
Abstract
The critical role of gut microbiota in human health and disease has been increasingly illustrated over the past decades, with a significant amount of research demonstrating an unmet need for self-monitor of the fecal microbial composition in an easily-accessible, rapid-time manner. In this study, we employed a tool for Smartphone Microbiome Evaluation and Analysis in Rapid-time (SMEAR) that uses images of fecal smears to predict microbial compositional characteristics in a human cohort. A subset of human fecal samples was randomly retrieved from the second wave of data collection in the Healthy Life in an Urban Setting (HELIUS) study cohort. Per sample, 16S rRNA gene sequencing data was generated in addition to an image of a fecal smear, spread on a standard A4 paper. Metagenomics-paired pictures were used to validate a computer vision-based technology to classify whether the sample is of low or high relative abundance of the 50 most abundant genera, and α-diversity (Shannon-index). In total, 888 fecal samples were used as an application of the SMEAR technology. SMEAR gave accurate predictions whether a fecal sample is of low or high relative abundance of Sporobacter, Oscillibacter and Intestinimonas (very good performance, AUC > 0.8, p-value < 0.001, for all models), as well as Neglecta, Megasphaera, Lachnospira, Methanobrevibacter, Harryflintia, Roseburia, and Dialister (good performance, AUC > 0.75, p-value < 0.001, for all models). Likewise, SMEAR could classify whether a fecal sample was of low or high α-diversity (AUC = 0.83, p-value < 0.001). Our study demonstrates that SMEAR robustly predicts microbial composition and diversity from digital images of fecal smears in a human cohort. These findings establish SMEAR as a new benchmark for rapid, cost-effective microbiome diagnostics and pave the way for its direct application in research settings and clinical validation.
Similar content being viewed by others

Introduction
The critical role of the gut microbiota in human health and disease has been increasingly illustrated over the past decades1. Strong associations between the human gut microbiota and obesity2, inflammatory bowel disease (IBD)3, liver cirrhosis4, and depression5 are established. These associations and potentially causal relationships would be better outlined through studying the fecal and mucosal-adherent gut microbiota in a more longitudinal, repetitive, and less cumbersome manner. In this study, we employed a previously presented machine-learning (ML) approach6, hereafter titled Smartphone Microbiome Evaluation and Analysis in Rapid-time (SMEAR), to monitor microbial genera and diversity in a human population cohort based on 16S rRNA gene sequencing.
Exploring the mucosal-adherent microbiota requires invasive and costly sampling during an endoscopy, and is thus limited to research purposes. Exploring the fecal microbiota is easier, sampling-wise, and has evolved from the traditional culture-dependent to culture-independent techniques using next-generation sequencing7. However, despite improvements regarding the overall microbial resolution, including within- and between-samples diversity, as well as the strain identification, both the wet-lab pre-processing of the fecal samples, and the dry-lab computational analysis of the output sequences remain time-consuming steps that limit their use in clinical settings8.
Currently, two main bioinformatics approaches are being employed to study associations between the human health or diseased conditions and the gut microbiota; a strain-level exploration that requires deep sequencing and sophisticated tools to succeed assembly of the gut microbial strains7, or a population-level comparison that requires large cohorts to define clusters of people depending on their projection on a reduced-dimensions principle coordinate plot, namely enterotypes9. Regardless of the approach of choice, the common confounding variable of the fecal microbiome remains the fecal water content, or differently, moisture content10.
In this regard, the fecal water content has been associated with the within-sample α-diversity, and the relative abundance of key enterotype-drivers, including the genus of Prevotella11. Moreover, it has been visually evaluated with the means of the Bristol Stool Form Scale (BSFS) for almost 30 years12. Here, we wish to elevate the usefulness of visual traits in microbial analysis using computer-vision (CV) techniques, namely deep-learning. Deep-learning has been increasingly adopted across healthcare domains13, allowing us to hypothesize that its use in fecal microbial analysis might be successful. If so, such a technology might improve the practicability of microbial analysis in real-world settings. As inferring a deep-learning model takes seconds, this can be done anywhere in the world with access to a computer, and it is several orders of magnitude more cost-effective than the traditional microbial analysis methods, this work serves as a stepping stone towards such a goal.
Recently, we reported the application of an image guided microbial profiling ML technology based on fecal samples that were profiled using shotgun metagenomics to assess proof of concept in rhesus macaques (Macaca mulatta)6. In the current paper, we expand on this study and aim to evaluate whether a digital image of a fecal smear can provide insight into elements of the human fecal microbial composition. To this end, we apply SMEAR on human fecal samples that were profiled using 16S rRNA gene sequencing. SMEAR extracts information from a digital image of a fecal smear, and classifies whether the sample is of low or high relative abundance of microbial taxa at genus level, and respectively of low or high α-diversity.
