
Abstract
Anopheles mosquitoes serve as a significant vector for the transmission of malaria and arboviruses holding great medical importance, but their phylogenetic relationships have not yet been clearly elucidated. This study was the first to sequence and analyze the mitochondrial genomic features of An. hyrcanus and An. messeae, and to incorporate them into a phylogenetic analysis with 102 other species within the genus Anopheles. Both genomes contain 37 genes, including 22 tRNAs, 13 protein-coding genes and 2 rRNAs, as well as one control region. The genome characteristics, codon usage, and tRNA secondary structure of the two species align with those observed in mitochondrial genomes of other mosquito species. Phylogenetic analysis revealed that the relationship among six subgenera was Lophopodomyia + ((Kerteszia + Stethomyia) + ((Cellia + Anopheles) + Nyssorhynchus)), providing strong support for their monophyly. Nevertheless, the phylogenetic relationships within subgenus Cellia were inconsistent with previous findings. The findings of this study will enrich the biological information repository for Anopheles mosquitoes, lay the foundation for further research on systematic classification, and hold significant implications for guiding the research, prevention and control of mosquito-borne diseases.
Similar content being viewed by others


Introduction
Mosquitoes, belonging to the order Diptera and family Culicidae, comprise 3,698 valid species and subspecies, 41 genera and 187 subgenera globally1. They show remarkable global distribution characteristics, primarily in tropical and temperate zones, with a few Arctic regions also present2. Malaria is regarded as one of the “Big Three” infectious diseases and is transmitted by Anopheles mosquitoes, posing a significant threat to human health3. The World Malaria Report 2023 indicates that in 2022, approximately 249 million malaria cases and an estimated 608,000 deaths were documented across 85 malaria-endemic countries4. While no indigenous malaria cases have been reported in China since 20175, there remain a substantial number of imported malaria infections annually6alongside the pervasive presence of Anopheles mosquito vectors. This scenario presents a considerable risk for secondary local transmission of these imported cases. Consequently, ongoing surveillance of Anopheles mosquito vectors is crucial to prevent the re-emergence of malaria in China7.
The genus Anopheles can be subdivided into eight subgenera: Anopheles, Baimaia, Cellia, Christya, Kerteszia, Lophopodomyia, Nyssorhynchus, and Stethomyia8. While only approximately 70 Anopheles species worldwide transmit the five Plasmodium species responsible for causing malaria in humans, an increasing number of Anopheles mosquitoes have been implicated in the transmission of arboviruses, including the Anopheles-exclusive viruses O’nyong-nyong transmitted in Africa by An. funestus Giles and An. gambiae Giles, as well as Guaroa virus disseminated in the Neotropics by An. neivai Howard, Dyar and Knab1. Anopheles hyrcanus (Pallas, 1771), which occurs in China only in Xinjiang9has been confirmed as a vector of malaria in the eastern Mediterranean region10. Additionally, Anopheles messeae is one of the six main malaria vectors within the Anopheles maculipennis group in Eurasia, with the widest distribution and highest genetic diversity among these vectors11. In China, it is primarily found in Inner Mongolia, Jilin, Xinjiang and other places9.
The phylogenetic relationships of the genus Anopheles have long been a contentious issue among taxonomists. However, these relationships remain unresolved due to inconsistent conclusions resulting from different data sets and classification methods. Early taxonomic studies of mosquitoes were carried out in terms of egg morphology, larval and pupal chaetotaxy, adult morphology, zymotaxonomy and hybridization experiments12. Cladistics analysis of 163 morphological characteristics from 64 mosquito species supported the monophyletic subgenera Kerteszia, Cellia, Nyssorhynchus, Stethomyia and Lophopodomyia13. Most of the findings based on morphological characters and molecular data supported the monophyly of subgenera Kerteszia, Cellia and Nyssorhynchus. Furthermore, they also indicated a sister group relationship between Nyssorhynchus and Kerteszia2,14,15,16. Although many attempts have been made to resolve the phylogeny of Anopheles, there are still subgenera, sections and series whose taxonomic status remains unclear. Therefore it is necessary to elucidate their phylogenetic relationships using the latest taxonomic methods and more species data.
The mitochondrial genome due to its simple structure, matrilineal inheritance, genetic conservation, rapid evolutionary rate and infrequent recombination has been extensively utilized in phylogeny, evolutionary biology and comparative genomics17,18,19. The first comprehensive analysis of mitochondrial genomes based on 50 mosquito species revealed the subgenera Anopheles, Nyssorhynchus, Keterszia and Cellia as monophyletic20. Phylogenetic analysis of 76 Anopheles species demonstrated that the subgenera Cellia, Nyssorhynchus, Anopheles, Kerteszia, Stethomyia and Lophopodomyia were monophyletic21.
