
Abstract
The GATA3 noncoding variant rs3824662 has been implicated in the pathogenesis of Ph-like B-ALL, where it is associated with extensive chromatin reorganisation, resulting in the dysregulation of multiple genes, including CRLF2 overexpression. Given the altered chromatin landscape and increased accessibility of GATA3 binding regions associated with the rs3824662 variant, we investigated the potential role of enhancer RNAs (eRNAs) located near the GATA3 locus in regulating CRLF2 expression. We found that the expression of eRNA_G3, located at chr10:8,443,562–8,449,563, was positively correlated with CRLF2 expression. Notably, eRNA_G3 was significantly upregulated in Ph-like ALL cases carrying the GATA3 rs3824662 variant. These findings suggest that both rs3824662 and eRNAs may cooperatively contribute to the regulatory mechanisms governing GATA3 and CRLF2 expression in Ph-like ALL.
Similar content being viewed by others

Introduction
The GATA3 single nucleotide polymorphism (SNP) risk allele (rs3824662) is strongly associated with an increased risk of developing BCR::ABL1-like (Ph-like) B-cell precursor acute lymphoblastic leukaemia (B-ALL)1. In 2022, Yang et al. conducted an elegant study demonstrating that this SNP is linked to numerous newly acquired open chromatin regions enriched for GATA3 binding sites, frequently located near chromosomal breakpoints observed in Ph-like ALL. This global reorganisation of the 3D genome led to a large-scale shift of chromatin regions from a repressive, compacted state to an active, open configuration. As a result, critical genes such as SEMA6A, PDGFRB, and IKZF1 showed altered expression in Ph-like ALL2. The study also provided compelling evidence that GATA3 can directly regulate CRLF2 and enhance JAK-STAT signalling during leukaemogenesis, thus establishing a mechanistic link between the GATA3 rs3824662 variant and CRLF2 overexpression (CRLF2-high).
Building on the observed changes in GATA3 binding sites and chromatin accessibility, we sought to investigate the potential role of enhancer RNAs (eRNAs) and GATA3 isoforms in the regulation of CRLF2. Recent studies have raised important questions regarding the roles of eRNAs in gene regulation, including the promotion of enhancer-promoter interaction and modulation of chromatin structure. Importantly, eRNAs can act independently of enhancer orientation, proximity, or linear distance to their target genes. The expression of these non-coding RNA molecules also serves as a useful proxy for identifying active enhancers within the genome3. In addition, recent studies have underscored the clinical relevance of eRNAs as potential therapeutic targets in cancer4,5.
In leukaemia, eRNAs regulate gene expression by facilitating enhancer-promoter interactions, with expression patterns that are often subtype-specific, suggesting their potential as biomarkers. For example, genome-wide downregulation of eRNAs and their associated target gene loci has been observed in ETV6::RUNX1 ALL, disrupting normal B lymphopoiesis and highlighting the essential role of eRNAs in maintaining gene expression patterns during blood cell development. Additionally, in KMT2A-rearranged (KMT2A-r) ALL, the long non-coding RNA LAMP5-AS1 has been associated with increased H3K79 methylation, promoting self-renewal programmes and differentiation blockage in KMT2A-r cells6,7,8. Together, these findings emphasise the potential involvement of eRNAs in leukaemogenesis.
Regarding the impact of GATA3 rs3824662 variant, this non-coding SNP may influence regulatory regions by altering chromatin conformation2. In a transcriptionally active chromatin state, GATA3 binding sites become more accessible, potentially promoting the expression of different GATA3 isoforms. Given this SNP plays a key role in the leukaemogenesis of B-ALLs lacking recurrent genetic alterations (B-others), we aimed to identify additional factors that could regulate CRLF2 expression.
Results and discussion
In the TARGET cohort, we identified 21 Ph-like ALL patients (16%). Interestingly, a male patient with KMT2A-r and a hyperdiploid karyotype, had previously been classified as Ph-like ALL with CRLF2 overexpression9. However, this sample clustered with the Ph + ALL signature (Suppl. Fig. S1). Additionally, two previously identified Ph-like ALL cases and two Ph + ALL samples did not cluster with their respective groups. These heterogeneous profiles have been discussed in prior studies, suggesting the existence of at least three distinct molecular subtypes within Ph + and Ph-like ALL10. This heterogeneity emphasises the need for accurate genetic classification of leukaemia subtypes to guide and optimise therapy decisions, as well as to improve patients’ prognoses.