Material and methods
Cohort
This study was conducted on a subset of samples from participants of the Healthy Life in an Urban Setting (HELIUS) study, a prospective multi-ethnic population study designed to investigate the causes of unequal disease burdens across ethnic groups living in Amsterdam, The Netherlands14,15,16. The study recruited participants aged 18–70 years by randomly sampling the municipal registry whilst stratifying for ethnicity. At baseline (2011–2015), participants, among others, underwent physical examination and were asked to collect a morning stool sample at home. The second wave of data collection took place between 2019 and 2022. All baseline participants who were still alive and living in the Netherlands were re-invited. Similar to the baseline data collection, the second wave consisted, amongst others, of a physical examination and the collection of fecal samples.
During the follow-up data collection, a total of 8,000 samples was collected. From this total pool, 888 stool samples were made available for the current study in a completely random selection manner. The HELIUS study is a population study. As such, it consists of both healthy and diseased participants.
Written informed consent was obtained from all participants, and the study received approval from the medical ethical review board of the Amsterdam UMC, location AMC. For more specific details regarding inclusion/exclusion of the HELIUS study, we would like to refer the reader to previous papers on the HELIUS study14,15,16. All research was performed in accordance with the relevant guidelines/regulations.
Fecal sample collection
After collection, all feces were temporarily stored at -20 °C until transportation to the Amsterdam University Medical Center, location AMC. Here, the samples were checked by a study nurse and stored at -80 °C. Eventually, a small fecal fraction of every sample was used to isolate DNA for the purposes of 16S rRNA gene sequencing analysis, and a second fraction was processed for image acquisition purposes. Images of the fecal samples were taken before the sequencing results were available, hence preventing bias in sample selection.
DNA extraction and 16S rRNA gene sequencing
DNA was extracted from 150 mg of every fecal sample and 16S rRNA gene amplicons were generated using a single step PCR protocol targeting the V4 region, as earlier reported14,16. For the purposes of the present CV tool evaluation, 16S rRNA gene sequencing was used as a benchmark method to define (a) the targets of SMEAR (i.e. the 50 most abundant taxa at genus level, see “Predicted microbes and diversity”), and (b) the labels of high or low (see “Thresholds definition”). The complete metabarcoding approach was earlier presented in detail by van der Vossen et al.16. For the purposes of the current study, it is important to note that the taxonomic annotation of the amplicon sequence variants (ASV) was performed using the Ribosomal Database Project (RDB) classifier and database (v18)17, and it was the count table at genus level that was further used.
Image acquisition and segmentation
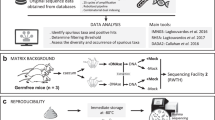
A small piece of frozen stool was thawed and carefully spread on a standard, white, pre-labelled sheet of A4 paper. The smear was created with the help of a long cotton tipped applicator (HARTMANN, 9670950, 14 × 11 mm) to fully cover a square of 36 cm2 (6 × 6 cm) in the middle of the paper, without empty spots in the coverage surface. Two subsequent pictures were taken using two different magnification levels with the means of a mobile phone camera (12 megapixels). The first picture captured the complete surface of the paper, including the label at the top right corner to facilitate the sample’s identification. The second picture was taken as such that it encapsulates as much of the 36 cm2 square with the fecal material spread as possible. The former was used to verify the match between smear images and participants, whilst the latter was used as input for SMEAR. All 888 fecal samples were processed in a random order by the same researcher to assure reproducibility in the smearing as well as the image acquisition steps.
In digital image processing and CV, segmentation refers to the process of extracting the desired subject, here referring to the fecal smear, from a medical image. It is an essential process for image analysis and generally considered an intermediate step of pattern recognition applications18. Here, after image acquisition, the images were segmented to isolate the fecal smear from the surrounding white paper background (Supplementary Fig. 1A). To limit inter-observer variability in the segmentations, we employed an automatic segmentation method using the “select subject” tool as made available in Adobe Photoshop 2022 to obtain a binary segmentation mask19. After obtaining the segmentations, the biggest possible square that can be selected from the smear was selected and then rescaled to 900 × 900 pixels with bilinear interpolation. Supplementary Fig. 1A illustrates the segmentation and preprocessing pipeline of an example fecal smear.
For the prediction of the microbial composition, all images were made of a uniform size of 224 × 224 pixels. For the training set, a random resized crop with random horizontal and vertical flip was used. For the test set, the images were resized in a deterministic manner by rescaling to 256 × 256 pixels and then center cropped to 224 × 224 pixels; normalization per (red–green–blue) RGB color channel used the mean and standard deviations (SD) of the original ImageNet dataset20.
Predicted microbes and diversity
As this study researches a novel approach of the application of CV to the field of human fecal microbiota, we focused on identifying which markers can be predicted using this technology, and where its limitations lay in as primary readout. We calculated the relative abundance of all taxa in the complete 16S rRNA gene sequencing-derived matrix count table (n = 888 human fecal samples) at genus level, and ranked the genera based on their average relative abundance per sample. Likewise, we defined which are the 50 most abundant genera in our human cohort, and performed an exhaustive search of SMEAR’s performance on these 50 genera. They should represent a large fraction of the variance within the microbiome and thus provide insight into the most important variations that occur within the general human population. Following up on the primary microbial taxa-based results, α-diversity was calculated with the means of the Shannon-index and explored as a secondary readout of SMEAR.
