulin polymerization via interaction with the colchicine binding site on tubulin, and activating c-Jun to induce caspase-mediated apoptosis28,40. Similarly, the cytotoxic activity of a related chroman, Trilexium, is attributed to its ability to inhibit tubulin polymerization in different cancer types including PDAC28. CRO-67’s mechanism of action appears to be similar to both chromans, as they have been shown to trigger mitotic arrest in cancer cells to induce apoptosis28. Further research is needed to elucidate the mechanism of action of CRO-67 in comparison to chromans. In addition, CRO-67’s potential cell cycle disrupting mechanism of action could be dependent on PDAC cell proliferation. This is evident in the orthotopic PDAC tumors in mice, where CRO-67 showed a significant decrease in tumor growth when restricting the analysis to tumors with > 150% growth at endpoint (i.e. tumors with actively proliferating cancer cells). Moreover, MiaPaCa-2 cells were more sensitive to CRO-67 single drug treatment compared to the relatively slower proliferating AsPC-1 cells. Our findings suggest that cancer cell proliferation status may influence their responsiveness to CRO-67.
We observed that anti-tumor and CAF reductions observed in vivo were substantially less than those observed in our initial evaluation in explants, despite using a dose of drug that was 15-fold higher in vivo than the highest dose used in explant cultures. Differences in tumor microenvironment and cancer cell biology between orthotopic PDAC tumors and patient-derived explants, especially given the heterogeneity of PDAC, could potentially explain the difference in sensitivity. Alternatively, this may have been a consequence of lower doses of drug reaching tumor cells in the in vivo setting due to the potential for drug absorption/metabolism by other organs. In support of the latter, lower CRO-67 dosing in explants (tenfold lower than the lowest concentration in our first screen) produced, on average, reductions in tumor cells and CAFs closer to those observed in vivo. Future studies will assess the pharmacokinetics of CRO-67 using in vivo PDAC models to determine if our observations were related to drug bioavailability.
Nevertheless, CRO-67 still alleviated physical barriers to tumor drug delivery across the whole cohort by reducing αSMA+ CAF frequency, loosening stromal fibrosis and normalizing tumor vasculature. Our in vivo results suggest that CRO-67 has the potential to enhance drug penetrance into PDAC tumors in a combination therapy setting through its stromal remodeling capability, highlighting the need to evaluate CRO-67 in combination with other chemotherapeutics. The most effective treatments for PDAC, albeit with limited survival benefit, are combination chemotherapy regimens41,42, reinforcing the need to select an ideal partner treatment with CRO-67 to maximize clinical translatability. We explored the potential of combining CRO-67 with standard-of-care chemotherapies to enhance its therapeutic effect. We demonstrated that CRO-67 treatment in combination with gemcitabine, 5-fluorouracil, or oxaliplatin significantly impaired cell proliferation of different PDAC cells (MiaPaCa-2 and/or AsPC-1) to a greater extent than single drug treatments alone in vitro. Based on anti-proliferative effects that were at least additive or greater, the most effective combination in either cell line was Gemcitabine or 5-fluorouracil combined with CRO-67 (MiaPaCa-2 only), and the most effective combination across both cell lines was Oxaliplatin combined with CRO-67. Future studies will explore these combinations in explants and in vivo.
CRO-67 is a novel dual-cell therapy with promising potential for further development as an effective therapeutic for pancreatic cancer. We demonstrated its unique ability to target both PDAC tumor cells and stromal αSMA+ CAFs in a clinically relevant PDAC explant model, revealing potent anti-cancer, anti-CAFs, and anti-proliferative effects, as well as activation of apoptosis. CRO-67 is now covered by the U.S. Orphan Drug Act (1983) and is recognized as a novel anti-cancer drug. The novel structure of CRO-67 and its counterparts in this study developed in collaboration with Noxopharm are detailed in the patent. To advance preclinical research on CRO-67, further studies with complementary preclinical models are needed to elucidate its mechanism of action and evaluate its efficacy both as a standalone treatment and in combination with chemotherapy or immunotherapy drugs. This will bolster our understanding of CRO-67’s unique dual-cell targeting effect and support its potential therapeutic use for pancreatic cancer.
