
Abstract
Healthy senior dogs were subjected for 77 days to a dietary regime with (or without) prebiotic + postbiotic mixture composed by short-chain fructo-oligosaccharides and yeast fractions (scFOS+). Their fecal microbiota was studied and the links between the microbiota and immune parameters were investigated after Lyme vaccination. All along the study results showed a clear modulation of the microbiota with a higher relative abundance (RA) for Megamonas spp., Bacteroidaceae, Bacteroidetes plebeius, Clostridiales, Phascolarctobacterium, Succinivibrionaceae, Fusobacterium spp. in feces from scFOS + dogs. An amplicon sequence variants (ASVs) index (Enterobacteriaceae + Clostridium spiroforme vs. Fusobacterium + Megamonas) was developed and found significantly different between the groups with scFOS + dogs showing a lower index suggesting a possible modulation of the physico-chemical environment in the gut, favoring the growth of strict anaerobes producing short-chain fatty acids. Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt2) analysis revealed stimulation of propionate and acetate production pathways, vitamin biosynthesis (vitamin B2, B5 and B9 precursors) and pathways related to tricarboxylic acid cycle in the scFOS + group. The RA of several ASVs from Bacteroidetes and Fusobacteria families were found moderately negatively correlated to IgA and IgG concentrations (P < 0.05). In particular, Megamonas and Phascolarctobacterium appeared as interest genera. In summary, scFOS + is a good candidate to support the health of elderly dogs through microbiota changes. Metabolomics or in vitro mechanistic experiments will be crucial to further understand the mechanisms at play.
Similar content being viewed by others


Background
As humans, elderly pets are subjected to immunosenescence1 and may present an altered immune response with a reduced ability to respond to new antigens and a constant presence of low-level inflammation, characterized by higher concentrations of pro-inflammatory cytokines. In recent years, the role of gut microbiota in modulating immunity has become an important research topic. Researchers have observed in rodents and humans that there is an association between gut microbiome changes and host age, although the precise mechanisms underlying the association of aging and gut microbiome are still unclear2. Xu et al.3 reported that in dogs gut microbiota structure differed according to their age and they observed that supplementation with probiotics may change this structure and the immune functions, especially in elderly dogs. One hypothesis is that the gut microbiota can influence several important physiological and metabolic functions of the host and drive immune homeostasis. A study performed by Thevaranjan et al.4 using young and old germ-free, and conventional mice revealed that mice under germ-free conditions are less prone to age-associated inflammation, exhibiting less pro-inflammatory cytokines in the blood than conventional mice. They also demonstrated that intestinal permeability increases with age due to age-related microbiota modifications. Microbial products that enter the blood stream trigger systemic inflammation ultimately altering macrophage function; illustrating that “aging” microbiota may promote inflammation. Thus, there is a strong interest to understand the interplay between gut microbiota and immunity to support health of elderly pets by mitigating adverse effects of immunosenescence and inflammation.
Diet is a key modulator that affects the gut microbiome. As the link between the hallmarks of immunosenescence and nutrition is increasingly studied, we are now entering into an era where animal health can be improved through dietary interventions. The primary focus of dietary strategies based on prebiotics is to enhance the immune fate of the intestinal mucosa by modulating the gut microbiome. We previously demonstrated in a study with old healthy dogs that a recently patented prebiotic and postbiotic mixture (Profeed ADVANCED®, from here scFOS+) was able to modulate several traits of immunosenescence, notably the serum ratio of CD4+:CD8+ T cells and the serum concentration of IgA5. No significant effect was found on the immune response after Lyme vaccination, or when comparing blood cytokine concentrations, potentially due to the vast intra-individual variability while using a limited number of dogs. The objective of the current article is to describe the effect of scFOS + on the fecal microbiota of the dogs from this previous study by Wambacq et al.5.
Materials and methods
Animals, diets, and experimental design
The analysis of the gut microbiota was performed from samples obtained in a previous study5. Written informed consent was obtained from the owners prior to their participation in the study. The research protocol was reviewed and approved by the Ethical Committee of the Faculty of Veterinary Medicine, Ghent University, Belgium (EC 2017/103) and was in accordance with institutional and national guidelines and regulations for the care and use of animals. All the methods reported were in accordance with the ARRIVE guidelines.
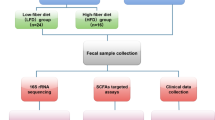
In brief, 22 client-owned senior dogs (following Fortney scale6) were randomly assigned to one of two groups (scFOS + versus Control), taking gender, age, body weight and body condition score into account (Supplemented Table 1). The first group of dogs received an extruded dry diet for adult dogs (formulated following National Research Council requirements7 as stated in Wambacq et al.5, with 21.1 ± 0.8 of crude protein, 8.0 ± 0.9 crude lipid, 2.0 ± 0.4 crude fiber, 6.6 ± 0.6 ash, 62.4 ± 1.1 Nitrogen-Free Extract and 1.6 ± 0.02 metabolizable energy (mean ± SD of dry matter %)), hereinafter referred to as ‘Control diet’, and the second group received the exact same diet supplemented before extrusion with 1.1% Profeed ADVANCED® (Beghin-Meiji, France), a compound feed combining scFOS prebiotic fibers with a new yeast postbiotic composed of various components from Saccharomyces and non-Saccharomyces strains (Beghin-Meiji; France), referred to as ‘scFOS+’ group for 14 consecutive weeks with a previous 7 days gradual transition to the new diet. Before the experiment started dogs were subjected to a complete health screening including a clinical examination, blood and urine analysis5, Lyme test was also performed (Canine Lyme Antibody Rapid Test, Abaxis Europe GmbH, Griesheim, Germany) and dogs were also dewormed (Caniquantel Plus, Fendigo sa/nv, Brussels, Belgium) 3 days before day 0. The inclusion criteria were: dogs should not be on any medication, they should be fed with dry food (kibbles) with body condition scores between 3 and 6 (0–9 score), have normal fasting blood outcomes and absence of protein in the urine, their core vaccination status should be completed from early life. Dogs accepted and tolerated very well the diets and there were no symptoms of gastrointestinal affection through the trial. The dogs were subjected to a first and a second Lyme vaccination at day 28 and 49, fecal and blood samples were collected at day 28 and day 77 (Fig. 1). Microbiota analyses were performed on a total of 16 dogs, due to five dogs could not finish the trial and few samples had low quality of DNA: 6 females and 2 males dogs for Control group, and 3 females and 5 males for scFOS + group.