Thresholds definition
SMEAR was developed to classify high versus low relative abundance samples of each of the inspected outcomes. As such, no direct prediction of the values of the relative abundance was calculated. The top and bottom 25% of all human fecal samples were categorized as being in the high or low group, respectively; samples falling in the middle 50% were excluded from the analyses.
In detail, after calculating the relative abundance of all genera, and defined the 50 most abundant in out cohort, we generated one density distribution plot per genus (for the 50 most abundant genera): the 16S rRNA gene sequencing-based relative abundance of every genus was plotted in a log-scale in the x axis, and the number of samples corresponding to every bin was corresponded in the y axis (distribution plots not shown). Per genus, the bottom 25% of the samples was categorized in Label 0 = Low and the top 25% of the samples was categorized in Label 1 = High. This threshold allowed us to capture a significant portion of samples with distinctly different microbial characteristics.
Computational analysis
Image analysis was conducted using the Tiny Vision Transformer, an efficient and compact model of the Vision Transformer (ViT) family, enhanced with Knowledge Distillation21, implemented using the PyTorch image library TIMM22. A pre-trained model on the ImageNet-21 k23 dataset, which consists of 14 million images, was used. The training of the model involved 50 stratified folds to ensure balanced classes. Each fold was independently cross-validated with 10% of samples set apart as independent test data only used as evaluation, allowing for a comprehensive performance assessment and standard deviation analysis which takes the stochasticity of the results into consideration. The training utilized the AdamW optimizer and binary cross-entropy loss function. Fine-tuning was focused on the final dense head layers, with performance measured via the Area Under the Receiver Operating Characteristic curve (AUC), a graph showing the performance of a classification model at all possible threshold values. To enhance the method’s robustness, generalizability, and prevent overfitting, several advanced techniques were employed. Early stopping was implemented to prevent excessive training, while dropout regularization (with a dropout rate of 50%) and ReduceLROnPlateau learning rate scheduling were utilized based on the training data, ensuring optimal convergence and generalization.
To evaluate the robustness of SMEAR predictions, we conducted permutation tests by randomly shuffling the labels of the fecal images while keeping the images themselves unchanged. This process was repeated 200 times to create a distribution of the model performance under the null hypothesis, where no genuine association exists between the fecal images and their corresponding labels (0 = Low; 1 = High). By comparing the actual model performance to this null distribution, we assessed the statistical significance of our results. This approach ensured that our model’s predictive power was not due to random chance but rather to meaningful patterns in the data.
Analysis and visualization were performed using Python 3.10, utilizing PyTorch, Scikit-learn, Scikit-image and Pillow.
Results
Participants
Eight hundred eighty adults were randomly selected from the second wave of data collection of the HELIUS study and used in the current analysis. Their age, sex, ethnicity, body mass index, diabetes and hypertension distributions are shown in Table 1. This sample (n = 888) is a cross selection of the general HELIUS population, rather than a patient or healthy selection.
Prediction of the microbial taxa
The average AUCs (higher is better) for the 50 most abundant genera are shown in Fig. 1A. Across the assessed genera, SMEAR showed the highest performance (AUC, mean ± SD) for Sporobacter (0.85 ± 0.03), Oscillibacter (0.84 ± 0.04) and Intestinimonas (0.81 ± 0.04). These performances can be regarded as very good. Other notable genera are Neglecta, Megasphaera, Lachnospira, Methanobrevibacter, Harryflintia, Roseburia & Dialister, which all showed average AUCs > 0.75, and can be regarded as good performances. Together, these ten genera with AUCs > 0.75 are hereafter termed Top10-performing genera.

SMEAR’s prediction of microbial taxa. (A) Average classification performance (AUC) for the 50 most abundant genera of the fecal image dataset. (B) ROC curves of the Top10-performing genera, with ROC curves of the permutation test results: Sporobacter, Oscillibacter, Intestinimonas, Neglecta, Megasphaera, Lachnospira, Methanobrevibacter, Harryflintia, Roseburia and Dialister.
The permutation test confirmed that the classification results were statistically significant for all Top10-performing genera (p-value < 0.001, for all models), verifying the robustness of SMEAR’s predictions (Fig. 1B). The hierarchical clustermap in Fig. 2A shows the co-occurrence patterns of the 50 most abundant genera based on Spearman correlations. Of note, six of the Top10-performing genera, namely Sporobacter, Oscillibacter, Methanobrevibacter, Harryflintia, Intestinimonas and Neglecta group together in a clear cluster; Dialister groups next to Megasphaera and Lachnospira next to Roseburia (all highlighted in red font in Fig. 2A). This indicates a common pattern in the fecal smear images that SMEAR is able to pick up on.