Our analyses identified the GATA3 rs3824662 A/A genotype as significantly associated with Ph-like ALL with a relative risk of 1.80 (95% CI 1.23–3.24) compared to non-Ph-like ALL (Suppl. Table S1), corroborating findings reported by Yang et al.2. However, we observed that the rs3824662 variant influences GATA3 transcription levels independently of the Ph-like ALL subtype (Suppl. Fig. S2-A). Since both our results and those of Yang et al. indicated that the A/C and A/A genotypes increase GATA3 expression, we compared wild-type (C/C) cases with those harbouring mutant alleles (A/C or A/A) for further analyses. Given that CRLF2-high is a hallmark of most Ph-like ALL cases, we assessed whether the GATA3 variant also affects CRLF2 expression within the TARGET cohort.
Due to the limited sample size, we divided the TARGET cohort into three groups based on GATA3 SNP status for comparison: (i) non-Ph-like (n = 59); (ii) Ph-like (n = 16); and (iii) B-ALL (n = 51). Both the non-Ph-like and Ph-like groups included cases with the GATA3 variant, while the B-ALL group consisted predominantly of wild-type cases, including 5 Ph-like and 46 non-Ph-like patients (Suppl. Table S2). Our analysis revealed that the association between GATA3 SNP and CRLF2-high is restricted to the Ph-like ALL subgroup (Suppl. Fig. S2-B).
The mechanistic landscape underlying CRLF2-high appears complex and includes CRLF2 mutations (e.g. F232C) and CRLF2 rearrangements (CRLF2-r), which were identified in most outlier cases with wild-type GATA3 genotype (Suppl. Fig. S2-B). Furthermore, we recently reported that IKZF1 deletions are also associated with CRLF2-high11. Together with the GATA3 variant, these distinct mechanisms likely contribute to CRLF2 dysregulation, particularly in Ph-like ALL cases.
To further characterise differences among the non-Ph-like, Ph-like, and B-ALL cohorts, we evaluated the expression patterns of additional genes included in the panel used to identify Ph-like ALL cases. As expected, the Ph-like group exhibited higher expression levels across all genes compared to non-Ph-like and B-ALL groups. This pattern remained consistent despite the presence of some Ph-like ALL cases within the B-ALL group, which appeared as outliers, this is likely because the B-ALL group was predominantly composed of non-Ph-like ALL cases (Suppl. Fig. S3).
To investigate the potential role of eRNAs in gene regulation, we analysed the expression of four eRNAs co-localised with the GATA3 locus and four eRNAs co-localised with CRLF2 locus (See Methods, Suppl. Fig. S4-A). We assessed their association with the GATA3 variant in Ph-like and non-Ph-like ALL samples compared with the B-ALL group (Fig. 1A and Suppl. Fig. S4-B). Our data showed that the enhancer associated with eRNA_G1 appears to regulate GATA3 expression regardless of ALL subgroups, with significantly increased expression in samples carrying the GATA3 rs3824662 variant (Suppl. Fig. S4-B).

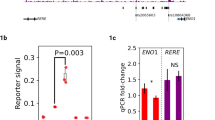
Interactions between GATA3 SNP and regulatory regions in chr10 and chrX. (A) eRNAs expression according to the occurrence of GATA3 rs3824662 SNP in non-Ph-like, Ph-like and other B-ALL patients. (B) Correlation analysis between eRNAs and ASMTL, CRLF2, GATA3 and P2RY8 genes. Positive correlations are displayed in blue and negative correlations in red. Colour intensity and the size of the circle are proportional to the correlation coefficients. Non-significant p-values are represented by a cross. (C) Schematic illustration of the GATA3 isoforms (GATA3-001, GATA3-002, GATA3-003 and GATA3-004, respectively), the eRNAs located on chr10 (eRNA_G1, eRNA_G2, eRNA_G3 and eRNA_G4), and the regulatory regions previously described in the GeneHancer database. GATA3 coding portions are illustrated in black, while the non-coding portions are shown in white. The promoter region is marked by a red rectangle and the enhancer regions by black squares. (D) GATA3 isoforms expression according to the GATA3 SNP. In all graphics, patients without the GATA3 variant were coloured in purple, and patients with the GATA3 variant were coloured in green. ns: p > 0.05; *: p ≤ 0.05; and **: p ≤ 0.01.