The management of mosquito-borne infectious diseases primarily relies on effective mosquito control strategies. The mitogenomes serves as a crucial molecular marker for the phylogenetic analysis of mosquitoes, offering significant insights for vector control efforts. To date, the complete mitogenomes of An. hyrcanus and An. messeae two key malaria vectors have not been documented. A comparative analysis was conducted between the mitogenomes of these two mosquito species and those of known Anopheles taxa, followed by phylogenetic assessment to advance research in mitochondrial molecular genetics and aid in malaria transmission prevention and control.
Materials and methods
Sample collection and total DNA extraction
The specimens of Anopheles hyrcanus and Anopheles messeae were collected in August 2023 from Kashgar (39° 37′ N, 75° 57′ E) and Yili (43° 74′ N, 81° 26′ E) Xinjiang, China, respectively. All samples were individually preserved in vials for transportation back to the laboratory, where they were subsequently identified by professionals based on their external morphological features9. Following identification, each specimen is labeled accordingly. The collected mosquitoes were archived in the Parasite Museum of Dali University, accompanied by their respective voucher numbers DLUP2464 and DLUP2501. The samples were stored at − 20 °C until DNA extraction. The total genomic DNA was extracted using the TIANamp Genomic DNA Kit (Tiangen, Beijing, China) following the manufacturer’s instructions. The extracted DNA samples were dispatched to Shenzhen Huitong Biotechnology Co. Ltd for the purpose of library construction and sequencing.
Genome sequencing annotation and bioinformatics analysis
After obtaining qualified DNA samples, 0.2 µg of total DNA per samples was utilized for 350 bp library preparation. Subsequently, paired-end (PE 150) sequencing of the DNA library was conducted on Illumina platform (HWI-ST1276). The raw data yielded 18.04 G (An. hyrcanus) and 18.07 G (An. messeae); while the filtered clean data amounted to 17.43 G (An. hyrcanus) and 17.08 G (An. messeae), respectively. Furthermore, the average sequencing depth was determined to be 1243.05 X (An. hyrcanus) and 136.83 X (An. messeae). The quality-controlled clean data were compared with published near-source mitochondrial data and protein-coding gene sequences using Blast, and de novo assembly was performed using SPAdes v3.14.122.
The protein-coding genes (PCGs), ribosomal RNA (rRNA) and transfer ribonucleic acid (tRNA) of the mitochondrial genomes were initially predicted through the MITOS Web Server (http://mitos.bioinf.uni-leipzig. de/index.py)23. These predictions were subsequently refined in Geneious v.9.0.224 by referencing published mosquito mitochondrial genomes to obtain a highly accurate set of conservative genes. The tRNA secondary structure was inferred in MITOS2 Web Server23. Using the formulas GC-skew = (G% - C%)/(G% + C%) and AT-skew = (A% - T%)/(A% + T%) to calculate the base composition bias25. The percentage of amino acid changes in Anopheles pullus (Genbamk ID: NC072942), Anopheles yatsushiroensis (GenBank ID: NC071885), Anopheles junlianensis (GenBank ID: NC071879), Anopheles atroparvus (GenBank ID: NC028213), An. hyrcanus (GenBank ID: PP803680) and An. messeae (Genbank ID: PP803681) were determined. The analysis tools utilized in this study included DNA star v.11.1 for base composition analysis, CodonW v.1.4.2 for the relative synonymous codon usage (RSCU) calculations, and DnaSP v.6.12.326 for the non-synonymous (Ka) to synonymous (Ks) rates in each PCG of 104 Anopheles species.
Phylogenetic analysis
The study utilized two newly sequenced complete mitochondrial genome sequences and 102 Anopheles mitogenome sequences from NCBI as ingroup taxa, with Chagasia fajardi (GenBank ID: MF381717) used as outgroup for phylogenetic analysis (Table S1). Phylogenetic relationships of the genus Anopheles were reconstructed based on a database of 13 PCGs using Maximum Likelihood (ML) in IQ-TREE v.1.6.1227 and Bayesian Inference (BI) methods in MrBayes v.3.2.728. The nucleotide sequences of 13 PCGs were aligned by Clustal W algorithm in MEGA 11 software29and the ambiguous sequences were removed. For ML, GTR + I + G was selected as the optimum model by Modeltest 3.730 and bootstrap values were estimated by applying 1000 replicates. The BI analysis involved two Markov Chain Monte Carlo (MCMC) runs with four chains per run for a total of 1,000,000 generations, with one sample per 100 generations, and a relative burn-in of 25% used to calculate Bayesian posterior probability. Finally edit and beautify the phylogenetic tree with iTOL v6.9.1 (https://itol.embl.de, accessed on 15 July 2024)31.