Experimental design of the trial, adapted from Wambacq et al. (2024). 16 dogs were included after passing a health screening (blood, urine and lyme antibody test) and other inclusion criteria, and were dewormed 3 days before day 0. From day 4 to day 7 dogs per group were adapted to the experimental diet (Ccontrol or scFOS+) for 3 weeks until lyme vaccination at day 28 (collecting fecal and blood samples right before), then booster vaccination at day 49. The final fecal and blood samples were collected at day 77.
DNA extraction and PCR of the fecal microbiota on V3−V4 region of the 16S rRNA genes
Fecal samples were collected by dog owners 24 hours before the consultation at D28 (prior to vaccination) and D77. The collection was done within 15 minutes after defecation and samples were promptly stored at −20 °C in sterile containers. The frozen samples were then transported to the Veterinary clinic of Ghent University in cooled condition and were subsequently stored at −80 °C until further processing. Previously to DNA extraction the 200 mg of samples were subjected to a lysis step (100 mM Tris pH 8, 100 mM Na EDTA pH 8, 100 mM NaCl, 1% (w/v) polyvinylpyrrolidone, 1% PVP40, and 2% (w/v) sodium dodecyl sulfate), with 0.3 g of zirconium beads (0.1 mm diameter) for 3 min at 2000 rpm in PowerLyzer (Qiagen, Venlo, the Netherlands), then DNA was extracted with phenol/chloroform extraction at the Center for Microbial Ecology and Technology (Ghent University, Belgium). DNA concentration and quality were verified with 260/280 and 260/230 ratios, using a DeNovix DS (Thermo Fisher Scientific, Waltham, MA, United States). PCR was done with universal primers 341F (5’-CCT ACG GGN GGC WGC AG -3’) and 785Rmod (5’-GAC TAC HVG GGT ATC TAA KCC-3’) according to Klindworth et al.8. PCR products were verified by gel electrophoresis, purified using the Promega Wizard PCR clean-up kit (Promega, Madison, WI, United States) following the manufacturer’s instructions, AMPure XP beads (Beckman-Coulter, Krefeld, Germany) and quantified with the QuantiFluor dsDNA System kit (Promega, Leiden, The Netherlands). High-throughput amplicon sequencing of the V3-V4 hypervariable region of the 16S rRNA genes were performed on a Illumina Miseq at LGC genomics GmbH (Berlin, Germany).
Bioinformatics
Raw fastq files were imported, demultiplexed and processed using QIIME 2 (version 2020.2)9. Paired-ends fastq files were truncated by primer length, quality filtered and dereplicated through high resolution sample inference with DADA210. MAFFT and FastTree2 were used for de-novo alignment and phylogeny construction11,12. Taxonomy was assigned to the resulting 16S rRNA marker genes against Greengenes (gg-13-8-99-nb-classifier, trained with naive-bayes for 341 F-785R region of the 16S) using sklearn classifier method to determine the taxonomy13. Low frequency amplicon sequence variants (ASVs; <100 reads in < 2 samples) were removed previously to taxonomical statistical analysis. Alpha and beta diversity were rarefied computed at 14,500 sequences depth and visualized by box plots (indexes) and non-metric multidimensional scaling (NMDS) ordination plots and data ellipses of significant groups at 0.75 SD using ggplot214 and unweighted UniFrac distances. Compositional beta-diversity based on Aitchison distances15 was used to identify the discriminant ASVs with DEICODE16, then QURRO17 was used to test specific log ratios of discriminant ASVs based on Morton et al.18. The Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt2) approach was used with default parameters to evaluate the functional potential of microbial communities19, obtaining the Kyoto Encyclopedia of Genes and Genomes (KEGG) abundance table and extracting the function with MetaCyc-Pathway-Classes database.
Statistical analysis
Differences obtained between alpha-diversity indexes were evaluated with non-parametric Kruskal-Wallis ranking test independently for each day and longitudinally from day 28 to day 77 and Beta-diversity with PERMANOVA. Taxonomical differential analysis was performed with ANOVA of the communities (ANCOM)20 at phyla level and with ALDEx221 and longitudinal Kruskal-Wallis test22 at ASV level to evaluate the significance of the effect. All analysis were made with QIIME2. Spearman correlations were made in Python (version 3.7.6) to study relationships between immune parameters and alpha-diversity indexes of discriminating ASVs from ALDEx2 and DEICODE analysis. Results are represented by the mean value and standard deviation (SD) and standard error (SE) for PICRUSt2 representation. P-value was considered significantly different when P < 0.05, and trends when 0.05 < P < 0.1.
Results
A total of 1,249,303 sequences of 373 ± 17 base pairs were obtained from 29 samples in high-throughput sequencing. After denoising and chimera filtering 744,329 sequences corresponding to 668 ASVs were kept for down-stream analysis. After a conservative filtration, 248 ASVs corresponding to 698,254 sequences were kept for the taxonomical analysis. Rarefactions curves showed a plateau in observed ASVs and Goods coverage close to 1 from 5000 sequences, reflecting that microbiota was fully sampled (supplementary Fig. 1). Table 1 summarizes the blood, immune and fecal pH values from each group used for microbiota analysis from Wambacq et al.5. Immune parameters followed normal physiological ranges with typical individual variability. Further experimental details and interpretation of immune parameters were reported elsewhere5.
Fecal microbiota alpha-diversity
No treatment effect was observed on the different alpha-diversity parameters at D28. However, Shannon index and the number of observed ASVs significantly decreased 7 weeks after vaccination (D77) in dogs fed scFOS + when looking at transversal analysis on each day independently, but longitudinally paired Kruskal-Wallis analysis showed no differences between diets from day 28 to day 77, only a trend of Shannon and Evenness in the scFOS + group (P = 0.08) (Table 2).