In contrast, eRNA_G3 was significantly more highly expressed in Ph-like ALL cases carrying the GATA3 variant (Fig. 1A), suggesting that this region may be particularly affected by chromatin reorganisation in Ph-like ALL. Regarding eRNAs near the CRLF2 gene, both eRNA_C1 and eRNA_C4 showed higher expression levels in Ph-like ALL cases with the GATA3 variant compared to wild-type B-ALL samples (Fig. 1A). Expression levels of these two eRNAs were strongly and positively correlated across all samples (Pearson’s r = 0.90, p < 0.01) and both correlated moderately with CRLF2 and neighbouring genes such as ASMTL and P2RY8 (Pearson’s r = 0.30–0.60, p < 0.01) (Fig. 1B). These results support a role for eRNA_C1 and eRNA_C4 in the regulation of CRLF2 and adjacent genes in B-ALL.
Interestingly, although eRNA_G3 expression did not correlate with GATA3 or CRLF2 levels, it was positively correlated with eRNA_C4 across all samples, suggesting a potential interaction between these two eRNAs (Pearson’s r = 0.18, p < 0.05, Fig. 1B). We observed that eRNA_C4 co-localises with the P2RY8 gene and the enhancer-promoter interaction region of CRLF2, as previously described by Yang et al., who reported increased enhancer activity in this region in Ph-like ALL cases carrying the GATA3 rs3824662 variant, which alters chromatin accessibility and the GATA3 binding patterns2. Further studies are needed to clarify the role of eRNA_C1 in this regulatory mechanism.
Additionally, we evaluated 144 Brazilian samples for the presence of the GATA3 rs3824662 variant and measured expression levels of both GATA3 and eRNA_G3 (Supp. Fig. S5). However, these results did not replicate the findings observed in the TARGET cohort. This discrepancy may be due to two main factors: (i) our Ph-like subgroup categorisation may have underestimated the actual number of cases, as it relied primarily on gene rearrangements associated with Ph-like ALL (see Methods section) and/or CRLF2 overexpression; and (ii) the risk allele ‘A’ was less frequent in the Brazilian cohort compared to the TARGET cohort (38.9% vs. 59.5%).
We hypothesised that the GATA3 risk allele might cause an imbalance in isoform expression, potentially affecting GATA3 protein binding. To test this, we analysed the expression patterns of four GATA3 splice isoforms (Fig. 1C) in Ph-like and non-Ph-like ALL subgroups carrying the rs3824662 variant. We found that three isoforms showed significantly higher expression in the presence of the GATA3 variant in both Ph-like and non-Ph-like ALL subgroups compared to wild-type B-ALL samples (Fig. 1D).
Our findings corroborate the evidence previously reported by Yang et al. and extend their observations by analysing a representative cohort of paediatric B-ALL. Notably, we demonstrate that the GATA3 rs3824662 risk allele influences GATA3 expression independently of B-ALL molecular subtype, whereas its association with CRLF2-high is restricted to Ph-like ALL. Moreover, we identified eRNAs as potential contributors to this regulatory network. Future studies are warranted to elucidate the precise roles of these eRNAs in Ph-like ALL pathogenesis and to explore their potential as therapeutic targets.
Methods
Data description
We characterised 126 B-ALL paediatric cases from the Therapeutically Applicable Research to Generate Effective Treatments project (TARGET) ALL Phase 2 cohort. Patients were selected based on the availability of whole genome sequencing (WGS) data to identify the presence or absence of the GATA3 variant, and RNA sequencing (RNA-seq) data to characterise the Ph-like ALL patients and to investigate the transcriptional levels of our target genes, isoforms, and eRNAs. The complete data generation protocol is available on the GDC Portal (see “TARGET Molecular Characterization Platforms” at https://www.cancer.gov/ccg/research/genome-sequencing/target). Information on B-ALL genetic alterations was obtained from publicly available clinical data on the same portal (https://portal.gdc.cancer.gov/projects/TARGET-ALL-P2).
