
Abstract
Bladder cancer has been considered as one of the most common urinary malignancies. Growing evidence has indicated that Centromere Protein F (CENPF) is a promising molecular biomarker for many human malignant tumors. However, the role of CENPF in bladder cancer (BC) proliferation, migration, invasion, cell cycle and apoptosis thereof remain unclear. In the present study, high-throughput sequencing technology and bioinformatics analysis were conducted to identify mRNAs profiles in 10 pairs of bladder cancer tissues and adjacent noncancerous tissues. CENPF was overexpression in bladder cancer tissues, and higher in MIBC than NMIBC. We investigated the expression of CENPF in 30 other pairs bladder cancers tissues, and the results were in accordance with the sequencing results. Furthermore, Immunohistochemical staining, showed that strong intensity of CENPF in BC tissues than normal tissues. Increased staining of CENPF was detected of tumor cells in MIBC compared with NMIBC. This suggests that CENPF might be highly expressed in aggressive and invasive tumor cells. Subsequently, in vitro functional experiments also demonstrated that the siRNA interference of CENPF expression significantly weakened the proliferation, migration, invasion and apoptosis of BC cells, and the cells were arrested in the G2/S phase in Cell cycle. Moreover, functional enrichment analyses, lncRNA-miRNA-mRNA, and protein-protein interaction networks revealed that CENPF was potentially involved in carcinogenesis and evolution of bladder cancer. Taken together, these results demonstrated that CENPF may serve as a potential biomarker of tumor occurrence, progression, and even prognosis for bladder cancer. However, further research is needed to be further clarified the pathway mechanisms of CENPF in bladder cancer.
Similar content being viewed by others

Introduction
Bladder cancer (BC) is one of the most common cancers worldwide. It is the seventh most frequently diagnosed carcinoma and the thirteenth leading cause of cancer-related deaths, with around 573,278 new cases reported each year1. BC is the predominant urinary malignant neoplasm in males, who are affected at a ratio of approximately 3:1 compared to females2. This malignancy originates from the urothelial layer of the bladder. BC is classified into two types based on the invasiveness of the urothelium: muscle-invasive bladder cancer (MIBC), which makes up about 25% of cases, and non-muscle-invasive bladder cancer (NMIBC), which accounts for approximately 75%3. These two forms of bladder cancer exhibit distinct clinical behaviors and require different treatment approaches4. NMIBC has a high recurrence rate, with around 15% of patients progressing to muscle-invasive bladder cancer (MIBC), which significantly increases the risk to their prognosis5. Nearly 50–70% of newly diagnosed NMIBC cases exhibit a significant risk of tumor progression and recurrence within five years6. Pathological staging T2, T3, and T4 are categorized as MIBC, with corresponding five-year survival rates of merely 45%, 40%, and 10%, respectively7. The molecular mechanisms that contribute to the pathogenesis and progression of bladder cancer (BC) remain inadequately understood. Therefore, understanding these mechanisms and identifying target genes involved in BC carcinogenesis is crucial. This knowledge will help develop new therapeutic strategies and enable clinicians to make informed decisions about diagnosis, treatment, pharmacotherapy, and patient follow-up. Transcriptome sequencing (RNA-seq) of tumor samples serves as a powerful methodology for investigating the carcinogenesis of cancer and for identifying novel biomarkers that may enhance the diagnostic, therapeutic, and prognostic approaches to these malignancies8,9.
Centromeric protein F (CENPF), also known as mitosin, is a nuclear antigen linked to the cell cycle. It acts as a microtubule-associated protein involved in both mitosis and cellular differentiation10. CENPF has low expression levels in G0/G1 phase cells, increases in the nuclear matrix during the S phase, and peaks during the G2/M phase11. An upregulation of CENPF has been documented in various human malignancies, including prostate cancer12, hepatocellular carcinoma13, breast cancer14 and other types of tumors15,16.
Functionally, CENPF has been shown to influence cell proliferation, apoptosis, metastasis, and poor prognostic outcomes17,18. However, the role of CENPF in bladder cancer has not been thoroughly investigated. In this study, we investigated how CENPF expression levels correlate with the proliferation, migration, invasion, apoptosis, and cell cycle of bladder cancer cells in vitro. Additionally, functional enrichment analyses, lncRNA-miRNA-mRNA interactions, and protein-protein interaction networks were employed to elucidate the potential involvement of CENPF in the carcinogenesis and progression of bladder cancer.