Methods
Chemical synthesis
Racemic CRO-05 1 was synthesized on multi-gram scale at Jubilant Biosys (Greater Noida, India) using a previously reported method (Patent #PCT/AU2023/050505) summarized in Fig. 1. In short, resorcinol 2 was acylated with 4-nitrobenzoyl chloride to obtain aryl ketone 3, which subsequently underwent cyclic condensation with 3,4-dimethylphenylacetic acid under heating in acetic anhydride in the presence of diisopropylethylamine (DIPEA) to form O-acylated coumarin derivative 4. This was deprotected and reduced to the substituted 2H-chromene 5 in a single step using borane dimethylsulfide. Hydrogenation of this intermediate with the use of catalytic palladium (0) on carbon produced CRO-05 1 as a racemic mixture of cis-enantiomers. This mixture was subsequently separated at ChemPartner Co., Ltd (Shanghai, China) via Supercritical Fluid Chromatography (SFC) to obtain enantiopure (+)-CRO-67 6 and (-)-CRO-68 7. Enantiomeric excess (ee) was 99.52% for CRO-67 and ee 100% for CRO-68. (Please refer to the Supplementary Methods and Supplementary Figures SM1-10 [within supplementary methods] for details on the synthesis of CRO-05, CRO-67, and CRO-68.)
Human PDAC 3D explant culture
Specimens of PDAC tumors were obtained fresh from patients undergoing pancreaticoduodenectomy (Whipple procedure) at Prince of Wales Hospital or Prince of Wales Private Hospital, Randwick, NSW, Australia. Written informed consent was given by these patients through the Health Science Alliance Biobank. The work was approved by UNSW human ethics committee (HC180973) and experiments were performed in accordance with regulations and guidelines. As previously described25, de-identified tumor samples were cut using a scalpel into explants of approximately 2 mm diameter. Explants were cultured on hemostatic gelatine sponges (Johnson & Johnson, Cat. JJ-12505) for 12 days with daily medium changes. Culture medium contains high-glucose Dulbecco’s Modified Eagle Medium (DMEM; ThermoFisher, Cat. 10313021), 10% fetal bovine serum (FBS; CellSera, Cat. AU-FBS/PG), 5 mM GlutaMAX (ThermoFisher, Cat. 35050061), 0.01 mg/mL hydrocortisone (Sigma-Aldrich, Cat. H0888-1G), 0.01 mg/mL insulin (NovoRapid, Cat. AUST R 133443), and 1 × antibiotic/antimycotic solution (Sigma-Aldrich, Cat. A5955-20ML)25. Between 2 and 4 explants from different regions of the patient’s tumor were cultured per treatment group. The explants were treated with vehicle (0.4% DMSO to match the amount used at the highest drug concentration; Sigma-Aldrich, Cat. D2650-100ML), or CRO-05 (10, 20 and 50 μg/mL), or CRO-67 (1, 2, 10, 20 and 50 μg/mL) or CRO-68 (10, 20 and 50 μg/mL) on days 0, 3, 6 and 9 by addition of the drug into the medium reservoir. A range of concentrations was used to assess drug potency in patient explants, which were based on well-tolerated in vivo doses from preliminary studies conducted by Noxopharm. Note that drug concentrations in the explant studies were expressed in µg/mL, as this unit is relevant to plasma concentration for drugs. On day 11, explants were treated with 10 mM BrdU substrate (BD Biosciences, Cat. 550891) and then fixed on day 12 in 4% paraformaldehyde (ProSciTech, Cat. EMS15712). All experiments were performed and are reported in accordance with ARRIVE guidelines.