SMEAR’s prediction of ɑ-diversity. (A) Clustered heatmap of the Spearman rho correlation coefficients of the top 50 most abundant genera. The Top10-performing genera lie close together in the clustermap, indicating that these genera co-occur together. (B) Principal Coordinate Analysis (PC0A) plots for ASV-level clustering colored by the log relative abundance of the Top10-performing genera. Samples with high levels of specific genera are placed lower and central in the plot. (C) Principal Coordinate Analysis (PC0A) plots for ASV-level clustering colored by ɑ-diversity (Shannon-index). Samples with higher ɑ-diversity are placed lower and central in the plot. (D) Scatterplot of the aggregated relative abundance of the Top10-performing genera and ɑ-diversity. Spearman rho = 0.503, p-value < 0.001. (E) ROC curve and AUC calculation of the ɑ-diversity experiment. SMEAR’s performance was similar to that of the Top10-performing genera from previously presented exhaustive search.
Top10-performing genera and α-diversity
Figure 2B shows a principal coordinate analysis (PCoA) scatter plot highlighting the distribution of the samples in a lower dimensional space whilst maintaining as much of the distances (Bray–Curtis dissimilarity) between samples as possible. Every dot is one sample and has been colored by the averaged relative abundance of the Top10-performing genera. As can be seen, the decomposition organizes the samples into a characteristic ‘chevron’ shape that is often observed in PCoA plots of fecal metagenomics data24. Samples with higher aggregated relative abundance of the Top10-performing genera are placed more downward and central, whilst samples with lower aggregated relative abundance are placed on the outer wings of the shape (Fig. 2B). This shows that, with the current high-low thresholding approach, the best performing model can distinguish between samples that are placed central from the ones placed in the outer wings of the chevron shape. Typically, this distribution of fecal microbial compositions on a PCoA plot (i.e., central versus outer) is aligned with α-diversity, which seemed to be the case in the present dataset as well (Fig. 2C). In Fig. 2C, the PCoA is recolored in a continuous scale by the levels of α-diversity, and indeed resembles Fig. 2B. Likewise, the direct Spearman rho correlation between the aggregated relative abundance of the Top10-performing genera and the α-diversity was found to be significant (Spearman rho = 0.503, p-value < 0.001), although the correlation coefficient was not exceptionally high (Fig. 2D).
Prediction of α-diversity
SMEAR was used to predict high-vs-low α-diversity based on the same set of fecal smear images. As expected, when considering the aforementioned results, the model performed (AUC, mean ± SD) very similarly to the ones trained on the Top10-performing genera (0.83 ± 0.02). Additionally, permutation testing gave similar results as for the Top10-performing genera (p-value < 0.001, Fig. 2E).
Relationship between clinical and microbial characteristics
In addition, to link predictions of SMEAR to general health, we investigated the association between Shannon diversity and the aggregated relative abundance of the Top10-performing genera to participant’s age and BMI. Shannon-index was significantly correlated with BMI (Spearman rho = -0.113, p-value < 0.001), but not with age (Spearman rho = 0.048, p-value = 0.150). Moreover, the aggregated relative abundance of the Top10-performing genera was not significantly correlated to BMI (Spearman rho = -0.054, p-value = 0.110) or age (Spearman rho = -0.034, p-value = 0.310). These results are visualized in Supplementary Fig. 2A-D.
Visual inspection of images of extreme samples
Although the AUCs show that SMEAR is capable of distinguishing between high and low α-diversity samples, it might prove useful to visually inspect what patterns can be observed by (human) visual scrutiny. It is worth noting that there can be many important features that the model might pick up on, including the number and size of clumps, and strokes, the color, and fecal water content. These and other characteristics collectively contribute to the model’s ability to differentiate between high and low diversity samples.
While color is an important factor, it is only one of many visual traits that might distinguish high and low diversity samples. The samples with the 10 highest and lowest α-diversity are shown in Supplementary Fig. 1B. As can be observed in the figure, samples with high α-diversity generally have a darker color. In contrast, samples with lower α-diversity appear wetter and have a lighter, more beige color. However, the combination of these various features, rather than color alone, aids in distinguishing the samples and reflects the complex nature of visual patterns in this context.
Discussion
This study supports an earlier application using image guided analyses to monitor the fecal microbiome composition determined through 16S rRNA gene sequencing. Here, we extend these observations in a large multi-ethnic human cohort for the first time. SMEAR could discriminate with a very good performance (AUCs > 0.80, p-value < 0.001), whether a fecal sample was of low or high relative abundance of Sporobacter, Oscillibacter, and Intestinimonas. Likewise, a good performance (AUCs > 0.75, p-value < 0.001) was observed for prediction of low or high relative abundance of Neglecta, Megasphaera, Lachnospira, Methanobrevibacter, Harryflintia, Roseburia and Dialister. These 10 genera together formed the so-called Top10-performing genera. Most Top10-performing genera compiled a clear co-occurrence cluster on an hierarchical clustered heatmap. When their relative abundance was aggregated, the enriched in Top10-performing fecal samples clustered separately at the lower end of the axis PC2, like the samples with high α-diversity, measured with the Shannon-index. To this end, SMEAR was challenged to predict samples with high or low α-diversity, and did so with a very good performance (AUC = 0.83, p-value < 0.001).