Materials and methods
Patients and samples
Tumor tissue specimens were procured from patients undergoing transurethral resection of the bladder or radical cystectomy at the Department of Urology, Shanghai Changhai Hospital, China. All specimens were fresh postoperative samples, cataloged by number and preserved at -80 °C. For immunohistochemistry, samples were fixed in formalin and embedded in paraffin for subsequent pathological examination. A total of 40 cases were included in this study, with 10 samples allocated for high-throughput sequencing and the remaining 30 designated for the validation of high-throughput sequencing results. Informed consent was obtained from all patients to utilize residual tissue for research purposes. The study received approval from the institutional ethical committee of Shanghai Changhai Hospital.
High-throughput transcriptome sequencing
Total RNA was extracted from 10 pairs of samples using TRIzol reagent (Thermo Fisher Scientific, Inc.), followed by quantification and purity assessment using a NanoDrop-2000 (Thermo Fisher Scientific, Inc., Wilmington, DE, USA). The integrity of the RNA was evaluated via formaldehyde denaturing 2% agarose gel electrophoresis (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The information and characteristics of these 10 patients were presented in Table 1. Of the 10 pairs of tissues, 5 pairs were classified as MIBC, while the other 5 were categorized as NMIBC. High-throughput whole transcriptome sequencing and data analysis were conducted in collaboration with the Beijing Genomics Institute (Shenzhen, China).
RNA extraction and reverse transcription quantitative PCR (RT-qPCR)
Total RNA was extracted from tumor tissues and healthy normal tissues utilizing Triazol reagent (Invitrogen). Micro-dissected tumor samples, containing at least 75% tumor cell content, along with healthy tissue, were lysed in 1 ml of Triazol, followed by phase separation, as described in the Triazol user manual. The integrity of RNA samples was assessed utilizing a UV spectrophotometer, and the extracted RNA was subsequently preserved at − 80 ℃. Reverse transcription was conducted employing the PrimeScriptTM RT reagent kit (Takara, Dalian, China) to synthesize complementary DNA (cDNA). Primers were synthesized by Sangon Biotech (Shanghai) Co. Ltd., and the information of their sequences was listed in Table 2. GAPDH acted as the internal control for the assessment of CENPF mRNA levels, and results were normalized against GAPDH. The relative expression of the gene was determined using the 2-ΔΔCt method.
Immunohistochemical (IHC)
Immunohistochemistry (IHC) was conducted on the tissue microarray (TMA) to assess the expression levels of CENPF proteins. Before the IHC procedure, the TMA was treated with xylene followed by 100% ethanol, and subsequently subjected to a gradual reduction in ethanol concentration. Following antigen retrieval, IHC was performed utilizing an anti-CENPF antibody (dilution of 1:500; Abcam Institute, USA) for both blocking and staining purposes. This was succeeded by incubation with secondary antibodies (diluted to 1:2000) and the execution of a standard avidin-biotinylated peroxidase complex assay. The immunostaining scores for CENPF were determined by summing the scores for the proportion of positively stained tumor cells (PP) and the staining intensity (SI) as assessed by two pathologists who were blinded to the clinical data. The PP was categorized into four levels: 0 (< 5%, negative), 1 (5–25%, sporadic), 2 (25–50%, focal), and 3 (> 51%, diffuse), while the SI was evaluated on a scale from 0 to 3 (0 indicating negative staining; 1 for weak staining; 2 for moderate staining; and 3 for strong staining). The overall staining score was derived by multiplying the SI score by the PP score, yielding a final score that ranged from 0 to 9. The IHC staining positivity was evaluated by two urologists, with patients classified into low-staining (0–3) and high-staining (4–9) groups based on their scores.