Results
Mitochondrial genome composition
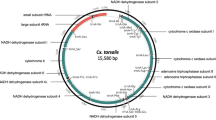
The newly sequenced mitochondrial genomes of An. hyrcanus (GenBank ID: PP803680) and An. messeae (Genbank ID: PP803681) in this study were found to be closed circular with full lengths of 15,406 bp and 15,453 bp, respectively (Fig. 1). The two consisted of 37 genes, including 13 PCGs, 22 tRNAs, and two rRNAs, as well as AT-rich region. Of these, 22 genes were situated on the heavy strand (H), while the remaining 15 genes were located on the light strand (L) (Table 1; Fig. 1). The gene order of trnR-trnA in both An. hyrcanus and An. messeae was consistent with that found in the mitogenomes of other mosquitoes. Furthermore, there were identified to be 14 overlapping genes in both mitochondrial genomes of An. hyrcanus and An. messeae; these overlaps had an overall length of 33 bp, ranging from 1 to 7 bp, with the longest occurring between ATP8 and ATP6 genes as well as between ND4 and ND4L genes (Table 1). Additionally, An. hyrcanus had eight intergenic regions, while An. messeae had nine intergenic regions, with the largest intergenic region existing between trnS and ND1 genes (Table 1).

Mitogenome circular map of An. hyrcanus and An. Messeae.
The A + T content of different genes in both An. hyrcanus and An. messeae was found to be higher than the G + C content, indicating a significant AT bias in the nucleotide organization of the two genes (Table 2). The AT-skew for the whole mitochondrial genes of An. hyrcanus and An. messeae were 0.024 and 0.035, respectively, while the GC-skew were − 0.155 and − 0.161, indicating a higher proportion of A compared to T, as well as a higher content of C compared to G (Table 2).
Protein coding genes
Both protein coding genes of both An. hyrcanus and An. messeae were 11,224 bp in length, with A + T content of 76.70% and 76.35%, respectively, indicating a significant prevalence of AT bases over GC bases. The AT-skew values for An. hyrcanus ranged from − 0.286 to −0.037, while for An. messeae they ranged from − 0.293 to −0.030, suggesting a slight bias towards T. The GC-skew values for both species varied from − 0.467 to 0.291 for An. hyrcanus and from − 0.482 to 0.297 for An. messeae showing no notable preference for G or C bases. In both An. hyrcanus and An. messeae, the majority of the PCGs utilized ATN as the initiation codons, in addition to TCG and GTG for COX1 and ND5. The complete TAA served as stop codon except for COX1, COX2, COX3 and ND4 which ended with an incomplete codon T.
The relative synonymous codon usage (RSCU) values for the 13 PCGs of An. hyrcanus and An. messeae are shown in Fig. 2; Table 3. It was observed that each species contained a total of 3,741 codons. Statistical analysis of RSCU values for all amino acid codons indicated that both species exhibited a strong preference for eight codon families (Ala, Gly, Leu2, Pro, Ser1, Ser2, Thr and Val). Specifically, UUA, AGA, GGA and GCU were identified as the most frequently used codons in both species with RSCU values greater than 2. Conversely, CGG, CUG, CGC and CUC were found to be used less frequently.

The relative synonymous codon usage (RSCU) of An. hyrcanus (A) and An. messeae (B).
The amino acid percentage values of An. hyrcanus were comparable to those of An. yatsushiroensis and An. junlianensis, whereas the values for An. messeae exhibited slight differences from those of An. atroparvus (Fig. 3). Nevertheless, the trend in amino acid percentages across the six mosquito species was largely consistent, with leucine (Leu) representing the highest proportion and arginine (Arg) constituting the lowest.

The percentage of amino acid changes An.hyrcanus and An.messeae relative to the reference sequence.
The Ka/Ks values were calculated for 13 protein-coding genes in 104 mosquito mitogenomes to evaluate the selection pressure on different PCGs. The results showed that all Ka/Ks values were below 1, indicating purifying selection across all genes. Notably, ND6 exhibited the highest value (0.2), followed by ATP8, ND2, ND5, ND4, ND4L and ND3 (values > 0.1), suggesting relatively relaxed evolutionary constraints on these genes. In contrast, ATP6, COX1-3, and CYTB showed particularly strong purifying selection (Ka/Ks < 0.05) (Fig. 4).