To validate our findings, we have retrospectively analysed an independent cohort of 144 paediatric B-ALL patients (0–18 years) from five cancer centres in Brazil: INCA-RJ, IPPMG/UFRJ-RJ, IMIP-PE, HEMORIO-RJ, and Hospital Amaral Carvalho-SP. Mononuclear cells were isolated from bone marrow (BM) or peripheral blood (PB) samples collected at diagnosis using Ficoll-Paque (Sigma-Aldrich) and screened for common B-ALL genetic abnormalities, including hyperdiploidy, ETV6::RUNX1, BCR::ABL1, TCF3::PBX1, and KMT2A-r, following standard procedures11. This study was approved by the Research Ethics Committee of the National Cancer Institute of Brazil (CAEE #87793418.0.1001.5274) and conducted in accordance with the ethical standards as laid down in the Declaration of Helsinki. Informed consent was obtained from all participants and/or their parents or legal guardians. All methods were performed in accordance with the relevant guidelines and regulations.
Identification of GATA3 rs3824662 variant
The genomic data in the TARGET cohort were previously analysed using the Complete Genomics Inc. (CGI) Assembly Pipeline12,13. We downloaded publicly available CGI-generated WGS files for somatic single nucleotide variants (SNVs), as well as short insertions, and deletions (indels) from the Data Matrix in the TARGET project portal (discontinued version). The files from each patient were merged into a single data frame. Subsequently, filters were applied to the region where the rs3824662 variant in GATA3 is located (chr10:8,104,208) to identify patients carrying the previously annotated variant. The SNP was classified as wild-type (C/C), heterozygous mutant (A/C), or homozygous mutant (A/A) based on the allele detected in the tumour sequencing compared to the reference allele.
For the validation cohort, genomic DNA was obtained from BM or PB mononuclear cells using the DNA MiniBlood kit (Qiagen) following the manufacturer’s standard protocol. DNA concentration and purity were assessed using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). Genotyping was performed using the C_27522049_10 TaqMan™ SNP Genotyping Assay, and the reactions were performed using the QuantStudio™ 3 Real-Time PCR (Thermo Fisher Scientific). To ensure genotyping accuracy, all samples were analysed in duplicate. Genotypes were determined by interpreting allelic discrimination plots, where the wild-type (C/C), heterozygous mutant (A/C), or homozygous mutant (A/A) patterns were identified by comparison with the reference controls for each allele using the QuantStudio™ Design & Analysis Software (Thermo Fisher Scientific).
eRNAs quantification and selection
We downloaded the fastq files obtained from Illumina paired-end RNA-seq (dbGAP-approved Project ID #19604) and applied them to the Pipeline for Enhancer Transcription (PET; https://github.com/fun-science-club-ntu/PET) to quantify the eRNA levels. This pipeline includes quality control using fastp, alignment with STAR, eRNA annotation based on the FANTOM5 project, and eRNA quantification using featureCounts. All detailed parameters defined by the developers are fully published14. In our work, to improve the eRNA annotation, we used a new annotation file (gtf) created based on the Human enhancer RNA Atlas (HeRA; https://hanlab.uth.edu/HeRA/). The HeRA database characterised the expression landscape of 45,411 eRNAs using 9,577 samples from 54 human tissues, incorporating enhancer data from FANTOM5, ENCODE, and the Roadmap Epigenomics Project15. All annotations and mappings were based on the human GRCh37/hg19 reference genome.