Cell culture and cell transfection
For cell culture and transfection, human bladder cancer cell lines (T24, 5637, EJ, J82, and T921) were maintained in RPMI‑1640 medium supplemented with 10% fetal bovine serum (FBS) at 37˚C in a 5% CO2 incubator. The T921 cell line was cultivated in the laboratory of the Department of Urology at Changhai Hospital, while the remaining cell lines were purchased in 2002. CENPF siRNA(small interfering RNA) and negative control siRNA (NC_siR) were sourced from Bioneer Co. Ltd. (Shanghai, China). Following the manufacturer’s guidelines, the siRNAs were transfected into T24 cells utilizing Lipofectamine™ 3000 (Invitrogen, USA). The siRNAs of CENPF and reference gene were synthesized by Sangon Biotech (Shanghai, China), and the information of their sequences was also listed in Table 1.
The effectiveness of the siRNA was evaluated by measuring the protein and mRNA levels of CENPF in cells 48 h post-transfection. Each group had at least 3 replicates.
Cell proliferation assays
Cell proliferation assays were conducted utilizing the Cell Counting Kit-8 (CCK-8) assay (70-CCK801, MultiSciences, China). Following transfection, bladder cancer cells were seeded in 96-well plates (5000 cells/well). At intervals of 24, 48, 72, 96, and 120 h, 10 µl of CCK-8 solution was added to each well and incubated at 37̊C for one hour. Subsequently, cell viability was assessed at a wavelength of 450 nm. All experiments were performed in triplicate.
Migration and invasion assays
To evaluate cell migration ability, a Transwell chamber (3422, Corning, USA) was inserted into the 24-well plates after transfection. Then cells (range of 3000 to 5000 cells)in serum-free medium were added to the upper chamber. A medium containing 20% serum was introduced into the lower chamber. After a 24-hour incubation, the cells adhered to the lower filter surfaces were fixed.Ethanol and subsequently stained with crystal violet. The number of cells that had migrated was then quantified using a microscope (TS100, Nikon, Japan). For the invasion assay, we used Matrigel (354248, Corning, USA) to block the pores in the membrane, and we conducted all experiments following the previously outlined methods in triplicate.
Cell cycle analysis and flow cytometry assay
Following trypsinization, cells were washed in ice-cold phosphate-buffered saline (PBS) and fixed in ice-cold 75% ethanol in PBS for an overnight period. A cell cycle assay kit (HaiGene, Harbin, China) facilitated the execution of the cell cycle assay. Subsequently, PI/RNase staining buffer (BD Biosciences) was introduced, and the cells were incubated at 4 °C for 30 min. For cell cycle analysis, a Guava® easyCyte flow cytometer equipped with GuavaSoft 2.5 (Millipore, MA, USA) was employed. In the context of apoptosis analysis, after a 48-hour transfection period, cells were collected, washed with PBS, stained using the FITC/Annexin V Apoptosis Detection Kit I (BD Biosciences), and underwent analysis via flow cytometry.
Western blot analysis
Following the trypsinization of the cells, protein extraction was performed utilizing radioimmunoprecipitation (RIPA) buffer (Beyotime Institute of Biotechnology, China), which was supplemented with protease inhibitors (Thermo Fisher Scientific Technology, USA). The quantification of proteins was conducted using a bicinchoninic acid (BCA) protein assay kit (Beyotime Institute of Biotechnology, China). Subsequently, total proteins were separated via SDS-PAGE on a 10% gel and subsequently transferred onto a polyvinylidene fluoride (PVDF) membrane (Millipore Sigma, USA). After a blocking step with 5% skim milk at ambient temperature for 2 h, primary antibodies targeting CENPF (1:1000; Abcam Institute, United States) were incubated overnight at 4 °C. Following three washes with Tris-buffered saline containing Tween 20 (TBST), the membrane was subjected to incubation with a secondary antibody (diluted to 1:2000), followed by the application of the standard avidin-biotinylated peroxidase complex assay for an additional 1.5 h at room temperature. Band signals were subsequently detected using a chemiluminescence system (Bio-Rad Laboratories Inc, USA) and analyzed utilizing Bio-Rad Image Lab for PC 6.1(https://www.bio-rad.com/ImageLab) software.