Microbial composition: taxonomy
The fecal microbiota was composed of 5 phyla: Firmicutes (RA of 67.8 ± 20.24%), Fusobacteria [RA of 12.8 ± 14.0%), Bacteroidetes (RA of 11.1 ± 10.2%), Actinobacteria (RA of 3.8 ± 4.7%), and Proteobacteria (RA of 4.5 ± 6.7%); all these phyla being observed in all dogs but with high inter individual variability. At family level, Lachnospiraceae (21.0 ± 17.4%), Fusobacteriaceae (12.5 ± 13.7%), Clostridiaceae (16.0 ± 12.5%), Erysipelotrichaceae (8.8 ± 12.6%), Veillonellaceae (8.1 ± 13.0%), Streptococcaceae (5.4 ± 16.8%), Bacteroidaceae (5.1 ± 6.9%) contributed to more than 75% of the RA whatever the sampling time and treatment, in lesser extent (RA < 5% on average) Lactobacillaceae, Paraprevotellaceae and Enterobacteriaceae. 21 ASVs detected with high frequency in our samples represented almost 60% of the microbiota. Among them, Clostridium hiranonis (14.7 ± 12.1% RA; 28/29 dog frequency), unidentified genus from Fusobacteriaceae (9.28 ± 9.60%; 28/29), Megamonas spp. (6.28 ± 11.7%; 23/29), Blautia producta (4.43 ± 4.07%; 29/29), Bacteroides (3.95 ± 5.94%; 24/29), Ruminococcus gnavus (3.69 ± 4.65%; 29/29), Dorea spp. (3.46 ± 3.78%; 28/29), unidentified genus from Lachnospiraceae (3.18 ± 5.29; 28/29), Blautia spp. (2.96 ± 4.19%; 28/29), Fusobacterium spp. (2.29 ± 3.87%; 24/29), Blautia spp. (1.86 ± 2.15%; 26/29), Collinsella stercoris (1.76 ± 1.94%; 27/29) and Faecalibacterium prausnitzii (1.54 ± 2.15%; 22/29).
Microbial composition: differential analysis
Remarkably, significant differences (P < 0.05) at phyla level were found between treatments at day 77 with higher RA of Fusobacteria (10.4 ± 13.3% vs. 18.7 ± 17.4%), Bacteroidetes (7.5 ± 6.35% vs. 18.3 ± 9.8%), and Proteobacteria (1.79 ± 2.36% vs. 6.08 ± 6.25%) and lower abundance of Firmicutes (72.7 ± 16.7% vs. 55.5 ± 16.6%) and Actinobacteria (7.47 ± 9.98% vs. 1.26 ± 1.45%) in the scFOS + group vs. Control (P < 0.05; Fig. 2). ASVs related to Bacteroidaceae, Bacteroides, Bacteroides plebeius, Fusobacterium, Lachnospiraceae and especially Megamonas were significantly impacted, as well as other ASVs trend for a higher RA of ASVs related to Phascolarctobacterium, Succinivibrionaceae and unidentified Clostridiales (Table 3). When looking at data independently of time, most of the effects detected at D77 were significant, in addition to a trend for a higher RA of Prevotella copri P < 0.10; Supplementary Table 2). At day 28 only few trends could be observed (P < 0.10), a decrease in the RA of Enterobacteriaceae ASV and an increase in the relative abundance of Megamonas ASV in scFOS + group. Longitudinal comparison from day 28 to day 77 showed some trends between treatments in Turicibacter (P = 0.087) from 2.41 ± 3.69 to 0.42 ± 0.8% RA for scFOS+ (P = 0.068) where Control did not change (from 0.29 ± 0.46 to 0.32 ± 0.43% RA; P = 0.465) and in Enterobacteriaceae (P = 0.066) from 6.22 ± 14.15 to 0.07 ± 0.15% RA for Control (P = 0.080) where in scFOS + did not change (from 0.33 ± 0.85 to 3.44 ± 7.19% RA; P = 0.345). These changes were basically because of 2 dogs: a giant Lerse Wolfshond dog for decreasing Turicibacter (3 to 0.3%RA) while increasing Enterobacteriaceae (3 to 19% RA) in scFOS + and a large Witte herder dog for decreasing notably the Enterobacteriaceae from day 28 to 77 (44 to 0.5% RA) in Control.

Relative abundance (%) of the phylum in Control and scFOS + at D28 and D77. Asterisk (*) represents significant higher relative abundance at phyla level at D77 between treatments using ANCOM analysis (P < 0.05).
Microbial composition: beta-diversity
At day 77 the composition of the communities significantly differed between treatments in unweighted distances (Fig. 3A). On the other hand, Aitchinson PCA analysis was used to obtain the discriminant ASVs to differentiate significantly the treatments with both Axis 1 (80.7% variability explained) and Axis 2 (19.3% variability explained) (Fig. 3B). Axis 1 was associated with more Fusobacterium spp., Megamonas spp., Phascolarctobacterium spp., Bacteroides spp., Prevotella spp. (Paraprevotellaceae family) and less Enterobacteriaceae and C. spiroforme. Opposite to Axis 2 that was characterized by an opposition between C. hiranonis and the families of Lachnospiraceae, Erysipelotrichaceae, Ruminococcaceae, and Eubacterium dolichum. We assessed the ratio between discriminant ASVs according to the treatment and the time point (Fig. 4). A significant lower ASV ratios of the log (Enterobacteriaceae + C. spiroforme)/(Megamonas spp. + Fusobacterium spp.) was found with the scFOS + supplementation whatever the time point (P < 0.05) and trends were observed at both time points (P < 0.10) (Fig. 4B). Similarly, the log (C. spiroforme/Fusobacterium spp.) was significantly lower with scFOS + all along the study (P < 0.01), at D77 (P < 0.05) and tended to be different at D28 (P < 0.10). The same observation was obtained for the log (Enterobacteriaceae/Megamonas spp.) at day 28 (data not shown). All those ratios were also significantly correlated with the first axis of the Aitchinson PCA, corroborating that only with the Axis 1 the differences in the treatments were explained (P < 0.0001; Fig. 4C), thus clearly showing 2 different microbiota profiles.

Non-metric multidimensional scaling (NMDS) of unweighted Unifrac distances betweenCcontrol and scFOS + during the experiment. Ellipses show 1.5 SD at D77 (P < 0.05) (A). Principal component analysis (PCA) of Aitchison distances of the treatments per time point with weight of the discriminant ASVs represented by arrows length from DEICODE. Data points represent individual samples and vectors are taxa driving differences in ordination space (B).

Ratio of taxonomic differences between Control and scFOS+. Ranking of principal component analysis (PCA) Axis 1 taxonomic loadings highlighting the 2 highest features loading amplicon sequence variants (ASVs) (A). Box plots of natural log ratios (Enterobacteriaceae + C. spiroforme)/(Megamonas spp.+Fusobacterium spp.) and (C. spiroforme/Fusobacterium spp.) per treatment time points (B) and significant correlated with PCA Axis 1 (ρ: Spearman correlation coefficient) (C). Horizontal bar with * shows significant differences between treatments (including both D28 and D77) and at D77 (P < 0.05).
Correlations between immune parameters and alpha-diversity index
We found moderate correlations (Spearman correlations between 0.30 and 0.45) between immune parameters and alpha-diversity index. More precisely, the CD4+:CD8 + ratio tended to be negatively correlated with Shannon index (trend, ρ = −0.34; P < 0.10), Faith’s phylogenetic diversity (faith_PD; ρ = −0.42; P < 0.05) and evenness (Pielou; ρ = −0.37; P < 0.05).