Earlier studies have succeeded in relating the visual traits of fecal samples to evaluate microbial alterations. BSFS for example was used to detect relevant fecal composition changes in irritable bowel syndrome (IBS), and could successfully differentiate between healthy volunteers and IBS-diarrhea predominant patient stools with an 81% accuracy25. Similarly, an earlier study from our group (Lee D, et al. iScience, 20246) showed the potential of image-based CV tools, applying an image-guided CV tool onto fecal samples from rhesus macaques that were profiled using shotgun metagenomics, respectively to how in the current study, we apply SMEAR onto human fecal samples that were profiled using 16S rRNA gene sequencing.
Important improvements were noted in our human 16S rRNA gene sequencing-based application of the SMEAR model, as we now successfully (AUC > 0.75, p-value < 0.001) predict whether a sample is of high or low relative abundance of 10 distinct genera (i.e. Top10-performing genera), where this was only possible for two genera in the earlier study6. Moreover, SMEAR can predict with a better performance (AUC = 0.83 vs. AUC = 0.72, for SMEAR vs. earlier study, respectively) whether a fecal sample is of high or low α-diversity. This discrepancy may be likely due to the smaller sample size, or different host species used. Irrespectively, this is the first time that accurate performance in a human cohort has succeeded.
Our primary read-out was that SMEAR can successfully predict whether a fecal sample is of low or high relative abundance of Sporobacter, Oscillibacter and Intestinimonas, a Gram-positive and two Gram-negative taxa, respectively, that harbor significant bile acid conversion ability26. Our results indicate that SMEAR may detect the conversion of primary to secondary bile acids by Gram-positive bacteria, a process that directly causes the darkening of fecal smears in high-diversity samples. This mechanistic insight confirms the predictive validity of our model. However, wet-lab experiments are required to prove this speculation. Considering that Sporobacter and Oscillibacter were previously found significantly enriched in fecal samples of rural populations, who consume high fiber diets, avoid processed foods, like the Hadza hunter-gatherers27, and that at functional level these bacteria, as well as Roseburia, are prominent butyrate producers, a short chain fatty acid essential for maintaining gut barrier integrity and immune responses28, it is very important to be able to track their relative abundance in a simple, non-costly way.
Our secondary read-out was that SMEAR can accurately classify patients with low or high α-diversity. This is a crucial asset of SMEAR, with significant potential for clinical practice, as highlighted by the recent multidisciplinary expert panel’s statement on microbiome testing in clinical practice: “Statement 15: The microbiome analysis should include alpha diversity metrics, including richness and evenness”. Albeit the long-lasting debate around the use and meaning of the term dysbiosis, it is still well appreciated that a low α-diversity associates with a worsened symbiosis, and several gastro- and extra-intestinal disorders are associated with a dysbiotic-like microbial composition2,3,4,5,29. In our dataset, we found a weak, yet significant correlation between α-diversity and BMI. This shows that α-diversity and BMI are associated, and with larger studies, SMEAR might be used to evaluate clinical phenotypes in addition to microbial biomarkers.
Fecal smear images with the highest and lowest α-diversity were visually assessed to explore traits SMEAR might identify. Importantly, variations in gut community structure directly correlate with total microbial abundance as shown by the quantitative framework linking gut microbiome composition to microbial load30. Also, low α-diversity samples appeared to have higher water content evidenced by prominent moisture marks that align with findings by Kim and Whon (2021), who linked fecal water content to stool consistency improvements following FMT in animals31. To test whether SMEAR predictions are influenced by water content, future experiments could include using SMEAR to predict BSFS scores. Additionally, controlled ‘water-spike’ experiments (adding known volumes of water to smears) could quantify how moisture alone drives AUC changes. However, we note that it is unlikely that spiking fecal samples with additional water would effectively mimic the fecal ecology well. Earlier studies focused on creating an automated system for stool recognition and characterization32, however, their approach involved inference of BSFS from whole stool images, rather than fecal smears that we do here.
Together, our study holds significant strengths. First, we evaluated the SMEAR model using a random sub-selection of a larger cohort of 888 fecal samples from the HELIUS cohort, which (a) is a multi-ethnic study population, capturing a large scale of the population living in The Netherlands, and (b) was compiled by an independent to the HELIUS cohort researcher, embracing no bias around the fecal samples selection. Second, SMEAR went through an exhaustive screening through the 50 most abundant genera, in order to evaluate its ability to classify between low and high relative abundances of profoundly -in human fecal samples- present taxa. Third, SMEAR used very simple wet-lab equipment, including a scalpel, long cotton bud, white A4 papers, and a mobile device, available in all laboratories, research and clinical, allowing for further use and validation of SMEAR based on 16S sequenced material.