In Silico analysis for potential RNAs regulated by CENPF
The differentially expressed mRNA data were analyzed using the R package clusterProfiler (version 3.2.11, http://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html). To assess the functional enrichment of pathways, we calculated the significance level (P-value) for each pathway19. The enrichment significance P-value was then adjusted using the BH method. The criteria for significance screening were as follows: a P-value of less than 0.05 and a count of at least 2. To identify candidate target genes or non-coding RNAs regulated by CENPF, we employed the STRING3 database (version 11) (https://string-db.org) for analyzing differentially expressed genes. The species was Homo sapiens, and the PPI score was set to 0.7, requiring that all protein nodes involved in the interactions were differentially expressed genes. The target gene CENPF was isolated to build a PPI network to identify its interacting genes. We also conducted a functional assessment of these genes as a cluster to evaluate the pathways and biological processes (BPs) involved TMEM131A, applying thresholds of P-value < 0.05 and count ≥ 2 to pinpoint upstream and downstream genes. We calculated the Pearson correlation coefficients for the differential lncRNAs associated with CENPF and TMEM132A expression. According to the known ceRNA mechanism, lncRNA-mRNA pairs should exhibit a positive correlation. We screened for pairs with a coexpression relationship of r > 0 and a p-value < 0.05 to identify the correlations between lncRNA, CENPF, and TMEM132A.Additionally, in the miRWalk 3.0 input analysis (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk3/miRretsys-self.html), we examined CENPF in the predicted genes module. We selected human as the species and generated a list of predicted miRNA-target regulatory pairs with scores above 0.95, categorizing them as significantly differential miRNAs. Finally, we co-expressed differentially expressed mRNAs from the screening with lncRNAs that had positive co-expression relationships to construct the lncRNA-miRNA-target gene ceRNA network, specifically the endogenous ceRNA network.
Statistical analysis
Data were presented as means ± standard deviation (SD) from at least three independent experiments. The SPSS 17.0 software (SPSS Inc., Chicago, IL, USA) was utilized for data analysis, and GraphPad Prism 8.0 (La Jolla, CA, USA) was employed for graph creation. The Student’s t-test was applied to compare differences between two groups, while one-way ANOVA was employed for comparisons involving multiple groups. A p-value of less than 0.05 was deemed statistically significant.
Results
The expression levels of CENPF were found to be elevated in tumor tissues compared to normal samples
Initial analysis of high-throughput sequencing data (with a threshold of fold change > 2 and probability > 0.8) revealed a total of 236 differentially expressed genes, which included 57 upregulated mRNAs and 179 downregulated mRNAs; 29 differentially expressed lncRNAs, comprised of 21 upregulated and 8 downregulated; and 8 differentially expressed miRNAs, with 5 upregulated and 3 downregulated (Fig. 1A–C). Following this, 21 mRNAs were selected based on a more stringent threshold (fold change > 4 and probability > 0.8) from the 57 mRNAs that exhibited significant differential expression in tumor versus normal tissues; notably, CENPF was identified among these 21 mRNAs. A hierarchical clustering heat map was generated to visualize the expression profiles of the most upregulated 21 mRNAs (Fig. 1D).
The gene expression analysis was performed using the web-based tool Morpheus (https://software.broadinstitute.org/morpheus/). Our findings indicated that, in 90% of the cases examined, CENPF expression was significantly elevated in tumor tissues compared to normal tissues (Fig. 1E). Additionally, CENPF levels were found to be higher in muscle-invasive bladder cancer (MIBC) compared to non-muscle-invasive bladder cancer (NMIBC) (Fig. 1F).

The results of high-throughput sequencing and the expression of CENPF in bladder cancer tissues. (A)(B)(C) The volcano plots of mRNA, lncRNA, and miRNA from high-throughput sequencing (with a fold change > 2.0) in tumor-adjacent normal bladder tissues (n = 10) and bladder cancer tissues (n = 10). Panel A shows differentially expressed mRNAs, Panel B shows differentially expressed lncRNAs, and Panel C shows differentially expressed miRNAs. (D)Hierarchical clustering analysis of 21 upregulated mRNAs with a fold change > 4.0 according to high‑throughput sequencing. CENPF is one of the 21 highly expressed mRNAs. (E) In 10 pairs of samples,9 paired CENPF expression were significantly higher in tumor tissues than the paired adjacent normal tissues. (F) CENPF expression was higher in MIBC than in NMIBC(P<0.05).