Orthotopic PDAC mouse model
Female BALB/c nude mice (Ozgene ARC) were used for the establishment of the orthotopic PDAC mouse model. All animal procedures were approved by the UNSW Animal Ethics Committee (ACEC 22/3A). All experiments were performed in accordance with the ethics guidelines and regulations. At 7-week-old, orthotopic PDAC tumors were established by co-injection of luciferase-expressing MiaPaCa-2 cells and immortalized human primary CAFs at a 1:1 ratio (106 cells each) to the pancreas, as previously described12,43. 4 weeks post-surgery, mice were randomized into the vehicle control or CRO-67 treatment group (10 mice/group), based on mouse tumor size via ultrasound measurements. Mice then received intraperitoneal injection (i.p.) of Vehicle Control (90% saline + 10% DMSO) or CRO-67 (60 mg/kg), twice daily, for 5 weeks. Tumor volumes were measured once a week by ultrasound to track tumor growth, and mice were weighed once weekly. At endpoint, mouse pancreatic tumors, spleen, liver, kidneys, intestines, heart and lungs, as well as any other organs or enlarged lymph nodes indicative of metastatic spread, were harvested, fixed in 4% paraformaldehyde, then sent for tissue processing and subsequent immunohistochemistry analyses. Ex vivo organ luminescence for metastasis detection was performed via bioluminescence imaging using IVIS SpectrumCT and confirmed by H&E (Supplementary Fig. S8c). All experiments were performed and are reported in accordance with ARRIVE guidelines.™
Immunohistochemistry staining and analysis
Paraformaldehyde-fixed explants and orthotopic mouse tumors were paraffin-embedded and sectioned (5 µm thickness) as previously described25. Hematoxylin and Eosin (H&E) staining was performed at the Histopathology Service of the Garvan Institute of Medical Research (Sydney, Australia) for the visualization of explant architecture (Supplementary Fig. S11). Explant sections were stained using immunohistochemistry as previously described25. For patient explants, immunohistochemistry for cytokeratin (DAKO, Cat. M3515; 1:100 overnight at 4 °C), αSMA (Sigma-Aldrich, Cat. A5228; 1:1000 1 h at room temperature) and bromodeoxyuridine (BrdU) (DAKO, Cat. M0744; 1:50 dilution overnight at 4 °C), was performed to characterize tumor cells, αSMA+ CAFs and cell proliferation, respectively. For mouse tumors, immunohistochemistry for αSMA, and CD31 (Taylor Bio-Medical, Cat. DIA-310) was performed to characterize αSMA+ CAFs, and CD31+ endothelial cells as a marker for blood vessels (manual count for open and closed vessels), respectively. The quantification of CD31+ blood vessels was performed on 20 × magnified fields of view (> 7/section) of mouse tumors, as described previously12,44. There was no significant difference in the total number of CD31+ blood vessels between treatment groups (Supplementary Fig. S8g), hence the number of open vessels was expressed as a percentage of total blood vessels. In all immunohistochemistry staining, hematoxylin (Agilent, Cat. S3309) was used as a counter stain, and 3,3′-diaminobenzidine tetrahydrochloride (DAB) substrate (Agilent, Cat. K346811-2) and VECTASTAIN ABC-Peroxidase Kit (VectorLabs, Cat. PK-4000) was used to visualize staining. All slides were scanned using an Olympus VS200 slide scanner. For quantification of staining in whole tissue explants and mouse tumors, excluding background staining in necrotic regions, the positive cell detection function was used on QuPath software (v0.5.1; open-source software for digital pathology image analysis). Cytokeratin and αSMA positive staining were expressed in number of positive cells per mm2, while BrdU and was expressed as a percentage of total cells. Figures summarizing the statistical analyses of tumor cells (cytokeratin), αSMA+ CAFs (αSMA), cell proliferation (BrdU), and cell death (TUNEL) in patient explants treated with CRO-05 or CRO-67 were expressed as an average of all patient explants for each treatment as a percentage of control. In our study, a total of sixteen PDAC patient explants were treated with CRO-05 or CRO-67 (Supplementary Table S1). However, 3/16 patients (Patients 9, 10, and 16) had partial response to neoadjuvant chemotherapy before explant treatments. These three patient explants were excluded from all final analyses to eliminate patient treatment history from confounding our assessment of CRO-67’s efficacy.