SMEAR has its limitations as well. First, our experimental design focused on the 50 most abundant genera in the dataset. Moreover, although shade and water content are important cues,41 showed that ablations of texture features incur a larger performance drop than grayscale conversion, underscoring the role of non-chromatic image attributes33. We cannot predict how effective SMEAR would be at lower taxonomic levels, such as species or strains. Nevertheless, deep taxonomic information requires costly in-depth sequencing, and most importantly trained bioinformaticians, with access to well-maintained servers and sophisticated tools, obstacles that SMEAR is aiming to overcome, and thus our current choice to compare SMEAR (a) against the well-established approach 16S rRNA gene amplicon sequencing, which currently constitutes the benchmark of the human gut microbial ecology; (b) at the taxonomic level of genus, for which most pipelines are successful to provide information. Second, all fecal smear images were generated and captured by the same researcher, aiming for uniformity within the dataset, yet reproducibility between different wet-lab researchers has not been confirmed.
To improve SMEAR further, future studies should evaluate SMEAR in less abundant genera of the human gut microbiome. For this, larger datasets would be required. Furthermore, instead of solely looking at extremes, SMEAR could be trained in a setting where the entire distribution of their microbial traits is included. Lastly, acknowledging the significant importance of gut microbiome in human health and disease, SMEAR could potentially be trained to predict directly a health status, bypassing the microbiome as a modulator of gut health and focusing directly on outcomes.
For instance, SMEAR demonstrates very good performance (AUC = 0.84, p-value < 0.001) in predicting whether a fecal sample has a high or low relative abundance of the genus Oscillibacter, which has been recently identified as a key metabolizer of cholesterol34. In the current version, SMEAR can rapidly and cost-effectively identify patients with low Oscillibacter abundance, potentially flagging individuals at higher risk for cardiovascular disease. Looking ahead, SMEAR could be further trained to predict stool cholesterol concentrations based on fecal smears. This would allow it to directly contribute to the diagnosis, monitoring, and evaluation of treatment responses in patients undergoing dietary interventions or medication regimens for cardiovascular diseases. Additionally, SMEAR could be adapted for other clinical applications. For example, it could be retrained to detect fecal calprotectin—a critical marker often used to assess inflammation in patients with IBD. Alternatively, SMEAR could identify the presence of highly resistant microorganisms that tend to overgrow after intensive antibiotic treatments, potentially causing significant harm. Notably, all these capabilities could be implemented for home use, ensuring minimal cost and convenience for patients.
The next steps in applying SMEAR in a clinical setting would require efforts specifically related to standardization of image protocols and data collection. For this purpose a multi-center study with independent research groups would be the most suitable. Collecting samples from different researchers or centers may introduce bias. To address this we plan to incorporate ArUco fiducial35 markers and standardized lighting rigs (as in Maaskant et al.33) to minimize inter-site variability before multicenter trials. This approach speeds up pre-processing and improves reproducibility by using a template instead of a blank piece of paper. We used these techniques in the rhesus macaques study and we are also currently employing them in the METARYL project, where patients with metabolic syndrome undergo bariatric surgery, providing us with fecal smears throughout the whole process, in order to evaluate the efficacy of SMEAR in a longitudinal fashion.
In conclusion, SMEAR offers a rapid, non-invasive, and cost-effective method for studying the human fecal microbiota at genus and diversity level. While promising, these findings demonstrate feasibility and prospective trials are needed to confirm performance across diverse patient populations and operators. SMEAR holds great potentials towards microbial diagnostics and therapeutics. Given that gut microbiota composition in populations with a Western lifestyle are steadily devolving into a more disrupted microbiome associated with a concomitant increase of disease burden3,24,29,30,36,37,38,39,40, it is becoming increasingly important for people to be able to track whether they are of low or high relative abundance of these important, steadily missing, taxa. Mass screening of large populations could be made much more cost-effective using SMEAR, and outpatient care may benefit from acquiring microbiota composition time-series in order to track the effect of certain interventions on the microbiome with much lower cost or effort. SMEAR complements the current high-throughput sequencing techniques to allow rather fast screening of the main microbial characteristics, making microbiome awareness more accessible to audiences.
Data availability
The HELIUS data are owned by the Amsterdam University Medical Centers, location AMC in Amsterdam, The Netherlands. To allow sharing of microbiome data collected in HELIUS with (inter)national researchers, 16S rRNA sequence analysis has been stored at the European genome-phenome archive (EGA; accession code EGAD00001004106). This requires that access needs to be granted, also because the HELIUS data are stored with relevant phenotypical variables. Any researcher can request the data by submitting a proposal to the HELIUS Executive Board as outlined at http://www.heliusstudy.nl/en/researchers/collaboration, by email: heliuscoordinator@amsterdamumc.nl. The HELIUS Executive Board will check proposals if they do not conflict with ethical approvals and informed consent forms of the HELIUS study.
References
Gacesa, R. et al. Environmental factors shaping the gut microbiome in a Dutch population. Nature 604(7907), 732–739 (2022).
Chanda, D. & De, D. Meta-analysis reveals obesity associated gut microbial alteration patterns and reproducible contributors of functional shift. Gut Microbes 16(1), 2304900 (2024).