Subsequently, we assessed CENPF expression in 30 additional pairs of bladder cancer tissues. Table 3 provides clinical information for 30 patients, comprising 23 males and 7 females; among them, 8 had low-grade, 22 had high-grade, 10 had NMIBC, and 20 had MIBC. Similarly, these results corroborated our sequencing findings, revealing that CENPF expression was notably greater in tumor tissues than in adjacent normal tissues, as well as being more pronounced in MIBC compared to NMIBC (Fig. 2A and B). Furthermore, to evaluate CENPF expression levels in bladder cancer, we performed an immunohistochemical (IHC) analysis on a tissue microarray. The CENPF protein predominantly localized to the nucleus in bladder cancer tissues, with some presence in the cell membrane and interstitial regions. In contrast, typical urothelium exhibited weak or absent staining, whereas tumor cell areas displayed moderate to strong staining (Fig. 2C). Notably, CENPF staining intensity was heightened in MIBC compared to the corresponding NMIBC samples (Fig. 2D).

The expression of CENPF in expanded 30 pairs of bladder cancer tissues and adjacent normal tissues and IHC staining of CENPF in patients. (A)(B) CENPF expression is higher in tumor tissues than in adjacent normal tissues, and higher in MIBC than NMIBC(P<0.05). (C) CENPF is not/weak stained in typical urothelium. CENPF is mainly moderate to strong stained in tumor cells(P < 0.05). (D) NMIBC, weak(rarely) to moderate(almost) stained; MIBC, moderate to strong stained. Increased staining of CENPF was detected of tumor cells in MIBC compared with NMIBC(P < 0.05).
The impact of reduced CENPF expression on the proliferation, migration, invasion, cell cycle, and apoptosis of bladder cancer (BC) cells
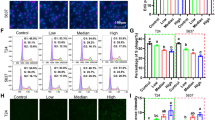
The expression levels of CENPF were quantified in five BC cell lines (T24, 5637, J82, EJ, and T921) using quantitative PCR. The T24 and J82 cell lines are classified as high-grade bladder cancer lines, recognized for their high invasiveness and infiltration. In contrast, the EJ and 5637 lines are classified as G2, exhibiting relatively lower levels of invasiveness and infiltration. The T921 cell line, developed in our laboratory, is an independently cultured bladder cancer line known for its high grade and rapid cell growth. The analysis revealed that CENPF was expressed across all five BC cell lines, with the T24 cell line exhibiting the highest expression level (Fig. 3A). Consequently, we selected the T24 cell line for further experimentation. The T24 cells were categorized into three groups: a mock control group, a normal control group, and an interference group, which was further divided into two subgroups using two distinct siRNAs, referred to as siRNA1 and siRNA2. Following a 48-hour transfection period, we assessed the expression levels of CENPF in the various groups via RT-PCR, setting the expression level in the mock control group to 1 for comparative purposes. The results indicated that the expression level in the normal control group was approximately 1. The interference efficiency of siRNA2 was approximately 70%, while siRNA1 achieved an interference efficiency of roughly 80%. Thus, siRNA1 demonstrated superior interference efficiency. This was further validated in a Western Blot experiment, which confirmed that the CENPF protein expression was significantly diminished in the siRNA-treated group, thereby validating the effectiveness of siRNA1 and the experimental methodology employed. We proceeded with the more effective siRNA1 for subsequent investigations (Fig. 3B).
To elucidate the effects of CENPF on the proliferation, migration, and invasion capabilities of the T24 bladder cancer cells, we conducted CCK-8 assays and transwell migration and invasion experiments post-transfection with siRNA1. The CCK-8 assay results indicated that, by the third day, the proliferative activity of the interference group was significantly lower than that of both the mock and normal control groups (Fig. 3C, P < 0.05). The transwell migration and invasion assays demonstrated a marked reduction in the migration and invasion abilities of the cells in the interference group by the first day (Fig. 3D, P < 0.05), highlighting the crucial role of CENPF in facilitating the proliferation, migration, and invasion of T24 BC cells.
To further investigate whether CENPF influences the cell cycle and apoptosis in BC cells, we performed cell cycle and apoptosis assays. The data from these experiments revealed that following siRNA-mediated interference in T24 cells, the proportion of cells in the G2 and S phase ratios were the highest, while the G1 phase ratio was the lowest than Mock and NC, indicating that decreased expression of CENPF leads to a reduction in cell division (Fig. 3E). After the downregulation of CENPF expression, the proportion of apoptotic cells exhibited a significant increase, whereas the distributions of cells in the four quadrants of the MOCK and NC control groups remained relatively consistent (Fig. 3F). This observation suggests that CENPF not only influences cell cycle progression but also facilitates the apoptosis of bladder cancer (BC) cells.