TUNEL staining to assess cell death
Fluorescent TUNEL staining (Sigma-Aldrich, Cat. 11684809910) or Fast Red (Abcam, Cat. ab64254) was performed according to the manufacturer’s instructions and as previously described25. Following incubation of tissue sections with TUNEL enzyme (diluted 1:2), samples were mounted with ProLong Gold antifade mounting medium with DAPI (Invitrogen, Cat. P36931) and whole explants were scanned on a Vectra Polaris (PerkinElmer) slide scanner. QuPath was used to calculate fluorescent TUNEL positive nuclei as a percentage of DAPI positive cells in whole tissue explants. As for immunohistochemistry staining for Fast Red substrate, positive cell detection function was used on QuPath.
Picrosirius red staining to assess total collagen content and fibril density (polarized light analysis)
Paraffin-embedded tumor sections were stained with 0.1% picrosirius red (Polysciences) according to manufacturer’s instructions. Polarized light imaging and intensity measurement were performed and analyzed using MATLAB programming, as described previously12,45,46. Collagen content was expressed as an average percent of total birefringence that was high (red), medium (yellow), and low (green).
In vitro PDAC and CAF cell culture
PDAC cell lines, MiaPaCa-2 (RRID: CVCL_0428), PANC-1 (RRID: CVCL_0480), and AsPC-1 (RRID: CVCL_0152) were obtained from the American Type Culture Collection (ATCC). The purity of cell lines was confirmed by short tandem-repeat profiling (CellBank Australia). MiaPaCa-2 cells were maintained in DMEM culture media, supplemented with 10% FBS, 2.5% horse serum (ThermoFisher, Cat. 16050122) and 2 mM GlutaMAX. PANC-1 cells were cultured in DMEM culture media, supplemented with 10% FBS and 2 mM GlutaMAX. AsPC-1 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 Medium (ThermoFisher, Cat. 11875093), supplemented with 10% FBS and 2 mM GlutaMAX. Patient-derived CAFs were isolated from surgical PDAC samples using the outgrowth method, as described12, and were used within 12 passages. CAFs were grown in Iscove’s Modified Dulbecco Medium (IMDM; ThermoFisher, Cat. 21056023), 10% FBS, and 4 mM GlutaMAX. Positive staining for glial fibrillary acidic protein (GFAP) and α-smooth muscle actin (αSMA) and negative staining for cytokeratin were used to assess the purity of CAFs, as described previously12. All cancer cell lines and CAFs were maintained at 37 °C in a humidified 5% CO2 incubator and monthly tested and found free from mycoplasma.
Cell viability assay
MiaPaCa-2 and PANC-1 cells were seeded in 6-well plates at 100,000 cells per well and then treated with 1.5 μM of vehicle DMSO control, CRO-05, CRO-67 or CRO-68, 24 h after cell seeding. Note that drug concentrations in all in vitro studies were expressed in µM, as this unit is relevant to the low dose range used in these assays. Cells were trypsinized with 0.25% Trypsin–EDTA (ThermoFisher, Cat. 25200072) 24 h post-treatment and resuspended 1:1 in culture medium and trypan blue staining solution (ThermoFisher, Cat. 15250061). Cell viability was measured by counting live cells (trypan blue negative) using a BioRAD automated cell counter47.
Apoptosis assay
Cells were treated with 1.5 μM of vehicle DMSO control, CRO-05, CRO-67 or CRO-68, 24 h after cell seeding in 6-well plates. 24 h post-treatment, adherent and floating cells were pelleted and resuspended in 1 × binding buffer containing Annexin V-PE (BD Biosciences, Cat. 559763) and 100 μg/mL DAPI (Sigma-Aldrich, Cat. D9542), incubated for 15 min in the dark at room temperature, according to the manufacturer’s instructions, then analyzed on a BD LSR Fortessa flow cytometer. Total apoptosis was quantified using FlowJo v10 software as the sum of cells in early, middle, and late apoptosis. Gating strategy is shown in Supplementary Fig. S12a.