Correlations between immune parameters and relative abundance of discriminant ASVs
Overall, the correlations between serum Ig and ASVs were moderate (0.30–0.60) to high (> 0.60), depending on the immune parameter considered.
Cytokines and ASVs
Negative correlations were found between IL6 and Lachnospiraceae (ρ = −0.592; P < 0.01), Blautia spp. (ρ = −0.561; P < 0.05), B. producta (ρ = −0.529; P < 0.05), Dorea (ρ = −0.465; P < 0.05) and Bacteroides (ρ = −0.426; P < 0.05).
Serum Ig and ASVs
Total serum IgG concentration was negatively correlated to Fusobacterium spp. (ρ = −0.421; P < 0.05), Bacteroidaceae (ρ = −0.487; P < 0.01), Bacteroides spp. (ρ = −0.485; P < 0.01), Prevotella spp. (ρ = −0.644; P < 0.01), while being positively associated with the log (C. spiroforme/Fusobacterium spp.) ratio (ρ = 0.606; P < 0.01). Serum Borrelia specific IgG was negatively associated with the Turicibacter (ρ = −0.676; P < 0.01). Total serum IgA was negatively correlated with Proteobacteria (ρ = −0.432; P < 0.05) and IgM positively correlated with Proteobacteria (ρ = 0.422; P < 0.05).
ELISPOT and ASVs
Total number of IgA ASC in serum was negatively associated with the Prevotella (ρ = −0.536; P < 0.05), Megamonas (ρ = −0.462; P < 0.05), Veillonellaceae family (ρ = −0.470; P < 0.05). When looking at the specific Borrelia IgA ASC, the % of covert area was positively correlated to Bacteroidetes (ρ = 0.591; P < 0.01), Bacteroidaceae (ρ = 0.640; P < 0.01), Bacteroides (ρ = 0.706; P < 0.01), Prevotella spp. (ρ = 0.713; P < 0.01), while the log (Enterobacteriaceae/Megamonas) ratio was negatively correlated (ρ = −0.627; P < 0.05). In addition, the ratio (Borrelia specific IgA ASC/ total number of IgA ASC) was negatively correlated with the log (Enterobacteriaceae/Megamonas) ratio (ρ = −0.975; P < 0.01), Blautia spp. (ρ = −0.706; P < 0.01) and B. producta (ρ = −0.633; P < 0.05) and positively associated with Veillonellaceae (ρ = 0.615; P < 0.05), Phascolarctobacterium (ρ = 0.686; P < 0.05) and Fusobacterium (ρ = 0.640; P < 0.05).
Fecal pH and ASVs
Fecal pH was positively correlated with Enterobacteriaceae (ρ = 0.487; P < 0.05) and negatively correlated with Phascolarctobacterium (ρ = −0.457; P < 0.05).
Microbial function: PICRUSt2
A total of inferential 306 distinct pathways were identified, with 32 showing significant differences between the microbiota of the Control and scFOS + groups (refer to Fig. 5). Among the 18 pathways upregulated with scFOS+, 4 were associated with vitamin B biosynthesis, 2 with vitamin K biosynthesis, 4 with the tricarboxylic acid cycle (TCA), 2 with propionate biosynthesis, and 1 with acetate biosynthesis. Conversely, among the 14 pathways overexpressed in the Control diet, 4 were involved in alcohol biosynthesis and 2 in formaldehyde metabolism.

Absolute abundance of significant different MetaCyc pathway classes found between treatments (including both D28 and D77) with PICRUSt2 (P < 0.05) using ALDEx2 analysis. Results are mean ± standard error.
Discussion
The addition of prebiotics and postbiotics in the diet of elderly pets is proposed as an interesting nutritional strategy to manage chronic inflammation and mitigate negative effects of immunosenescence23,24. However, to our knowledge, little is known on their effects on the gut microbiota of elderly dogs, in relation to their immune parameters. We thus studied the impact of an original combination of a prebiotic and postbiotic (Profeed ADVANCED®) on the fecal microbiota and the link with previously reported effects on immune parameters of healthy elderly dogs. We hypothesized that scFOS + could improve some hallmarks of immunosenescence through modulation of the gut microbiota and/or through direct immune-stimulation.
Our study allowed characterizing the gut microbiota of old dogs and shaping 2 different profiles
The fecal microbiota of the dogs was composed of five main phyla across the treatments. The phylum of Firmicutes emerged as the most dominant phylum, comprising 68% of relative abundance, followed by Fusobacteria at 13%, Bacteroidetes at 11%, and approximately 4% for Proteobacteria and Actinobacteria. This is consistent with recent studies involving dogs of varying ages25,26,27,28,29. The predominant families identified were Lachnospiraceae, Fusobacteriaceae, Clostridiaceae, Erysipelotrichaceae, Veillonellaceae, Streptococcaceae, Bacteroidaceae, Lactobacillaceae, Paraprevotellaceae, and Enterobacteriaceae. Notably ASVs associated with C. hiranonis, Fusobacteriaceae, Megamonas, Allobaculum, Bacteroides spp., Blautia (B. producta) and Lachnospiraceae accounted for over 50% of the total relative abundance and were prevalent among nearly all dogs. Despite the lack of extensive investigation into the microbiota of aging dogs to date30,31, a recent study involving 29 healthy companion dogs ranging from 3 to 13 years of age and representing various breeds28 reported a core microbiota consistent with our findings in elderly dogs.
We used a new approach, Aitchinson distances, in order to investigate the influence of ASVs on composition and interrelationship of fecal microbiota. Our findings suggest that Fusobacterium spp./Fusobacteriaceae, C. hiranonis, Lachnospiraceae, Erysipelotrichaceae, Blautia spp., Prevotella spp., Megamonas spp., Phascolarctobacterium spp. and Enterobacteriaceae are key contributors. Remarkably, our data revealed distinct bacterial profiles. Notably, C. hiranonis was an important contributor that was negatively associated with Erysipelotrichaceae, Lachnospiraceae and Ruminococcaceae and E. dolichum ASVs, while being positively associated with Blautia spp., B. producta and Fusobacteriaceae. Interestingly, these opposed bacteria may compete for the same substrates and share overlapping functions, like short-chain fatty acid (SCFA) production from carbohydrates or proteins. Furthermore, in humans E. dolichum has been linked to frailty32, while in dogs, C. hiranonis, Blautia spp., and Fusobacteriaceae are considered as key bacteria to assess microbial changes in subjects suffering from chronic inflammatory enteropathy33. In addition, C. hiranonis is associated with normal bile acid metabolism34. Bile acid metabolism is an important pathway not just for lipid digestion, but also for regulating intestinal inflammation, which is commonly altered in chronic gastrointestinal diseases, making this bacterium an interesting biomarker and a potential new probiotic candidate for dogs. Importantly, some bacteria belonging to the Erysipelotrichaceae family have also been described as key microbes in bile acid metabolism35,36. Those observations suggest that we shaped two profiles of microbiota, potentially competing for the same substrates and involved in similar functions, maybe depending on the phenotype/genetics/metabolism of the host. More research is needed to better understand the biological significance of these microbiota profiles on the host, as well as the impact of the host characteristics on those profiles.