After obtaining the raw counts of eRNAs, we selected those located near the GATA3 and CRLF2 genes. To identify potential GATA3 regulatory regions, we defined a genomic range of approximately 640 kbp (chr10:8,085,705–8,457,148), covering both upstream (~ 193 kbp) and downstream (~ 427 kbp) regions. This selection revealed only four eRNAs annotated near GATA3, all overlapping with regulatory regions previously identified in the GeneHancer database16 (Track illustrated in Suppl. Fig. S3-A). The positions and codifications of these eRNAs are as follows: (i) chr10:8,136,909–8,142,910—eRNA_G1; (ii) chr10:8,207,762–8,213,763—eRNA_G2; (iii) chr10:8,443,562–8,449,563—eRNA_G3; and (iv) chr10:8,446,470–8,452,471—eRNA_G4. For the CRLF2 gene, we initially searched within a 200 kbp window (chrX:1,200,000–1,400,000) but did not identify any annotated eRNAs in this region. We then expanded our search to chrX:1,600,000–1,800,000 region (200 kbp), encompassing the P2RY8 gene. This region was highlighted by Yang et al. as a distal super-enhancer involved in a novel chromatin loop with the CRLF2 promoter in the presence of the GATA3 SNP2. Within this region, we identified four eRNAs, designated as follows: (i) chrX:1,597,593–1,603,594—eRNA_C1; (ii) chrX:1,599,492–1,605,493—eRNA_C2; (iii) chrX:1,600,392–1,606,393—eRNA_C3; and (iv) chrX:1,637,992–1,643,993—eRNA_C4 (Suppl. Fig. S3-A).
Quantification of gene and isoform expressions
The fastq files obtained from RNA-seq data were processed using a standardised pipeline for gene and isoform expression quantification. Quality control was assessed with FastQC, and adapter trimming was performed using Trimmomatic in paired-end mode, removing low-quality bases and sequencing artefacts (Parameters: ILLUMINACLIP:PE-adapters.fa:2:30:10:2:keepBothReads, SLIDINGWINDOW:4:20, MINLEN:36, LEADING:3, TRAILING:3). Reads were aligned to the human GRCh37/hg19 reference genome, and gene/isoform expression quantification was conducted using RSEM, with STAR as the aligner within RSEM under default settings. Expression levels were measured in transcripts per million (TPM) and log2-transformed (log2(TPM + 1)) for downstream analysis.
Quantification of CRLF2, GATA3 and eRNA_G3 in validation cohort
CRLF2, GATA3 and eRNA_G3 transcript levels were analysed by reverse transcription quantitative real-time PCR (RT-qPCR). The commercial TaqMan™ Gene Expression Assays Hs00845692_m1 and Hs00939627_m1 (Thermo Fisher Scientific) were used to target CRLF2 and the reference gene GUSB, respectively. GATA3 and eRNA_G3 gene expression were evaluated using the following primers: GATA3.forward: CACCACGGGAGCCAGGTG, GATA3.probe: CGCCCGCCTCTGCTTCATGG, GATA3.reverse: GGTGGATGGACGTCTTGGAG; eRNA_G3.forward: CATCTTGGAGCAGCATTCGC, eRNA_G3.probe: TAGTGCCCTCCCTGGTGCCC and eRNA_G3.reverse: CGTACATGAGCACCTGGTGT. cDNA material from all samples were tested in duplicates using TaqMan™ Fast Advanced Master Mix protocol (Applied Bio-systems, Foster City, CA). The amplification efficiency of the oligonucleotides was calculated for each transcript and evaluated based on the quantification cycle (Cq) values obtained from a set of five serial tenfold dilutions using high-quality cDNA. To evaluate the relative gene expression, the fold change was quantified by the 2 − ΔΔCt method referred to the median ΔCt of this set. RNA extraction, cDNA synthesis, qPCR assays, and subsequent analyses were performed following the quality control standards recommended by The Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines11,17. All experiments were performed in the QuantStudio™ 3 Real-Time PCR (Thermo Fisher Scientific) and analysed using the QuantStudio™ Design & Analysis Software Genotyping software (Thermo Fisher Scientific).