Cell proliferation assays
To measure real-time cell proliferation for PDAC cell lines (MiaPaCa-2 and PANC-1) and patient-derived CAFs, cells were treated with vehicle DMSO control, CRO-05, CRO-67 or CRO-68 at a concentration of 1.5 μM (CRO-05 IC50 for MiaPaCa-2), 24 h after seeding. Immediately following drug treatment in vitro, cells were placed in an IncuCyte S3 Live-Cell Analysis System (Essen BioScience), and each well was scanned every hour for a total of 48 h. For evaluating the anti-proliferative effect of CRO-67 in combination with PDAC standard-of-care chemotherapy drugs, PDAC cell lines (MiaPaCa-2 and AsPC-1) were treated with vehicle DMSO control, CRO-67 (0.5 µM for MiaPaCa-2 and 1.2 µM for AsPC-1), chemotherapy drugs [gemcitabine (GEM, Sigma Aldrich, Cat. G6423-10MG; 0.02 µM for MiaPaCa-2 and 0.05 µM for AsPC-1), 5-fluorouracil (5-FU, Accord Healthcare Pty Ltd, Cat. 285,803; 5 μM), oxaliplatin (OXAL; 2.5 μM), or irinotecan (IRINO; 2.5 μM)], or CRO-67 in combination with each of the chemotherapy drugs, 24 h after seeding. Cells were immediately placed in the IncuCyte S3 Live-Cell Analysis System following drug treatments, and each well was scanned every 4 h up to 72 h. IncuCyte S3 software (v2018B) was used to calculate cell proliferation by using phase-contrast images and integrated cell confluence metrics.
Cell cycle analysis
4 h and 24 h after CRO-67 treatment, cells were lifted and fixed in 80% ice-cold ethanol (Sigma-Aldrich, Cat. 1.11727) for 30 min. After fixation, cells were washed twice with PBS (ThermoFisher, Cat. 14,190,144), then stained with 1 μg/mL DAPI in PBS with 1% TWEEN-20 (Sigma-Aldrich, Cat. P1379-250ML) for 15 min at 37 °C. Cells were analyzed using a violet laser (V450) on BD LSR Fortessa cytometer47, and the proportion of cells in the G1, S, and G2/M phases was quantified using FlowJo v10 software. Gating strategy is shown in Supplementary Fig. S12b.
Statistical analyses
Statistical analyses were performed using GraphPad Prism software (v10.4.1). Normality of data was assessed by Shapiro–Wilk test, confirming normal distribution for all data. To calculate statistical significance (p < 0.05), two-tailed student t-tests (2 groups) and one-way ANOVA (≥ 3 groups) followed by Bonferroni’s multiple comparison tests were performed.
Data availability
The data generated in this study are available within the article and its supplementary data files. Raw images and data related to patient explants are available upon reasonable request from corresponding authors.
References
Siegel, R. L., Giaquinto, A. N. & Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 74, 12–49. https://doi.org/10.3322/caac.21820 (2024).
Kokkinos, J. et al. Does the microenvironment hold the hidden key for functional precision medicine in pancreatic cancer?. Cancers (Basel) https://doi.org/10.3390/cancers13102427 (2021).
Zeng, S. et al. Chemoresistance in pancreatic cancer. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20184504 (2019).
Kokkinos, J. et al. Targeting the undruggable in pancreatic cancer using nano-based gene silencing drugs. Biomaterials 240, 119742. https://doi.org/10.1016/j.biomaterials.2019.119742 (2020).
Raghavan, S. et al. Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell 184, 6119-6137.e6126. https://doi.org/10.1016/j.cell.2021.11.017 (2021).
Huber, M. et al. The immune microenvironment in pancreatic cancer. Int. J. Mol. Sci. https://doi.org/10.3390/ijms21197307 (2020).
Phillips, P. Pancreatic Cancer and Tumour Microenvironment (ed Trivandrum) Ch. 3, (Transworld Research Network, 2012).
Apte, M. V. et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 29, 179–187 (2004).
McCarroll, J. A. et al. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front. Physiol. 5, 141. https://doi.org/10.3389/fphys.2014.00141 (2014).
Murakami, T. et al. Role of the tumor microenvironment in pancreatic cancer. Ann. Gastroenterol. Surg. 3, 130–137. https://doi.org/10.1002/ags3.12225 (2019).