The effects of reduced CENPF expression on bladder cancer cells. (A) The expression levels of CENPF in different bladder cell lines (T24,5637, J82, EJ and T921 cells) were measured by qRT-PCR. (B) The siRNA1 was better transfection efficiency through qRT-PCR and the Western Blot experiment. (C) The proliferation capacity of T24 cells was measured by CCK-8 assays after interference with MOCK, NC and siRNA1 at 0 d, 1 d, 2d,3d,4d and 5d(P<0.05). (D) The cell migration and invasion of T24 cells were evaluated by transwell assay at 100x after 24 h. Numbers of migration and invasive cells compared to mock, NC groups were measured in the right charts. Data were presented as means ± SD; n = 3. SD: standard deviation. (E) Cell cycle assays of T24 cells. The proportions of cells in the G1, G2, and S phase for the three groups were shown in the right charts. (F) Flow cytometry analysis of apoptosis of cells. The apoptosis rates of T24 cells in the three groups were shown in the right charts (P < 0.05).
KEGG pathway enrichment for CENPF, focusing on the lncRNA-miRNA-mRNA interactions and protein-protein interactions (PPI)
Initially, the analysis identified sets of differentially expressed mRNAs, both upregulated and downregulated, using a stringent threshold of a corrected P-Value < 0.05. The findings indicated that these genes predominantly participated in specific biological pathways, with the upregulated mRNAs being enriched in 5 pathways, and among them, the two most abundant terms were “Metabolism of xenobiotics by cytochrome P450” and “Retinol metabolism”.The downregulated mRNAs were associated with 19 pathways, and of them, the four most enriched terms were “Vascular smooth muscle contraction”, “Complement and coagulation cascades”, “Focal adhesion”and “Regulation of actin cytoskeleton” (Fig. 4A).
Following this, a PPI network was established, comprising 129 nodes and 305 interaction pairs. Notably, CENPF and TMEM13A were linked to a higher number of nodes, and gene sequencing results indicated that both were highly expressed mRNAs. We then constructed the PPI network for two target genes, CENPF and TMEM132A, resulting in a network with 16 nodes and 58 relationship pairs, as illustrated in Fig. 4B. Subsequently, we analyzed the genes with reciprocal relationships to CENPF and TMEM132A as a cluster. Our functional analysis identified 13 KEGG pathways and 140 biological processes (BPs), as depicted in Fig. 4C. Of these pathways, the most abundant terms were “Legionellosis”, “Platinum drug resistance”, “EGFR tyrosine kinase inhibitor resistance” and “Toll − like receptor signaling pathway”. Among these BPs, the most enriched terms were “Post − translational protein modification”, “Nuclear division”, “Organelle fission” and “Mitotic nuclear division”.
We obtained co-expressed differential lncRNA-CENPF and TMEM132A co-expression relationship pairs of 9 (positively correlated), including 8 lncRNAs and 2 mRNAs. Lastly, predictions concerning key miRNA-related genes were performed, resulting in the identification of six target gene pairs. However, none of these pairs corresponded to differentially expressed miRNAs, and no miRNA-target interactions were observed. Consequently, the co-expression relationships among miRNA-lncRNA and differentially expressed mRNAs with their corresponding lncRNAs (considering only positive co-expression links) were integrated. This synthesis culminated in the establishment of a lncRNA-miRNA-Gene network, which comprised 5 miRNAs, 13 lncRNAs, 2 target genes, and 19 regulatory relationships, as illustrated in Fig. 4D.