Mosquito mitochondrial genome substitution rates, showing Ka, Ks, and Ka/Ks values per PCG for 104 species.
Transfer and ribosomal RNA genes
The total length of 22 tRNAs in both An. hyrcanus and An. messeae was 1,475 bp, with individual tRNA genes ranging from 64 to 72 bp (Table 1). The AT contents of the total tRNA genes were 78.24% and 78.85%, respectively, with positive shifts in both AT and GC (Table 2). In both species, twenty-one out of the 22 tRNAs exhibited a typical cloverleaf-like structure, including amino acid acceptor (AA), TΨC arm, dihydrouridine (DHU), anticodons (AC) and loops; only the trnS1 gene lacked the DHU arm and instead had a DHU loop (Figure S1).
The two ribosomal RNA genes in both mitogenomes occupied the identical position within the light chain: rrnL between trnL1 and trnV and rrnS between trnV and the AT-rich region (Table 1). The AT contents of rRNA genes were similar in both species, with a negative shift in AT but a positive shift in GC (Table 2).
Phylogenetic analysis
Based on the pairwise distance analysis of 13 PCGs presented in Table S2, the pairwise distances among 104 species of Anopheles mosquitoes varied from 0.001 to 0.200. The minimum pairwise distance observed between An. hyrcanus and both An. yatsushiroensis and An. junlianensis was recorded at 0.029, whereas the lowest pairwise distance between An. messeae and An. atroparvus was noted at 0.038.
We constructed phylogenetic trees based on a joint dataset of 13 PCGs using BI and ML methods to infer phylogenetic relationships within the genus Anopheles. The two trees were roughly the same at subgenus level, as shown in Figs. 5 and 6. Both analyses favored monophyly of six subgenera Nyssorhynchus, Cellia, Lophopodomyia, Kerteszia, Stethomyia and Anopheles, with Bayesian posterior probability (BPP) of 1 for each subgenus (Fig. 5) and bootstrap values (BV) ranging from 90 to 100 (Fig. 6). However, there were still some differences between the two trees, such as the change in position of the series Cellia. In the BI tree, the series Cellia was identified as sister group of the series Pyretophorus; whereas in the ML tree, series Cellia was nested in the series Pyretophorus. The results also suggested a close relationship between An. hyrcanus and ((An. pullus + An. yatsushiroensis) + An. junlianensis) (BPP = 1.0, BV = 100), while An. messeae was more closely associated with An. atroparvus (BPP = 1.0, BV = 95). Both species examined are classified within the Anopheles subgenus, while An. hyrcanus is categorized under the Laticorn section Myzorhynchus Series and An. messeae within the Angusticorn section Anopheles series.

Phylogenetic relationships of the mitochondrial genomes of 104 species of the genus Anopheles constructed based on 13PCGs using Bayesian inference; the numbers on the nodes are Bayesian posterior probabilities. The mitochondrial genomes of the two species newly sequenced in this study are indicated by black triangles, and the Genbank sequence imformation of the 105 mitochondrial genomes is shown in Table S1.

Phylogenetic relationships of the mitochondrial genomes of 104 species of the genus Anopheles constructed based on 13PCGs using maximum likelihood method; the numbers on the nodes are bootstrap values. The mitochondrial genomes of the two species newly sequenced in this study are indicated by black triangles, and the Genbank sequence imformation of the 105 mitochondrial genomes is shown in Table S1.
Within the genus Anopheles, the phylogeny of the six subgenera is as follows: Lophopodomyia + ((Kerteszia + Stethomyia) + (Nyssorhynchus + (Cellia + Anopheles))). Within the subgenus Anopheles, it was found that the sections of Angusticorn and Laticorn were paraphyletic. However, it was observed that the series Arribalzagia (BPP = 1.0, BV = 94) and series Myzorhynchus (BPP = 1.0, BV = 100) in the section Laticorn were monophyletic. Moving on to the subgenus Nyssorhynchus, it was discovered that the sections Albimanus, Argyritarsis and Myzorhynchella were also paraphyletic, along with the series Oswaldoi, Albitarsis and Argyritarsis. In terms of subgenus Cellia, it was determined that both series Myzomyia (BPP = 1.0, BV = 100) and Neocellia (BPP = 1.0, BV = 100) were monophyletic; however, the series Neomyzomyia was found to be paraphyletic. In addition, the series Pyretophorus appeared to be monophyletic (BPP = 0.7) in the BI tree, but it showed as paraphyletic due to the embedding series Cellia in the ML tree.