Pereira, B. A. et al. CAF subpopulations: A new reservoir of stromal targets in pancreatic cancer. Trends Cancer 5, 724–741. https://doi.org/10.1016/j.trecan.2019.09.010 (2019).
Sharbeen, G. et al. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma determine response to SLC7A11 inhibition. Cancer Res. 81, 3461–3479. https://doi.org/10.1158/0008-5472.Can-20-2496 (2021).
Vaish, U., Jain, T., Are, A. C. & Dudeja, V. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma: An update on heterogeneity and therapeutic targeting. Int. J. Mol. Sci. https://doi.org/10.3390/ijms222413408 (2021).
Menezes, S., Okail, M. H., Jalil, S. M. A., Kocher, H. M. & Cameron, A. J. M. Cancer-associated fibroblasts in pancreatic cancer: New subtypes, new markers, new targets. J. Pathol. 257, 526–544. https://doi.org/10.1002/path.5926 (2022).
Rebelo, R., Xavier, C. P. R., Giovannetti, E. & Vasconcelos, M. H. Fibroblasts in pancreatic cancer: Molecular and clinical perspectives. Trends Mol. Med. 29, 439–453. https://doi.org/10.1016/j.molmed.2023.03.002 (2023).
Öhlund, D. et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 214, 579–596. https://doi.org/10.1084/jem.20162024 (2017).
Biffi, G. et al. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 9, 282–301. https://doi.org/10.1158/2159-8290.Cd-18-0710 (2019).
Elyada, E. et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 9, 1102–1123. https://doi.org/10.1158/2159-8290.Cd-19-0094 (2019).
Geng, X. et al. Cancer-Associated Fibroblast (CAF) heterogeneity and targeting therapy of CAFs in pancreatic cancer. Front. Cell Dev. Biol. 9, 655152. https://doi.org/10.3389/fcell.2021.655152 (2021).
Gopinathan, A., Morton, J. P., Jodrell, D. I. & Sansom, O. J. GEMMs as preclinical models for testing pancreatic cancer therapies. Dis. Model Mech. 8, 1185–1200. https://doi.org/10.1242/dmm.021055 (2015).
Bengtsson, A. et al. Organoid technology for personalized pancreatic cancer therapy. Cell Oncol. (Dordr.) 44, 251–260. https://doi.org/10.1007/s13402-021-00585-1 (2021).
Boj, S. F. et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 160, 324–338. https://doi.org/10.1016/j.cell.2014.12.021 (2015).
Driehuis, E. et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. U. S. A. 116, 26580–26590. https://doi.org/10.1073/pnas.1911273116 (2019).
Frappart, P. O. & Hofmann, T. G. Pancreatic Ductal Adenocarcinoma (PDAC) organoids: The shining light at the end of the tunnel for drug response prediction and personalized medicine. Cancers (Basel) https://doi.org/10.3390/cancers12102750 (2020).
Kokkinos, J. et al. Ex vivo culture of intact human patient derived pancreatic tumour tissue. Sci. Rep. 11, 1944. https://doi.org/10.1038/s41598-021-81299-0 (2021).
Decker-Farrell, A. R., Ma, A., Li, F., Muir, A. & Olive, K. P. Generation and ex vivo culture of murine and human pancreatic ductal adenocarcinoma tissue slice explants. STAR Protoc. 4, 102711. https://doi.org/10.1016/j.xpro.2023.102711 (2023).
Saif, M. W., Heaton, A., Lilischkis, K., Garner, J. & Brown, D. M. Pharmacology and toxicology of the novel investigational agent Cantrixil (TRX-E-002-1). Cancer Chemother. Pharmacol. 79, 303–314. https://doi.org/10.1007/s00280-016-3224-2 (2017).
Stevenson, A. J. et al. Mechanism of action of the third generation benzopyrans and evaluation of their broad anti-cancer activity in vitro and in vivo. Sci. Rep. 8, 5144. https://doi.org/10.1038/s41598-018-22882-w (2018).