Characterisation of BCR::ABL1-like (Ph-like) patients
To characterise BCR::ABL1-like (Ph-like) patients in the TARGET cohort, we performed an unsupervised hierarchical cluster analysis based on the expression of target genes associated with primary molecular ALL subgroups, including BCR::ABL1 (Ph +), ETV6::RUNX1, hyperdiploidy, KMT2A-r, and TCF3::PBX1. The gene list was obtained from previous studies18,19. Some of TARGET samples were already characterised as non-Ph-like (n = 35) or Ph-like (n = 10) ALL cases using Predictive Analysis of Microarrays (PAM) as previously described9,20. To identify other Ph-like ALL cases, we evaluated the expression of eight genes strongly associated with the BCR::ABL1 (Ph+) ALL signature, i.e. JCHAIN (a.k.a. IGJ), SPATS2L, CA6, NRXN3, MUC4, CRLF2, ADGRF1 (a.k.a. GPR110), BMPR1B19. We also evaluated the gene expression profile associated with ETV6::RUNX1 (FUCA1, PTP4A3, CRMP1, EPOR, TCFL5, TNS1, TUSC3, BIRC7, KCNN1, and CLIC5), Hyperdiploidy (IL1B, PLP2, SH3BP5, HNRNPH2, MSN, ATP6AP2), KMT2A-r (HOXA9, MEIS1, MBNL1, KLRK1) and TCF3::PBX1 (FAT1, TRIB2, and ADARB1) to classify non-Ph-like ALL cases. These gene signatures were determined based on their overexpression profiles. The data standardisation process included a log2(x + 1) transformation of each gene expression value (TPM), followed by scaling (z-score normalisation). Genes were then hierarchically clustered using distance as 1-cor, being ‘cor’ the Pearson correlation coefficient, implemented through the stats CRAN R package v4.3.0. The unsupervised hierarchical clustering of samples was performed using Euclidean distance by the ‘pheatmap’ CRAN R package v1.0.1218. The number of clusters was determined based on visual inspection of the dendrogram, ensuring that the resulting groups captured distinct expression patterns relevant to the analysis. The clusters were refined using the ‘cutree’ parameter of the ‘pheatmap’ function. Therefore, we classified B-ALL patients as follows: i) Ph-like ALL if they clustered with groups associated with the Ph + gene signature but lacked the Ph + translocation (BCR::ABL1); and ii) non-Ph-like ALL if they clustered with groups associated with other B-ALL subtypes.
To characterise Ph-like patients in the validation cohort, B-other ALL patients were screened by fluorescence in situ hybridisation (FISH) for the detection of rearrangements associated with Ph-like-ALL, including CRLF2, ABL1, ABL2, JAK2, and IGH (ZytoVision), as well as PDGFRB, CSF1R and EPOR genes (Cytocell) using commercial break-apart FISH probes21. Both probe hybridisation and fluorescence signal detection were performed according to the manufacturer’s recommendations. Samples harbouring any of these gene rearrangements were considered Ph-like positive. Additionally, patients with available CRLF2 expression data from RT-qPCR were categorised into quartiles based on fold change values, with samples in the fourth quartile comprising the CRLF2-high subgroup and the remaining samples categorised as CRLF2-low. Therefore, B-other ALL patients showing CRLF2 overexpression were also considered Ph-like positive, even in the absence of one of the rearrangements mentioned above.
Statistical analysis
The Relative Risk (RR) Wald Test was used to evaluate the significance of the association between the GATA3 SNP risk allele (rs3824662) and BCR::ABL1-like (Ph-like) B-ALL. This analysis was achieved using the DescTools CRAN R package version 0.99.50. To investigate the association between genes and eRNAs, a correlation matrix was computed. The expression values underwent a log2(x + 1) transformation and the Pearson correlation coefficients with p-values were calculated using the Hmisc CRAN R package version 5.1–0. Boxplots were created using the ggplot2 CRAN R package v3.4.3. The Wilcoxon test was employed to compare gene, isoform and eRNA expressions between two groups at a time, with p-values computed using ggpurb CRAN R package v0.6.0. All analyses were conducted using R version 4.3.0 and p-values ≤ 0.05 were considered statistically significant.
Data availability
The results published here are wholly or partly based on data generated by the TARGET initiative, ALL Expansion Phase 2 (phs000464). The data used in this analysis are publicly available at https://portal.gdc.cancer.gov/projects.
References
Madzio, J. et al. GATA3 germline variant is associated with CRLF2 expression and predicts outcome in pediatric B-cell precursor acute lymphoblastic leukemia. Genes Chromosom. Cancer 58, 619–626 (2019).
Yang, H. et al. Noncoding genetic variation in GATA3 increases acute lymphoblastic leukemia risk through local and global changes in chromatin conformation. Nat. Genet. 54, 170–179 (2022).