The combination of prebiotic and postbiotic modulated the gut microbiota of aged dogs
ScFOS + diet led to microbial composition modulation, especially after 77 days of supplementation and following vaccination. At this time point we observed higher RA of Fusobacteria, Bacteroidetes and Proteobacteria and lower RA of Actinobacteria and Firmicutes phyla. Still, it remains difficult to interpret these results which could be partially attributed to the lower alpha-diversity. The proportion of Firmicutes respect to Bacteroidetes decrease with scFOS at both Day 28 and 77. The Firmicutes/Bacteroidetes ratio is considered as a potential biomarker of health in humans, this ratio has been reported to increase in humans fed with high levels of fibre37, tend to increase with age38, and be higher in obese and type 2 diabetes mice fed with live yeast39. In puppies Bacteroidetes was found to be associated with puppies weight at birth, being minimum in low birth weight puppies26. In addition, a decrease in fecal Bacteroidetes from 11 to 9% has been observed in dogs with chronic enteropathy40. We also reported a significant decrease in RA of Actinobacteria and an increase of Fusobacteria in ScFOS+. Interestingly a recent study performed to compare the microbiota of young adult vs. old dogs reported that despite the fact the main phyla remained unaffected, an increase in RA of Actinobacteria was associated with memory failures, and the RA of Fusobacteria phylum was negatively associated with age28.
At a deeper taxonomic level, all along the study and particularly at day 77, Megamonas spp., Bacteroidaceae, B. plebeius, Clostridiales, Phascolarctobacterium, Succinivibrionaceae, Fusobacterium spp. were found in higher RA in feces from scFOS + dogs. Most of these taxa are present in high abundance in fecal microbiota of healthy adult dogs from 0 to 10 years old26,27,41. In humans, some authors associated Bacteroides with celiac or IBD disease, however, the association must be done at a species level (and even at strain level)42 rather than at genus level. Indeed, infants with high genetic risk to develop celiac disease had a reduced prevalence of B. ovatus, B. plebeius, and B. uniformis, and an increased prevalence of B. vulgatus compared with infants with low genetic risk43, underlining the necessity to associate the microbiota evolution with metabolic/phenotypic traits of the microbes or the host. Interestingly, the genomes of Bacteroidetes contain polysaccharide utilization loci that encode the tools required to utilize complex carbohydrates, and certain Bacteroides encode proteins predicted to display α-mannosidase or α-mannanase activity, making them able to metabolize the major α-mannose-containing glycans from different yeast species44. This could explain why in our study Bacteroidetes harboured higher relative abundance in microbiota from scFOS + fed dogs.
As the beta-diversity significantly changed with scFOS+, we identified key bacteria that could explain change in the community structure. We discriminated the fecal microbiota structure from dogs fed scFOS + and Control diets by identifying which bacteria contributed to the discrimination. Fusobacterium spp. and Megamonas spp. on the one side (denominator), Enterobacteriaceae and C. spiroforme on the other side (numerator) significantly opposed the scFOS + and the Control groups, respectively. Genera Fusobacterium and Megamonas were associated with Phascolarctobacterium, Fusobacteriaceae and Prevotella spp. In addition, even if scFOS + did not affect the relative abundance of F. prausnitzii (1.2% and 1.9% for Control and scFOS+, respectively), the species was found to be associated with Fusobacteriaceae (data not shown). Unlikely to what is observed in humans, Fusobacterium in dogs is associated to a healthy microbiota31 and can produce SCFA from different amino acids. The genus Phascolarctobacterium spp. produces high amounts of acetate and propionate and can use succinate produced by other bacteria45,46. It is interesting to underline that this genus was associated with B. plebeius and Succinivibrionaceae, which are potential succinate producers. Wu et al.46) found a decrease in relative abundance of Phascolarctobacterium in elderly humans; while this genus has been associated with a positive mood47. Interestingly, a decrease in several species of Megamonas, P. copri and B. plebeius has been observed in dogs suffering from canine anal furunculosis48. Megamonas is an important propionate and acetate producer and is a common gut commensal microbe of carnivorous animals. Increased abundance of Megamonas have been found in healthy compared to diarrheic cats49 and in dogs consuming inulin-rich diets50. Based on these results, a greater emphasis on Veillonellaceae family and its potential impact on dog gastrointestinal health may be justified in the future.
The significant opposition obtained between the (Enterobacteriaceae + C. spiroforme) and (Megamonas + Fusobacterium) and the crosstalk existing between the different ASV to produce SCFAs, support the hypothesis that scFOS + could modulate the physico-chemical environment in the gut, leading to better conditions for the growth of strict anaerobes producing SCFAs, even after vaccination. The family of Enterobacteriaceae is composed of facultative anaerobe bacteria so that they take advantage of the oxygen available in the intestine. In addition, they efficiently use various substrates among which sugars and proteins. In agreement with our hypothesis, we obtained a positive correlation between the relative abundance of Enterobacteriaceae and fecal pH while being negatively correlated with Phascolarctobacterium. Taken all together, our results suggest that scFOS + resulted in stabilizing microbiota by favouring SCFAs producing bacteria using different substrates and cross-feeding. Although we did not measure SCFAs concentration, propionate is primarily produced by Bacteroidetes and some Firmicutes, among which Phascolarctobacterium51 and Megamonas spp. Of note, PICRUSt2 analysis revealed the stimulation of propionate and acetate production pathways in the scFOS + group.