Functional enrichment analyses, protein-protein interaction networks, and lncRNA-microRNA-mRNA (ceRNAs) interactions. (A) The KEGG pathway analyses for both upregulated and downregulated genes are shown. The left side displays the pathway enrichment outcomes for upregulated mRNAs, while the right side presents the pathway enrichment results for downregulated mRNAs. The x-axis denotes the quantity of enriched genes, and the y-axis identifies the KEGG pathway names. The p-value indicates the significance threshold, with the color gradient transitioning from blue to red signifying a reduction in p-values and an increase in significance. (B) The protein-protein interaction (PPI) network and module analysis is depicted. The left side illustrates the PPI network established from 129 overlapping target genes, whereas the right side focuses on the PPI network involving the target gene CENPF and TMEM132A. The green downward triangles signify downregulated mRNAs, while the red circles denote upregulated mRNAs. The size of the nodes correlates with the degree value magnitude. (C) Enrichment analysis of CENPF and TMEM132A gene cluster. The left side reveals the pathway enrichment results, and the right side showcases the Biological Process (BP) enrichment outcomes. The x-axis represents the number of enriched genes, while the y-axis specifies the names of the pathways or biological processes. The p-value serves as the significance threshold, with colors ranging from blue to red indicating decreasing p-values and increasing significance. (D) The lncRNA-miRNA-mRNA (ceRNAs) network for the CENPF gene is illustrated. Red circles indicate upregulated genes, orange upward triangles represent upregulated miRNAs, green upward triangles denote downregulated miRNAs, blue squares signify downregulated lncRNAs, and purple squares represent upregulated lncRNAs.
Discussion
The underlying mechanisms contributing to the onset and progression of bladder cancer remain inadequately elucidated. Sequencing analyses have indicated a high degree of mutational heterogeneity within bladder cancer, with novel mutations accumulating throughout carcinogenesis and advancement, thus complicating the investigation of bladder cancer mechanisms20,21. In the present study, we performed high-throughput transcriptome sequencing on 10 pairs of bladder cancer tissue samples, revealing noteworthy differential gene expression between tumor and normal tissues. These identified genes are likely intricately linked to carcinogenic processes, including cancer pathways and cell cycle regulation, offering valuable insights into the pathogenesis of bladder cancer.
Among the differentially expressed genes, CENPF was found to be significantly overexpressed in bladder tumor tissues compared to adjacent normal tissues, with its expression levels being higher in invasive bladder cancer than in non-invasive counterparts. The elevated expression of CENPF in bladder cancer tissues was validated through RT-PCR and immunohistochemistry in an additional cohort of 30 bladder cancer tissue pairs, demonstrating higher mRNA and protein levels compared to normal tissues. Furthermore, we noted a correlation between CENPF expression and tumor staging, which aligned with initial sequencing results. Notably, the upregulation of CENPF has also been corroborated in multiple other human malignancies, including gastric cancer17, lung cancer22, hepatocellular carcinoma13 and prostate cancer23. In prostate cancer, the study by Aytes et al.24 established CENPF as a significant co-regulatory factor and a negative prognostic marker for survival and metastasis. These findings underscore the clinical relevance of CENPF expression, suggesting its potential role in tumor pathogenesis. However, the precise expression patterns and biological functions of CENPF in bladder cancer remain to be comprehensively understood.
CENPF displays distinctive expression and distribution characteristics and possesses multifunctional roles. Over the last two decades following its identification, extensive research has elucidated the pivotal role of CENPF in a multitude of cellular functions, encompassing the regulation of mitosis, microtubule dynamics, gene expression, myogenic differentiation, and histone methylation10,25. As a critical modulator of cell division, CENPF is implicated in the mechanisms underpinning carcinogenesis26. Experimental investigations into cell functionality have demonstrated that the knockout of CENPF markedly impedes cell proliferation, while alterations in CENPF expression result in a G2/S phase arrest in various cell types. Furthermore, the study conducted by Dai et al.27 provides evidence that CENPF serves to inhibit the G2/M phase transition in hepatic cancer cells through the downregulation of the cell cycle regulatory protein cyclin B1. Collectively, these findings suggest that CENPF is a promoter of cellular proliferation. Additional literature suggests a correlation between CENPF and cellular differentiation, characterized by an enhancement in proliferative capacity alongside a decline in differentiation, reflecting the transition from normal cells to tumorigenic cells28,29.