Discussion
Anopheles is the main vector of malaria but is also capable of transmitting filaria and arboviruses32. Due to their significant medical importance, taxonomic studies of the genus Anopheles have become increasingly crucial. This study represented the first analysis of the mitochondrial genes of An. hyrcanus and An. messeae, utilizing the data for comparative genomics, relative synonymous codon usage and phylogenetic analysis. Both species under scrutiny are classified within the subgenus Anopheles; specifically, An. hyrcanus is categorized in the Laticorn section of the Myzorhynchus Series, whereas An. messeae is positioned within the Angusticorn section of the Anopheles series.
The differences in mitochondrial gene length between An. hyrcanus and An. messeae are primarily attributed to the size of control region. The genetic order of trnR-trnA in An. hyrcanus and An. messeae is opposite to that of trnA-trnR in the mitogenome of insect ancestor (Drosophila yakuba)33but consistent with other mosquitoes34. Both species preferentially utilize bases A and C, as indicated by the base composition and skew values, which is associated with selection pressure and asymmetric mutation of four bases during replication and transcription19,35. The PCGs initiate with ATN codons, except for the COX1 and ND5 genes, which use TCG and GTG as initiation codons, respectively, consistent with previous reports for other mosquitoes, such as Aedes aegypti, Ochlerotatus caspius, and Anopheles belenrae, etc34,36. Most PCGs terminate with complete or incomplete termination codons TA or T; this phenomenon exist in all metazoan mitochondrial genes37with post-translational modifications converting incomplete termination codon to the complete termination codon TAA38. Only the trnS1 gene fails to fold into a typical cloverleaf structure, as its DHU arm is replaced by a DHU loop, consistent with previous reports39,40. By analyzing the Ka/Ks ratio, it is possible to determine whether protein-coding genes are subject to selection pressure. A Ka/Ks ratio greater than 1 indicates a positive selective effect, while a ratio less than 1 denotes a purifying selective effect41. Thirteen PCGs exhibited a purifying selective effect, in line with previous reports on mosquitoes20,21,42. The observed pattern matches expectations for oxidative phosphorylation (OXPHOS) genes: core respiratory chain subunits evolve under stringent functional constraints due to their essential roles in election transport and energy production.
The six subgenera of the genus Anopheles investigated in this study are all monophyletic, the phylogenetic relationships of the six subgenera are as follows: Lophopodomyia + ((Kerteszia + Stethomyia) + (Nyssorhynchus + (Cellia + Anopheles))). While early morphological analyses supported the monophyly of Nyssorhynchus, Kerteszia, and Cellia, they also suggested that Kerteszia and Nyssorhynchus were sister taxa and that the subgenus Anopheles was polyphyletic13,16,43. In contrast, molecular studies using limited markers initially supported the monophyly of the subgenus Anopheles14,44. Subsequent analyses based on mitochondrial PCGs indicated a close relationship between Cellia, Anopheles, Nyssorhynchus, and Kerteszia, though the monophyly of these subgenera required further validation with expanded mitogenomic data45. The phylogenies inferred from the mitochondrial genomes of 50 mosquito species and 1085 single-copy orthologs of 18 species provided support for the monophyly for subgenera Anopheles, Nyssorhynchus and Cellia, with the subgenus Nyssorhynchus being sister to the clade (Anopheles + Cellia)20,46. Recent phylogenetic findings, based on extensive mitochondrial genomic data from mosquitoes, indicated that all six subgenera of the genus Anopheles (Anopheles, Nyssorhynchus, Lophopodomyia, Kerteszia, Stethomyia and Cellia) are monophyletic and the phylogenetic relationships at the subgenus level were consistent with the results of this study21,42. Thus, while morphological and molecular data historically conflicted, large-scale molecular datasets now consistently support subgeneric monophyly.
Despite the general monophyly observed at the subgenus level, lower taxonomic ranks (sections and series) frequently exhibit conflicting phylogenetic patterns. Within the subgenus Anopheles, sections Laticorn and Angusticorn have consistently been identified as polyphyletic across multiple studies. Conversely, certain series, such as Arribalzagia and Myzorhynchus, demonstrate stronger evidence of monophyly. Early phylogenetic analyses supported the monophyletic status of the Arribalzagia series, whereas the Anopheles series was found to be paraphyletic13,14. Subsequent morphological investigations confirmed the paraphyly of section Angusticorn and the Anopheles series, while supporting the monophyly of section Laticorn and the Arribalzagia and Myzorhynchus series16. However, conflicting results have also emerged, with some morphological analyses recovering Arribalzagia as monophyletic but identifying series Anopheles and Myzorhynchus as paraphyletic47.