Beside the activation of some acetate and propionate production pathways, functional inference revealed that adding scFOS + resulted in increased RA of KEGG pathways related to B vitamin biosynthesis, especially B2 (riboflavin), B9 precursor (pterin) and B5 and different pathways related to the TCA cycle. In healthy adult dogs on complete and balanced diets, vitamin B deficiencies are not common as commercial diets contain enough B vitamins. However, in case of chronic gastrointestinal disease (e.g. pancreatic insufficiency), dysbiosis or anemia, vit B12 and folic acid deficiency are common which could be the case of Senior dogs52. Bacterial vitamin B2 exists as free riboflavin and is directly absorbed in the large intestine. Vitamin B2 levels are important for T cell differentiation53. A vitamin B2 deficiency suppresses the activity of acylCoA dehydrogenases, involved in the oxidation of fatty acids to generate acetyl-CoA54; while B5 vitamin, pantothenate and phosphopantothenate are precursors of acetyl CoA55. Fatty acid oxidation is involved in the activation, differentiation and proliferation of immune cells through the generation of acetyl CoA and its entry into the tricarboxylic acid cycle54. In addition, the same authors reported how the balance between B2 and B9 vitamins was key to understand immune homeostasis. The commensal bacteria are both providers and consumers of B vitamins. Interestingly, P. copri, F. varium express factors essential for vitamin B2 and B9 synthesis, suggesting that these bacteria are important sources of microbial vitamin B2 and B9 in the large intestine. In addition, recent evidence indicates that some species in Bacteroidetes phylum produce more riboflavin (B2 vitamin) than Firmicutes or Actinobacteria56. Those observations are consistent with our findings. However, given the limitations of PICRUSt2 as solely a predictor of metagenomic function, a true experimental validation through is necessary to confirm these predicted functions.
How scFOS + could play a role to shape immune parameters through a gut microbiota modulation?
The last part of our work was dedicated to the analysis of some statistical correlations between different immune parameters and ASVs. Total serum IgM concentration was positively, while total serum IgA was negatively, correlated with higher Proteobacteria RA. The number of IgA secreting cells was negatively associated with Veillonellaceae family, Megamonas spp. and Prevotella spp. Finally, total serum IgG concentration was negatively associated with RA of Bacteroidaceae, Bacteroides, Prevotella and Fusobacterium while positively associated with the ratio between C. spiroforme and Fusobacterium. Overall, the microbiota as shaped with the scFOS + supplementation was correlated to less concentrations of total serum IgG and IgA, the latter being found significantly lower5. Total serum IgA and IgG contents increased with age and chronic inflammatory disorders in humans57,58, but also with age in dogs59. Thus, we could speculate that the supplementation with scFOS + allowed decreasing the pro-inflammatory status, in agreement with the numerically lower concentrations of IL6 (an Ig synthesis inducer) and IL17, and the significant increase in CD4+:CD8+ ratio5.
The specific immune response against B. burgdoferi vaccine was also investigated. In this case, serum Borrelia specific IgG concentration was negatively correlated with the genus Turicibacter, a genus formerly described as a butyrate-producer, decreasing with hemorrhagic diarrhea in dogs60 and in dogs suffering from chronic enteropathy33, as it seems the case of the giant dog fed with scFOS + in the present experiment. Interestingly, the Borrelia specific IgA covert area was positively correlated with Bacteroidetes, Bacteroidaceae, Bacteroides, Fusobacterium, Prevotella and negatively (ρ=-0.975) with the ratio between Enterobacteriaceae and Megamonas. We can hypothesize that the production of specific Borrelia IgA, but not the proliferation of the cells, was stimulated by the ASVs that were significantly modulated with scFOS+, even if the biological causation remains unclear. No correlation was found between the number of specific Borrelia IgA secreting cells and ASVs while the ratio between the number of IgA secreting cells over the number of Borrelia specific IgA secreting cells was positively associated with Veillonellaceae, Phascolarctobacterium and Fusobacterium; however being negatively associated with Blautia and B. producta. Those results confirm the potential interest of deeper investigating the functional role of Megamonas spp. and Phascolarctobacterium spp. not only for gut health indices, but also for more systemic health parameters in the canine species.
It is worth to note that this work is preliminary, and the interpretation should be taken cautiously. First, even if we applied strict exclusion and inclusion criteria, our canine cohort was small, resulting in a small sample size (due to the difficulty to recruit and follow a high number of elderly dogs) for statistical analysis. Second, the study was focused around Lyme disease vaccination, however the study lacks on microbiota baseline data at day 0. Thirdly, due to our limited sample size, the correlations we performed were Spearman correlations not adjusted by the multi-comparisons, so that we cannot exclude the presence of false-positives. Lastly, several of those correlations – but not all – were considered as moderate, thus comprised between 0.30 and 0.60. We therefore need to go further to deepen our conclusions, notably by increasing the sample size, and, ultimately, by going from a statistical relationship to a causation relationship, by, for example, designing specific in vitro models to decipher possible links between some ASVs and inflammation or even Ig induction effect. However, such kind of data obtained in elderly owned dogs is still very scarce, and we think that this work, although imperfect, paves the way for a better understanding between the microbiota, the inflammation and the immune response of elderly dogs.
Conclusion
This study showed how a new ingredient composed of prebiotic combined with specific yeast fractions (Profeed ADVANCED®) can shape the gut microbiota composition and community structure of elderly dogs and modulate the CD4+:CD8+ ratio and total serum IgA, known to decrease and increase with age respectively. To our knowledge, this is the first time that such a change in microbiota has been demonstrated in old dogs. There are some limitations to our study that could potentially impact on the interpretation of the findings among which the low number of dogs, absent of fecal microbiota day 0 and the lack of “sentinel” animals. Further research is required to better understand the impact on specific immune responses, but also on the functional impact of the change in microbiota. Notably, we could imagine going further immune cell phenotyping, to be able to see what type of cells have been changed with the prebiotic and postbiotic mixture, as well as metabolomics in both blood and feces. Also, validating the putative role of bacteria on immunity would deserve more attention by performing classical microbiology and in vitro mechanistic experiments. Having a complete picture of what happens in the holobiont will be crucial to further optimize the use of nutritional strategies to support elderly dogs.
Data availability
Sequence data that support the findings of this study have been deposited in the NCBI with the primary accession code PRJNA1183820.
References
Day, M. J. & Ageing Immunosenescence and inflammageing in the dog and Cat. J. Comp. Pathol. 142, S60–S69. https://doi.org/10.1016/j.jcpa.2009.10.011 (2010).
Claesson, M. J. et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 488 (7410). https://doi.org/10.1038/nature11319 (2012).
Xu, H. et al. Oral administration of compound probiotics improved canine feed intake, weight gain, immunity and intestinal microbiota. Front. Immunol. https://doi.org/10.3389/fimmu.2019.00666 (2019). 10https://www.frontiersin.org/journals/immunology/articles/
Thevaranjan, N. et al. Age-Associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell. Host Microbe. 23 (4), 570. https://doi.org/10.1016/j.chom.2018.03.006 (2018).