Ceramella, J. et al. A look at the importance of chirality in drug activity: Some significative examples. Appl. Sci. 12, 10909 (2022).
von Ahrens, D., Bhagat, T. D., Nagrath, D., Maitra, A. & Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 10, 76. https://doi.org/10.1186/s13045-017-0448-5 (2017).
Behera, P., Ramakrishna, D. S., Chandrasekhar, M. M. & Kothakapu, S. R. A concise review on recent advances in catalytic asymmetric hydrogenation. Chirality 35, 477–497. https://doi.org/10.1002/chir.23559 (2023).
Nurmik, M., Ullmann, P., Rodriguez, F., Haan, S. & Letellier, E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer 146, 895–905. https://doi.org/10.1002/ijc.32193 (2020).
Özdemir, B. C. et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734. https://doi.org/10.1016/j.ccr.2014.04.005 (2014).
Rhim, A. D. et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25, 735–747. https://doi.org/10.1016/j.ccr.2014.04.021 (2014).
Steele, N. G. et al. Inhibition of Hedgehog signaling alters fibroblast composition in pancreatic cancer. Clin. Cancer Res. 27, 2023–2037. https://doi.org/10.1158/1078-0432.Ccr-20-3715 (2021).
Murray, E. R. et al. Disruption of pancreatic stellate cell myofibroblast phenotype promotes pancreatic tumor invasion. Cell. Rep. 38, 110227. https://doi.org/10.1016/j.celrep.2021.110227 (2022).
Kim, M. S. et al. Cucurbitacin D induces G2/M phase arrest and apoptosis via the ROS/p38 pathway in Capan-1 pancreatic cancer cell line. Evid. Based Complement Alternat. Med. 2020, 6571674. https://doi.org/10.1155/2020/6571674 (2020).
Kumar, S. et al. Targeting pancreatic cancer by TAK-981: A SUMOylation inhibitor that activates the immune system and blocks cancer cell cycle progression in a preclinical model. Gut 71, 2266–2283. https://doi.org/10.1136/gutjnl-2021-324834 (2022).
Orth, J. D., Loewer, A., Lahav, G. & Mitchison, T. J. Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol. Biol. Cell 23, 567–576. https://doi.org/10.1091/mbc.E11-09-0781 (2012).
Alvero, A. B. et al. TRX-E-002-1 induces c-jun-dependent apoptosis in ovarian cancer stem cells and prevents recurrence in vivo. Mol. Cancer Ther. 15, 1279–1290. https://doi.org/10.1158/1535-7163.Mct-16-0005 (2016).
Di Costanzo, F., Di Costanzo, F., Antonuzzo, L., Mazza, E. & Giommoni, E. Optimizing first-line chemotherapy in metastatic pancreatic cancer: Efficacy of FOLFIRINOX versus nab-paclitaxel plus gemcitabine. Cancers (Basel) https://doi.org/10.3390/cancers15020416 (2023).
Kolbeinsson, H. M., Chandana, S., Wright, G. P. & Chung, M. Pancreatic cancer: A review of current treatment and novel therapies. J. Invest. Surg. 36, 2129884. https://doi.org/10.1080/08941939.2022.2129884 (2023).
Vonlaufen, A. et al. Pancreatic stellate cells: Partners in crime with pancreatic cancer cells. Cancer Res. 68, 2085–2093. https://doi.org/10.1158/0008-5472.Can-07-2477 (2008).
Ruscetti, M. et al. Senescence-induced vascular remodeling creates therapeutic vulnerabilities in pancreas cancer. Cell 181, 424-441.e421. https://doi.org/10.1016/j.cell.2020.03.008 (2020).
Vennin, C. et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat. Commun. 10, 3637. https://doi.org/10.1038/s41467-019-10968-6 (2019).
Conway, J. R. W. et al. Three-dimensional organotypic matrices from alternative collagen sources as pre-clinical models for cell biology. Sci. Rep. 7, 16887. https://doi.org/10.1038/s41598-017-17177-5 (2017).
Sharbeen, G. et al. MutY-Homolog (MYH) inhibition reduces pancreatic cancer cell growth and increases chemosensitivity. Oncotarget 8, 9216–9229. https://doi.org/10.18632/oncotarget.13985 (2017).