In the subgenus Nyssorhynchus, the three sections Albimanus, Argyritarsis and Myzorhynchella, as well as all the series contained therein, were paraphyletic. Most of the findings were consistent with the results of the present study, substantiating the three sections Albimanus, Argyritarsis and Myzorhynchella are polyphyletic13,21.
In the subgenus Cellia, strong support was found for the monophyly of the series Myzomyia and Neocellia (BPP = 1.0, BV = 100), as well as for the monophyly of the series Neomyzomyia (BPP = 1.0, BV = 100) when excluding Anopheles nili species. The embedding of the series Cellia resulted in the series Pyretophorus behaving as polyphyletic in the ML tree. This study only included one species of the series Cellia, and additional mitochondrial sequences will contribute to resolving the phylogenetic status of the series Cellia. Morphological studies indicated that only the series Cellia exhibited monophyly13whereas molecular studies demonstrated that the four series Myzomyia, Neocellia, Pyretophorus and Neomyzomyia displayed monophyly14,20,21. These inconsistencies underscore the complexity of phylogenetic relationships within the genus Anopheles and suggest that taxonomic revisions may be warranted. While higher-level groupings are generally well-supported as monophyletic, finer-scale classifications (e.g., sections and series) exhibit varying degrees of resolution. This variability is likely attributable to factors such as morphological convergence, incomplete lineage sorting, or insufficient phylogenetic signal.
Notably, phylogenetic analyses based on nuclear genomes have provided enhanced resolution, corroborating the monophyly of major subgenera46. However, the placement of certain clades occasionally varies due to discrepancies in lineage sorting or introductions. These investigations underscore the constraints of relying solely on mitochondrial data, as such data may not fully capture the complex evolutionary history of Anopheles. In future studies, the integration of nuclear genome data will be indispensable for elucidating deep phylogenetic relationships and reconciling morphological and molecular hypotheses.
Conclusion
In this study, we conducted comprehensive analysis of the mitochondrial genomes of An. hyrcanus and An. messeae, revealing their genomic features, codon usage and selection pressure on each PCG gene. Our findings are consistent with previously reported mosquito mitogenomes. We utilized the ML and BI methods to construct phylogenetic trees for 104 Anopheles species, yielding novel insights into the phylogenetic relationships within the genus Anopheles. However, more mitochondrial genome data from Anopheles species is needed to further assess the phylogeny of different taxonomic orders within the genus.
Data availability
All data and materials of the study are included in the manuscript and the supplementary material. The complete mitochondrial genome data of An. hyrcanus and An. messeae have been stored on the NCBI website (https://www.ncbi.nlm.nih.gov/) under the accession number PP803680 and PP803681.
References
Wilkerson, R. C., Linton, Y. M. & Strickman, D. Mosquitoes of the WorldVol. 1 (Johns Hopkins University, 2021).
Harbach, R. E. & Kitching, I. J. The phylogeny of Anophelinae revisited: inferences about the origin and classification of Anopheles (Diptera: Culicidae). Zool. Scr. 45, 34–47. https://doi.org/10.1111/zsc.12137 (2015).
Makam, P. & Matsa, R. Big three infectious diseases: tuberculosis, malaria and HIV/AIDS. Curr. Top. Med. Chem. 21, 2779–2799. https://doi.org/10.2174/1568026621666210916170417 (2021).
Venkatesan, P. The 2023 WHO world malaria report. Lancet Microbe. 5, e214. https://doi.org/10.1016/s2666-5247(24)00016-8 (2024).
Feng, J. et al. Ready for malaria elimination: zero Indigenous case reported in the people’s Republic of China. Malar. J. 17, 315. https://doi.org/10.1186/s12936-018-2444-9 (2018).
Zhang, L., Yi, B. Y., Zhou, S. S., Xia, Z. G. & Yin, J. H. Epidemiological characteristics of plasmodium malariae malaria in china: a malaria that should not be neglected post elimination. Infect. Dis. Poverty. 12, 101. https://doi.org/10.1186/s40249-023-01156-2 (2023).
Feng, X., Levens, J. & Zhou, X. N. Protecting the gains of malaria elimination in China. Infect. Dis. Poverty. 9 https://doi.org/10.1186/s40249-020-00661-y (2020).
Harbach, R. E. An Anopheles by any other name …. J. Med. Entomol. 55, 1069–1070. https://doi.org/10.1093/jme/tjy108 (2018).