Wambacq, W. A. et al. A new combination of a prebiotic and postbiotic mitigates Immunosenescence in vaccinated healthy senior dogs. Front. Vet. Sci. 11, 1392985. https://doi.org/10.3389/fvets.2024.1392985 (2024).
Fortney, W. D. Implementing a successful senior/geriatric health care program for veterinarians, veterinary technicians, and office managers. Veterinary Clinics: Small Anim. Pract. 42 (4), 823–834. https://doi.org/10.1016/j.cvsm.2012.04.011 (2012).
National Research Council. Nutrient Requirements of Dogs and Cats (National Academies, 2006).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41 (1), e1. https://doi.org/10.1093/nar/gks808 (2013).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible Microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. https://doi.org/10.1038/s41587-019-0209-9 (2019).
Callahan, B. J. et al. DADA2: High-resolution sample inference from illumina amplicon data. Nat. Methods. 13 (7), 581–583. https://doi.org/10.1038/nmeth.3869 (2016).
Katoh, K., Misawa, K., Kuma, K., Miyata, T. & MAFFT A novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 30 (14), 3059–3066. https://doi.org/10.1093/nar/gkf436 (2002).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PloS One. 5 (3), e9490. https://doi.org/10.1371/journal.pone.0009490 (2010).
Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6 (1), 90. https://doi.org/10.1186/s40168-018-0470-z (2018a).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer-, 2016). https://ggplot2.tidyverse.org
Aitchison, J. The statistical analysis of compositional data. J. Roy. Stat. Soc.: Ser. B (Methodol.). 44 (2), 139–160. https://doi.org/10.1111/j.2517-6161.1982.tb01195.x (1982).
Martino, C. et al. A novel sparse compositional technique reveals microbial perturbations. mSystems 4 (1). https://doi.org/10.1128/mSystems.00016-19 (2019). e00016-19.
Fedarko, M. W. et al. Visualizing ’omic feature rankings and log-ratios using Qurro. NAR Genomics Bioinf. 2 (2), lqaa023. https://doi.org/10.1093/nargab/lqaa023 (2020).
Morton, J. T. et al. Establishing microbial composition measurement standards with reference frames. Nat. Commun. 10 (1), 2719. https://doi.org/10.1038/s41467-019-10656-5 (2019).
Douglas, G. M. et al. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38 (6), 685–688. https://doi.org/10.1038/s41587-020-0548-6 (2020).
Mandal, S. et al. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Disease. 26, 27663. https://doi.org/10.3402/mehd.v26.27663 (2015).
Fernandes, A. D. et al. Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2, 15. https://doi.org/10.1186/2049-2618-2-15 (2014).
Bokulich, N. A. et al. q2-longitudinal: longitudinal and Paired-Sample analyses of Microbiome data. mSystems 3:10.1128/msystems.00219 – 18. (2018). https://doi.org/10.1128/msystems.00219-18
Akatsu, H. Exploring the effect of probiotics, prebiotics, and postbiotics in strengthening immune activity in the elderly. Vaccines 9 (2). https://doi.org/10.3390/vaccines9020136 (2021). Article 2.
Sharma, R. & Padwad, Y. Probiotic bacteria as modulators of cellular senescence: emerging concepts and opportunities. Gut Microbes. 11 (3), 335–349. https://doi.org/10.1080/19490976.2019.1697148 (2020).
Coelho, L. P. et al. Similarity of the dog and human gut microbiomes in gene content and response to diet. Microbiome 6 (1), 72. https://doi.org/10.1186/s40168-018-0450-3 (2018).
Garrigues, Q. et al. Composition and evolution of the gut microbiota of growing puppies is impacted by their birth weight. Sci. Rep. 13 (1). https://doi.org/10.1038/s41598-023-41422-9 (2023).
Garrigues, Q. et al. The supplementation of female dogs with live yeast Saccharomyces cerevisiae var. Boulardii CNCM I-1079 acts as gut stabilizer at whelping and modulates immunometabolic phenotype of the puppies. Front. Nutr. 11, 1366256. https://doi.org/10.3389/fnut.2024.1366256 (2024).
Kubinyi, E., Bel Rhali, S., Sándor, S., Szabó, A. & Felföldi, T. Gut Microbiome composition is associated with age and memory performance in pet dogs. Animals: Open. Access. J. MDPI. 10 (9), 1488. https://doi.org/10.3390/ani10091488 (2020).
Mizukami, K. et al. Age-related analysis of the gut Microbiome in a purebred dog colony. FEMS Microbiol. Lett. 366 (8), fnz095. https://doi.org/10.1093/femsle/fnz095 (2019).
Fernández-Pinteño, A. et al. Age-associated changes in intestinal health biomarkers in dogs. Front. Veterinary Sci. 10, 1213287. https://doi.org/10.3389/fvets.2023.1213287 (2023).
Pilla, R. & Suchodolski, J. S. The role of the canine gut Microbiome and metabolome in health and Gastrointestinal disease. Front. Veterinary Sci. https://doi.org/10.3389/fvets.2019.00498 (2020). 6https://www.frontiersin.org/articles/
Jackson, M. A. et al. Signatures of early frailty in the gut microbiota. Genome Med. 8 (1), 8. https://doi.org/10.1186/s13073-016-0262-7 (2016).
AlShawaqfeh, M. et al. A dysbiosis index to assess microbial changes in fecal samples of dogs with chronic inflammatory enteropathy. FEMS Microbiol. Ecol. 93 (11), fix136. https://doi.org/10.1093/femsec/fix136 (2017).
Blake, A. B. et al. Altered microbiota, fecal lactate, and fecal bile acids in dogs with Gastrointestinal disease. PloS One. 14 (10), e0224454. https://doi.org/10.1371/journal.pone.0224454 (2019).
Kaakoush, N. O. Insights into the role of Erysipelotrichaceae in the human host. Front. Cell. Infect. Microbiol. https://doi.org/10.3389/fcimb.2015.00084 (2015). 5https://www.frontiersin.org/articles/
Martínez, I. et al. Diet-Induced alterations of host cholesterol metabolism are likely to affect the gut microbiota composition in hamsters. Appl. Environ. Microbiol. 79 (2), 516–524. https://doi.org/10.1128/AEM.03046-12 (2013).
Hirata, S. & Kunisawa, J. Gut microbiome, metabolome, and allergic diseases. Allergology Int. 66 (4), 523–528. https://doi.org/10.1016/j.alit.2017.06.008 (2017).