Arnold, P. R., Wells, A. D. & Li, X. C. Diversity and emerging roles of enhancer RNA in regulation of gene expression and cell fate. Front. Cell Dev. Biol. 7, 377 (2019).
Léveillé, N., Melo, C. A. & Agami, R. Enhancer-associated RNAs as therapeutic targets. Expert Opin. Biol. Ther. 15, 723–734 (2015).
Zhang, Z. et al. Transcriptional landscape and clinical utility of enhancer RNAs for eRNA-targeted therapy in cancer. Nat. Commun. 10, 4562 (2019).
Teppo, S. et al. Genome-wide repression of eRNA and target gene loci by the ETV6-RUNX1 fusion in acute leukemia. Genome Res. 26, 1468–1477 (2016).
Heuts, B. M. H. & Martens, J. H. A. Understanding blood development and leukemia using sequencing-based technologies and human cell systems. Front. Mol. Biosci. 10, 1266697 (2023).
Wang, W.-T. et al. The lncRNA LAMP5-AS1 drives leukemia cell stemness by directly modulating DOT1L methyltransferase activity in MLL leukemia. J. Hematol. Oncol. 13, 78 (2020).
Roberts, K. G. et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 371, 1005–1015 (2014).
Kim, J. C. et al. Transcriptomic classes of BCR-ABL1 lymphoblastic leukemia. Nat. Genet. 55, 1186–1197 (2023).
Maciel, A. L. T. et al. IKZF1 deletions associate with CRLF2 overexpression leading to a poor prognosis in B-cell precursor acute lymphoblastic leukaemia. Transl. Oncol. 15, 101291 (2022).
Ma, X. et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 555, 371–376 (2018).
Carnevali, P. et al. Computational techniques for human genome resequencing using mated gapped reads. J. Comput. Biol. 19, 279–292 (2012).
Wu, Y. et al. Multi-omics analysis reveals the functional transcription and potential translation of enhancers. Int. J. Cancer 147, 2210–2224 (2020).
Zhang, Z. et al. HeRA: an atlas of enhancer RNAs across human tissues. Nucleic Acids Res. 49, D932–D938 (2021).
Fishilevich, S. et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford) 2017, bzx028 (2017).
Bustin, S. A. et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622 (2009).
Den Boer, M. L. et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: A genome-wide classification study. Lancet Oncol. 10, 125–134 (2009).
Reshmi, S. C. et al. Targetable kinase gene fusions in high-risk B-ALL: A study from the Children’s Oncology Group. Blood 129, 3352–3361 (2017).
Roberts, K. G. et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell 22, 153–166 (2012).
Schwab, C. J. et al. Genetic characterisation of childhood B-other-acute lymphoblastic leukaemia in UK patients by fluorescence in situ hybridisation and multiplex ligation-dependent probe amplification. Br. J. Haematol. 196, 753–763 (2022).
Acknowledgements
We thank the Multi-user platform of Bioinformatics and Computational Biology Laboratory for their help with data downloading and pre-processing. We are also grateful to Cristovão Antunes de Lanna, Thamyris Figueiredo Amaral and Marianna Dantas Villares for their invaluable contributions to some of the final analyses and data acquisition. The authors would like to extend their gratitude to the patients and their families who agreed to participate in this study, as well as to the clinicians who contributed to our research efforts. This study was supported by research grants from the Brazilian National Council for Scientific and Technological Development-CNPq (406641/2022-6; PQ-310982/2023-5) and Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro-FAPERJ (E-26/201.113/2021 and E-26/204.227/2024).
Author information
Authors and Affiliations
Contributions
TCB, CPP and ALTM performed the analyses, interpreted the results and wrote the paper. CBB and RLS performed the validation analyses and interpreted the results. MB and MBM supervised research and wrote the paper. ME supervised research, conceived and designed the experiments, and wrote the paper. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
da Conceição Barbosa, T., Poubel, C.P., Maciel, A.L.T. et al. Enhancer RNAs contribute to genome reprogramming driven by a GATA3 noncoding variant in leukaemia. Sci Rep 15, 29153 (2025). https://doi.org/10.1038/s41598-025-10262-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-10262-0