Baolin, L. Fauna Sinica, Insecta Vol. 9: Diptera, Culicidae II (Vol. Insecta (Science, 1997).
Zahar, A. R. Review of the ecology of malaria vectors in the WHO Eastern mediterranean region. Bull. World Health Organ. 50, 427–440 (1974).
Artemov, G. N. et al. New cytogenetic photomap and molecular diagnostics for the cryptic species of the malaria mosquitoes Anopheles messeae and Anopheles daciae from Eurasia. Insects 12, 835, (2021). https://doi.org/10.3390/insects12090835
Linton, Y. M. et al. Morphological and molecular characterization of Anopheles (Anopheles) maculipennis meigen, type species of the genus and nominotypical member of the maculipennis complex. Syst. Entomol. 28, 39–56. https://doi.org/10.1046/j.1365-3113.2003.00198.x (2003).
Sallum, M. A. M., Schultz, T. & Wilkerson, R. Phylogeny of Anophelinae (Diptera Culicidae) based on morphological characters. Ann. Entomol. Soc. Am. 93, 745–775. https://doi.org/10.1603/0013-8746(2000)093[ (2000). 0745:POADCB]2.0.CO;2.
Sallum, M. A. M. et al. Phylogeny of Anophelinae (Diptera: Culicidae) based on nuclear ribosomal and mitochondrial DNA sequences. Syst. Entomol. 27, 361–382. https://doi.org/10.1046/j.1365-3113.2002.00182.x (2002).
Collucci, E. Phylogenetic analysis of the subgenus Kerteszia of Anopheles (Diptera: culicidae: Anophelinae) based on morphological characters. Insect Syst. Evol. 34, 361–372. https://doi.org/10.1163/187631203x00018 (2003).
Harbach, R. E. & Kitching, I. J. Reconsideration of anopheline mosquito phylogeny (Diptera: culicidae: Anophelinae) based on morphological data. Syst. Biodivers. 3, 345–374. https://doi.org/10.1017/S147720000500174X (2005).
Lin, X. L. et al. Mitogenomes provide new insights of evolutionary history of boreheptagyiini and Diamesini (Diptera: chironomidae: Diamesinae). Ecol. Evol. 12, e8957. https://doi.org/10.1002/ece3.8957 (2022).
Solorzano-Garcia, B., Hernandez-Mena, D. I. & Choudhury, A. Perez-Ponce de leon, G. The complete mitochondrial genome of three species of allocreadiids (Digenea, Allocreadiidae): characterization and phylogenetic position within the order Plagiorchiida. Parasitology 1–26. https://doi.org/10.1017/S0031182024000064 (2024).
Cameron, S. L. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117. https://doi.org/10.1146/annurev-ento-011613-162007 (2014).
Hao, Y. J. et al. Complete mitochondrial genomes of Anopheles stephensi and An. dirus and comparative evolutionary mitochondriomics of 50 mosquitoes. Sci. Rep. 7, 7666 (2017).
Guo, J. et al. Complete mitogenomes of Anopheles peditaeniatus and Anopheles nitidus and phylogenetic relationships within the genus Anopheles inferred from mitogenomes. Parasit. Vectors. 14, 452. https://doi.org/10.1186/s13071-021-04963-4 (2021).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. https://doi.org/10.1089/cmb.2012.0021 (2012).
Bernt, M. et al. MITOS: improved de Novo metazoan mitochondrial genome annotation. Mol. Phylogenet Evol. 69, 313–319. https://doi.org/10.1016/j.ympev.2012.08.023 (2013).
Kearse, M. et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. https://doi.org/10.1093/bioinformatics/bts199 (2012).
Perna, N. T. & Kocher, T. D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358 (1995).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. https://doi.org/10.1093/molbev/msx248 (2017).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. https://doi.org/10.1093/molbev/msu300 (2015).
Ronquist, F. et al. MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. https://doi.org/10.1093/sysbio/sys029 (2012).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Posada, D. & Crandall, K. A. MODELTEST: testing the model of DNA substitution. Bioinformatics 14, 817–818 (1998).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. https://doi.org/10.1093/nar/gkae268 (2024).
Forattini, O. P. Culicidologia médica: identificação, biologia, epidemiologia Vol. 2Edusp,. (1996).
Clary, D. O. & Wolstenholme, D. R. The mitochondrial DNA molecular of Drosophila yakuba: nucleotide sequence, gene organization, and genetic code. J. Mol. Evol. 22, 252–271. https://doi.org/10.1007/BF02099755 (1985).