Yoshida, N. et al. Average gut flora in healthy Japanese subjects stratified by age and body mass index. Biosci. Microbiota Food Health. 41 (2), 45–53. https://doi.org/10.12938/bmfh.2021-056 (2022).
Everard, A., Matamoros, S., Geurts, L., Delzenne, N. M. & Cani, P. D. Saccharomyces boulardii administration changes gut microbiota and reduces hepatic steatosis, low-grade inflammation, and fat mass in obese and type 2 diabetic db/db mice. mBio 5 (3), e01011–01014. https://doi.org/10.1128/mBio.01011-14 (2014).
Minamoto, Y. et al. Fecal short-chain fatty acid concentrations and dysbiosis in dogs with chronic enteropathy. J. Vet. Intern. Med. 33 (4), 1608–1618. https://doi.org/10.1111/jvim.15520 (2019).
Burton, E. N. et al. Characterization of the urinary Microbiome in healthy dogs. PLOS ONE. 12 (5), e0177783. https://doi.org/10.1371/journal.pone.0177783 (2017).
Singh, R. P. Glycan utilisation system in Bacteroides and bifidobacteria and their roles in gut stability and health. Appl. Microbiol. Biotechnol. 103 (18), 7287–7315. https://doi.org/10.1007/s00253-019-10012-z (2019).
Sánchez, E. et al. Influence of environmental and genetic factors linked to Celiac disease risk on infant gut colonization by Bacteroides species. Appl. Environ. Microbiol. 77 (15), 5316–5323. https://doi.org/10.1128/AEM.00365-11 (2011).
Cuskin, F. et al. Human gut Bacteroidetes can utilize yeast Mannan through a selfish mechanism. Nature 517 (7533). https://doi.org/10.1038/nature13995 (2015).
Watanabe, Y., Nagai, F. & Morotomi, M. Characterization of Phascolarctobacterium succinatutens sp. Nov., an asaccharolytic, Succinate-Utilizing bacterium isolated from human feces. Appl. Environ. Microbiol. 78 (2), 511–518. https://doi.org/10.1128/AEM.06035-11 (2012).
Wu, F. et al. Phascolarctobacterium faecium abundant colonization in human Gastrointestinal tract. Experimental Therapeutic Med. 14 (4), 3122–3126. https://doi.org/10.3892/etm.2017.4878 (2017).
Li, L. et al. Gut microbes in correlation with mood: case study in a closed experimental human life support system. Neurogastroenterology Motil. 28 (8), 1233–1240. https://doi.org/10.1111/nmo.12822 (2016).
Maldonado-Contreras, A. et al. Dysbiosis in a canine model of human fistulizing crohn’s disease. Gut Microbes. 12 (1), 1785246. https://doi.org/10.1080/19490976.2020.1785246 (2020).
Suchodolski, J. S. et al. The fecal Microbiome in cats with diarrhea. PLOS ONE. 10 (5), e0127378. https://doi.org/10.1371/journal.pone.0127378 (2015).
Beloshapka, A. N. et al. Fecal microbial communities of healthy adult dogs fed Raw meat-based diets with or without inulin or yeast cell wall extracts as assessed by 454 pyrosequencing. FEMS Microbiol. Ecol. 84 (3), 532–541. https://doi.org/10.1111/1574-6941.12081 (2013).
Reichardt, N. et al. Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J. 8 (6). https://doi.org/10.1038/ismej.2014.14 (2019).
Stanley, E., Appleman, E., Schlag, A. & Siegel, A. Relationship between cobalamin and folate deficiencies and anemia in dogs. J. Vet. Intern. Med. 33 (1), 106–113. https://doi.org/10.1111/jvim.15348 (2019).
Shimizu, J. et al. Relative abundance of Megamonas hypermegale and Butyrivibrio species decreased in the intestine and its possible association with the T cell aberration by metabolite alteration in patients with behcet’s disease (210 characters). Clin. Rheumatol. 38 (5), 1437–1445. https://doi.org/10.1007/s10067-018-04419-8 (2019).
Yoshii, K., Hosomi, K., Sawane, K. & Kunisawa, J. Metabolism of dietary and microbial vitamin B family in the regulation of host immunity. Front. Nutr. https://doi.org/10.3389/fnut.2019.00048 (2019). 6https://www.frontiersin.org/articles/
Leonardi, R. & Jackowski, S. Biosynthesis of pantothenic acid and coenzyme A. EcoSal Plus. 2 (2). https://doi.org/10.1128/ecosalplus.3.6.3.4 (2007).
Tastan, C. et al. Tuning of human MAIT cell activation by commensal bacteria species and MR1-dependent T-cell presentation. Mucosal Immunol. 11 (6), 1591–1605. https://doi.org/10.1038/s41385-018-0072-x (2018).
Blanco, E. et al. Defects in memory B-cell and plasma cell subsets expressing different Immunoglobulin-subclasses in patients with CVID and Immunoglobulin subclass deficiencies. J. Allergy Clin. Immunol. 144 (3), 809–824. https://doi.org/10.1016/j.jaci.2019.02.017 (2019).
Gonzalez-Quintela, A. et al. Serum levels of Immunoglobulins (IgG, iga, IgM) in a general adult population and their relationship with alcohol consumption, smoking and common metabolic abnormalities. Clin. Exp. Immunol. 151 (1), 42–50. https://doi.org/10.1111/j.1365-2249.2007.03545.x (2008).
Glickman, L. T., Shofer, F. S., Payton, A. J., Laster, L. L. & Felsburg, P. J. Survey of serum iga, igg, and IgM concentrations in a large beagle population in which IgA deficiency had been identified. Am. J. Vet. Res. 49 (8), 1240–1245 (1988).
Ziese, A. L. et al. Effect of probiotic treatment on the clinical course, intestinal microbiome, and toxigenic Clostridium perfringens in dogs with acute hemorrhagic diarrhea. PLOS ONE. 13 (9), e0204691. https://doi.org/10.1371/journal.pone.0204691 (2018).
Author information
Authors and Affiliations
Contributions
M.H., E.A., C.L.B., F.B., E.C. and W.W. designed the experiment and protocols, W.W. collected the samples and performed DNA extraction. A.R. performed DNA-based bioinformatics and statistical analyses. A.R. and E.A. wrote the first draft, A.R. designed the figures and wrote the final manuscript after reviewing. All authors contributed to the writing and accepted the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rodiles, A., Wambacq, W., Le Bourgot, C. et al. Supplementation of a new combination of prebiotic and postbiotic shapes fecal microbiota of old dogs while influencing immune parameters. Sci Rep 15, 28447 (2025). https://doi.org/10.1038/s41598-025-10280-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-10280-y