Vieira-Silva, S. et al. Quantitative microbiome profiling disentangles inflammation- and bile duct obstruction-associated microbiota alterations across PSC/IBD diagnoses. Nat. Microbiol. 4(11), 1826–1831 (2019).
Shen, T. D. et al. The mucosally-adherent rectal microbiota contains features unique to alcohol-related cirrhosis. Gut Microbes 13(1), 1987781 (2021).
Marx, W. et al. Diet and depression: exploring the biological mechanisms of action. Mol. Psychiatry 26(1), 134–150 (2021).
Lee, D., Maaskant, A., Ngo, H., Montijn, R. C., Bakker, J., Langermans, J. A. M., & Levin, E. (2024). A rapid, affordable, and reliable method for profiling microbiome biomarkers from fecal images. iScience, 27(12), 111310. https://doi.org/10.1016/j.isci.2024.111310
Carter, M. M. et al. Ultra-deep sequencing of Hadza hunter-gatherers recovers vanishing gut microbes. Cell 186(14), 3111 (2023).
Galloway-Peña, J. & Hanson, B. Tools for analysis of the microbiome. Dig. Dis. Sci. 65(3), 674–685 (2020).
Arumugam, M. et al. Enterotypes of the human gut microbiome. Nature 473(7346), 174–180 (2011).
Tito, R. Y. et al. Microbiome confounders and quantitative profiling challenge predicted microbial targets in colorectal cancer development. Nat. Med. 30(5), 1339–1348 (2024).
Vandeputte, D. et al. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 65(1), 57–62 (2016).
O’Donnell, L. J., Virjee, J. & Heaton, K. W. Detection of pseudodiarrhoea by simple clinical assessment of intestinal transit rate. BMJ 300(6722), 439–440 (1990).
Tran, K. A. et al. Deep learning in cancer diagnosis, prognosis and treatment selection. Genome Med. 13(1), 152 (2021).
Deschasaux, M. et al. Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat. Med. 24(10), 1526–1531 (2018).
Snijder, M. B. et al. Cohort profile: the Healthy Life in an Urban Setting (HELIUS) study in Amsterdam, The Netherlands. BMJ Open 7(12), e017873 (2017).
van der Vossen, E. W. J. et al. Gut microbiome transitions across generations in different ethnicities in an urban setting-the HELIUS study. Microbiome 11(1), 99 (2023).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73(16), 5261–5267 (2007).
Comaniciu, D. & Meer, P. Mean shift: a robust approach toward feature space analysis. IEEE Trans. Pattern Anal. Mach. Intell. 24(5), 603–619 (2002).
Inc. A. Adobe Photoshop Available at: https://www.adobe.com/products/photoshop.html. (2019).
Deng, J., Dong, W., Socher, R., Li, L.-J., Li, K., & Fei-Fei, L. (2009). ImageNet: A large-scale hierarchical image database. In 2009 IEEE Conference on Computer Vision and Pattern Recognition (CVPR) (pp. 248–255).
Dosovitskiy, A., Beyer, L., Kolesnikov, A., Weissenborn, D., Zhai, X., Unterthiner, T., Dehghani, M., Minderer, M., Heigold, G., Gelly, S., Uszkoreit, J., & Houlsby, N. (2020). An Image is Worth 16×16 Words: Transformers for Image Recognition at Scale. arXiv preprint arXiv:2010.11929
Wightman, R., Raw, N., Soare, A., Arora, A., Ha, C., Reich, C., … Uchida, Y. (2023). rwightman/pytorch-image-models: v0.8.10dev0 Release [Software]. Zenodo. https://doi.org/10.5281/zenodo.761883
Ridnik T, Ben-Baruch E, Noy A, Zelnik-Manor L. ImageNet-21K pretraining for the masses. arXiv. (2021).
Wang, J. et al. Enterotype bacteroides is associated with a high risk in patients with diabetes: A pilot study. J. Diabetes Res. 2020, 6047145 (2020).
Blake, M. R., Raker, J. M. & Whelan, K. Validity and reliability of the Bristol stool form scale in healthy adults and patients with diarrhoea-predominant irritable bowel syndrome. Aliment. Pharmacol. Ther. 44(7), 693–703 (2016).
Majait, S., Nieuwdorp, M., Kemper, M. & Soeters, M. The black box orchestra of gut bacteria and bile acids: Who is the conductor?. Int. J. Mol. Sci. 24(3), 1816 (2023).
Schnorr, S. L. et al. Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 5, 3654 (2014).
Louis, P. & Flint, H. J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 19(1), 29–41 (2017).
Vieira-Silva, S. et al. Statin therapy is associated with lower prevalence of gut microbiota dysbiosis. Nature 581(7808), 310–315 (2020).
Vandeputte, D. et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature 551(7681), 507–511 (2017).
Kim, H. S. et al. Longitudinal evaluation of fecal microbiota transplantation for ameliorating calf diarrhea and improving growth performance. Nat. Commun. 12(1), 161 (2021).