Ma, X. X. et al. First description of the mitogenome and phylogeny:Aedes Vexansand Ochlerotatus caspius of the tribe Aedini (Diptera: Culicidae). Infect. Genet. Evol. 102, 105311. https://doi.org/10.1016/j.meegid.2022.105311 (2022).
Francino, M. P. & Ochman, H. Strand asymmetries in DNA evolution. Trends Genet. 13, 240–245 (1997).
Dong, H. et al. Phylogenetic analysis of some species of the Anopheles hyrcanus group (Diptera: Culicidae) in China based on complete mitochondrial genomes. Genes (Basel). 14, 1453. https://doi.org/10.3390/genes14071453 (2023).
Wolstenholme, D. R. Animal mitochondrial DNA: structure and evolution. Int. Rev. Cytol. 141, 173–216. https://doi.org/10.1016/s0074-7696(08)62066-5 (1992).
Ojala, D., Montoya, J. & Attardi, G. tRNA Punctuation model of RNA processing in human mitochondria. Nature 290, 470–474. https://doi.org/10.1038/290470a0 (1981).
da Silva, F. S. et al. Mitochondrial genome sequencing and phylogeny of Haemagogus albomaculatus, Haemagogus leucocelaenus, Haemagogus spegazzinii, and Haemagogus tropicalis (Diptera: Culicidae). Sci Rep 10, 16948, (2020). https://doi.org/10.1038/s41598-020-73790-x
Martinez-Villegas, L. et al. Characterization of the complete mitogenome of Anopheles aquasalis, and phylogenetic divergences among Anopheles from diverse geographic zones. PLoS One. 14, e0219523. https://doi.org/10.1371/journal.pone.0219523 (2019).
Hurst, L. D. The ka/ks ratio: diagnosing the form of sequence evolution. Trends Genet. 18, 486. https://doi.org/10.1016/s0168-9525(02)02722-1 (2002).
Chen, D. H. et al. Mitogenome-based phylogeny of mosquitoes (Diptera: Culicidae). Insect Sci. 31, 599–612. https://doi.org/10.1111/1744-7917.13251 (2024).
Harbach, R. E. In Anopheles mosquitoes-New Insights into Malaria Vectors (IntechOpen, 2013).
Freitas, L. A. et al. Diversification of the genus Anopheles and a Neotropical clade from the late cretaceous. PloS One. 10, e0134462. https://doi.org/10.1371/journal.pone.0134462 (2015).
Hua, Y. Q. et al. The complete mitochondrial genome of Anopheles minimus (Diptera: Culicidae) and the phylogenetics of known Anopheles mitogenomes. Insect Sci. 23, 353–365. https://doi.org/10.1111/1744-7917.12326 (2016).
Neafsey, D. E. et al. Mosquito genomics. Highly evolvable malaria vectors: the genomes of 16 Anopheles mosquitoes. Science 347, 1258522. https://doi.org/10.1126/science.1258522 (2015).
Collucci, E. & Sallum, M. A. Cladistic analysis of the subgenus Anopheles (Anopheles) Meigen (Diptera: Culicidae) based on morphological characters. Mem. Inst. Oswaldo Cruz. 102, 277–271. https://doi.org/10.1590/s0074-02762007005000026 (2007).
Funding
This study was supported by the Special Basic Cooperative Research Programs of Yunnan Provincial Undergraduate Universities’ Association (Grant No.202401BA070001-005), the Yunnan Fundamental Research Projects (grant NO. 202401AT070084), the Special Basic Cooperative Research Programs of Yunnan Provincial Undergraduate Universities’ Association (grant NO. 202301BA070001-045) and Scientific Research Fund of Yunnan Education Department (grant NO.2024J0857).
Author information
Authors and Affiliations
Contributions
Conceptualization, MD, ZL and JW; Data curation, MD, SL and ST; Funding acquisition, JW, DJ and XY; Methodology, GS and DJ; Project administration, QZ, WG and XY; Resources, ZL and GS; Supervision, QZ, WG and XY; Writing – original draft, MD, ZL and JW; Writing – review & editing, GS, SL, ST, DJ, QZ, WG and XY.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethics statement
No specific permits were required for this study. The study did not involve endangered or protected species. Therefore, the local ethics committee deemed that approval was unnecessary.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Duan, M., Lin, Z., Wu, J. et al. Comparative mitochondrial genome and phylogenetic analysis of malaria mosquitoes Anopheles hyrcanus and Anopheles messeae. Sci Rep 15, 23959 (2025). https://doi.org/10.1038/s41598-025-09815-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-09815-0