Acknowledgements
The authors would like to acknowledge the Katharina Gaus Light Microscopy Facility (KGLMF) at UNSW, Sydney, Australia, for their technical support on flow cytometry and biomedical imaging. The authors sincerely thank the patients who kindly consented to donate their tumor samples for research. They would like to thank Prof. Koroush Haghighi for acquiring biospecimens used for the 3D explants, and Dr. Carmel Quinn and Dr. Anusha Hettiaratchi of the HSA Biobank for their support in managing clinical samples and patient consent. The authors would also like to acknowledge their community consumer Mr. Gino Iori for his invaluable input on the project and grant applications. This research was made possible by major contract funding from Noxopharm Ltd and was also supported by: NHMRC Ideas Grant (P.A. Phillips, G. Sharbeen, APP2002707), Maridulu Budyari Gumal Sydney Partnership for Health, Education, Research and Enterprise [SPHERE] Cancer Clinical Academic Group Senior Research Fellowship (Funded by Cancer Institute NSW Translational Cancer Research Capacity Building Grant, 2021/CBG0003, G. Sharbeen), Cancer Institute NSW Translational Program Grant (P.A. Phillips, D Goldstein), Pankind Accelerator Grants (x2; P.A. Phillips, G. Sharbeen, D Goldstein, KS Haghighi).
Author information
Authors and Affiliations
Contributions
S. Chiang, K. Garcia Netto and J. Kokkinos: Conceptualization, formal analysis, investigation, validation, visualization, methodology, supervision, writing-original draft, project administration. K.S. Haghighi: Resources, funding acquisition, methodology. A.S. Raina: Formal analysis, investigation, validation, visualization, methodology, supervision, project administration. J. Youkhana: Investigation, visualization, methodology. O. Pitiyarachchi: Investigation. Q. Truong: Conceptualization, formal analysis, supervision, funding acquisition, investigation, methodology, writing–original draft, project administration. D. Wenholz: Conceptualization, resources, formal analysis, supervision, funding acquisition, investigation, methodology, writing–original draft, project administration. J. Wilkinson: Conceptualization, formal analysis, supervision, funding acquisition, investigation, methodology, writing–original draft, project administration. O. Laczka: Investigation, methodology. X. Li: Investigation, methodology. V.R. Aldilla: Resource, methodology. N. Kumar: Conceptualization, resources, formal analysis, funding acquisition, validation, investigation, methodology. D. Goldstein: Conceptualization, resources, formal analysis, funding acquisition, investigation, methodology. P.A. Phillips and G. Sharbeen: Conceptualization, formal analysis, supervision, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, project administration. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
As part of an industry partnership research contract with UNSW, Noxopharm provided the novel compounds to the Pancreatic Cancer Translational Research Group at UNSW led by P.A. Phillips. The therapeutic assessment of these compounds was conducted independently by the group, and the results were reported to Noxopharm as stipulated in the contract. In addition, Quach Truong, Daniel Wenholz, John Wilkinson, Olivier Laczka, and Xiang Li are paid employes of Noxopharm. All other authors have no competing interests to declare.
ARRIVE statement
The study is reported in accordance with ARRIVE guidelines.
Authors disclosures
N. Kumar (patents: US20080306140, US20040152761)) synthesized the series of novel chromans, CRO-05, CRO-67, and CRO-68, in collaboration with Noxopharm Limited, and report a composition of matter patent for WO2023235929. As part of an industry partnership research contract with UNSW, Noxopharm provided the novel compounds to the Pancreatic Cancer Translational Research Group at UNSW led by P.A. Phillips. The therapeutic assessment of these compounds was conducted independently by the group, and the results were reported to Noxopharm as stipulated in the contract. No disclosures were reported by the other authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chiang, S., Garcia Netto, K., Kokkinos, J. et al. CRO-67 has anti-cancer activity in pancreatic tumor cells and stromal cancer-associated fibroblasts. Sci Rep 15, 24488 (2025). https://doi.org/10.1038/s41598-025-09411-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-09411-2