Hachuel, D., Jha, A., Estrin, D., Martinez, A., Staller, K., & Velez, C. (2019). Augmenting gastrointestinal health: A deep learning approach to human stool recognition and characterization in macroscopic images. arXiv preprint arXiv:1903.10578. https://doi.org/10.48550/arXiv.1903.10578
Maaskant, A. et al. AI for rapid identification of major butyrate-producing bacteria in rhesus macaques (Macaca mulatta). Anim. Microbiome 7(1), 39 (2025).
Li, C. et al. Gut microbiome and metabolome profiling in Framingham heart study reveals cholesterol-metabolizing bacteria. Cell 187(8), 1834–52.e19 (2024).
Garrido-Jurado, S., Muñoz-Salinas, R., Madrid-Cuevas, F. J. & Marín-Jiménez, M. J. Automatic generation and detection of highly reliable fiducial markers under occlusion. Pattern Recogn. 47(6), 2280–2292 (2014).
Essex, M., Rios Rodriguez, V., Rademacher, J., Proft, F., Löber, U., Markó, L., Pleyer, U., Strowig, T., Marchand, J., Kirwan, J. A., Siegmund, B., Kirke Forslund, S., & Poddubnyy, D. (2022). Spondyloarthritis, acute anterior uveitis, and Crohn’s disease have both shared and distinct gut microbiota [Preprint]. medRxiv. https://doi.org/10.1101/2022.05.13.22275044
Gu, Y. et al. Gut and vaginal microbiomes in PCOS: Implications for women’s health. Front. Endocrinol. 13, 808508 (2022).
Jie, Z. et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 8(1), 845 (2017).
Liu, P. et al. Altered microbiomes distinguish Alzheimer’s disease from amnestic mild cognitive impairment and health in a Chinese cohort. Brain Behav. Immun. 80, 633–643 (2019).
Reynders, T. et al. Gut microbiome variation is associated to multiple sclerosis phenotypic subtypes. Ann. Clin. Transl. Neurol. 7(4), 406–419 (2020).
Maaskant, A., Lee, D., Ngo, H., Montijn, R. C., Bakker, J., Langermans, J. A. M., & Levin, E. (2025). AI for rapid identification of major butyrate-producing bacteria in rhesus macaques (Macaca mulatta). Animal Microbiome, 7, Article 39. https://doi.org/10.1186/s42523-025-00410-2
Acknowledgements
We would like to thank Tristan de Waard for his valuable contributions during the initial stages of this project.
Funding
The HELIUS study is conducted by the Amsterdam University Medical Centers, location AMC and the Public Health Service of Amsterdam. Both organizations provided core support for HELIUS. The HELIUS study is also funded by the Dutch Heart Foundation [2010T084], the Netherlands Organization for Health Research and Development (ZonMw) [200500003], the European Union (FP-7) [278901], and the European Fund for the Integration of non-EU immigrants (EIF) [2013EIF013]. We are most grateful to the participants of the HELIUS study and the management team, research nurses, interviewers, research assistants and other staff who have taken part in gathering the data of this study. This study was funded by HORAIZON BV, which specifically supported image acquisition, analysis, computations, and the salary costs of scientists affiliated with HORAIZON BV throughout this project. JD and KZ are supported by a Vidi-stipendium awarded from ZonMw (016.196.343), and an Amsterdam UMC stipendium awarded from Amsterdam University Medical Centers (200402). KZ is supported by Health Holland TKI funding “MICROPIAL”. MN is supported by a personal Vici-stipendium awarded from ZonMw 2020 (09150182010020) and an ERC-Advanced grant 2023 (101141346). WdeJ is funded through METARYL, an EU Joint Progamming Initiative (JPI) of Health diet Healthy Life.
Author information
Authors and Affiliations
Contributions
KZ: generated all fecal smears and captured the images, produced the draft and final version of the manuscript; BV: conducted computational analysis, produced the draft and final version of the manuscript; HN, JM & DL: performed pre-processing, conducted final simulations and performed model adaptations during the rebuttal stages; JD, ML, AZ & MN: led the samples collection; MN: provided access to HELIUS microbiome cohort and analysis of sequenced data; MdG: contributed to the evolution of the study; WJ: co-supervised the study, conception of study, writing of manuscript, and funding; EL: conceived and designed the study, trained and supervised the machine-learning researchers and edited the first draft of the study. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
MN is a member of the Scientific Advisory Board of Caelus Pharmaceuticals and Advanced Microbiome Interventions, the Netherlands (however both of these bear no relations to the content of the current paper). WJ has received speaker’s fees from Alimentiv Bv, and Janssen Pharmaceuticals, over the last 3 years. EL is a board member and employee of HORAIZON BV. HORAIZON BV owns a patent into the technology used under international patent number (WO2023055238A1). The rest of the authors have no conflicts of interest to disclose.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zafeiropoulou, K., Voermans, B., Ngo, H. et al. Application of image guided analyses to monitor fecal microbial composition and diversity in a human cohort. Sci Rep 15, 26237 (2025). https://doi.org/10.1038/s41598-025-10629-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-10629-3