Moreover, the knockout of CENPF has been shown to significantly diminish the migratory and invasive capabilities of T24 cells, a finding that aligns with clinical observations indicating a positive correlation between CENPF expression levels and the progression of bladder cancer. These findings further substantiate the critical function of CENPF in the invasion and metastasis of bladder cancer, corroborating earlier studies by Dai et al.27 regarding its role in migration and invasion across liver and prostate cancers. Given that patients with non-muscle invasive bladder cancer (NMIBC) face an enduring risk of recurrence and disease progression, CENPF presents as a promising biomarker for evaluating the invasiveness of bladder cancer. Shi et al.30 reported that CENPF serves as a robust prognostic marker for NMIBC, with its expression levels closely associated with progression-free survival (PFS) and overall survival (OS). Apoptosis, a genetically regulated biological process, facilitates the elimination of damaged, infected, or surplus cells. Disruption of this apoptotic mechanism may precipitate autoimmune disorders, including malignancies. In our apoptosis assays, we observed that interference with CENPF expression resulted in enhanced apoptosis in T24 bladder cancer cells. Previous studies have indicated that the depletion of CENPF may instigate premature chromosomal decondensation, culminating in increased apoptotic rates29,31. Additionally, we have demonstrated that siRNA targeting CENPF effectively curtails the proliferation and invasive potential of cancer cells, positioning CENPF as a potential candidate for anticancer therapeutic development.
In subsequent analyses, we conducted GO enrichment and KEGG pathway assessments to further investigate the roles of the identified genes, which included 57 upregulated mRNAs and 179 downregulated mRNAs. This analysis revealed significant enrichment in several cancer-related pathways, such as pathways in cancer, Cytochrome P450-related metabolic activities, coagulation cascades, vascular smooth muscle contraction, regulation of the actin cytoskeleton, and proteoglycans in cancer, which are known to influence cancer susceptibility, cellular growth, invasion, and metastasis32,33,34,35. A growing body of research suggests that Focal Adhesion is an independent factor in cell migration, especially in oncology research36. Regulation of the actin cytoskeleton is significantly associated with tumor infiltration and metastasis35.
TMEM132A is a transmembrane protein that influences the aggressiveness of gastric cancer cells and affects the overall survival rates of patients with bladder cancer37,38. Furthermore, it has been reported in prostate adenocarcinoma, renal cell carcinoma, and other types of tumor39,40,41. Interestingly, our investigation revealed a significant correlation between CENPF and TMEM132A. The sequencing results indicated that both CENPF and TMEM132A were highly expressed, with numerous associated pairs and nodes. we constructed PPI networks for CENPF and TMEM132A, comprising 16 nodes and 58 interaction pairs. Additionally, we conducted KEGG pathway enrichment and biological process (BP) analyses, resulting in the identification of 13 KEGG pathways and 140 biological processes (BP) associated with tyrosine kinase inhibitors (TKI), SUMOylation, and nuclear division. The analysis indicates that CENPF and TMEM132A may jointly contribute to proliferation, invasion, metastasis, and drug resistance in bladder cancer cells. Ultimately, we established a competing endogenous RNA (ceRNA) network concerning CENPF and TMEM132A consisting of 5 miRNAs, 13 lncRNAs, 2 target genes, and 19 regulatory interactions. Notably, several miRNAs, including miR-21, miR-34a, and miR-155, emerged as key regulators within this network, impacting the expression of target genes related to cancer progression and responses to therapy.
The microRNAs hsa-miR-145-5p, hsa-miR-1, hsa-miR-143-3p, and hsa-miR-192-5p, among others, have been identified as either tumor promoters or suppressors in various malignant neoplasms42,43,44. It is essential to explore further whether CENPF and TMEM132A exert a synergistic effect in the initiation and progression of bladder cancer. CENPF and TMEM132A may be potential diagnostic markers for the progression, metastasis, treatment resistance, and prognosis of bladder cancer.
Conclusion
In conclusion, through high-throughput transcriptome sequencing analysis, we have identified the upregulation of CENPF in bladder cancer. This finding was subsequently validated by increasing the sample size, confirming that CENPF is linked to tumor staging. In vitro studies demonstrated that CENPF facilitates the proliferation, migration, and invasion of bladder cancer cells while also inhibiting their apoptosis. Thus, CENPF holds potential as a valuable biomarker and therapeutic target in the diagnosis, treatment, and prognosis of bladder cancer. Nonetheless, additional research is required to elucidate the underlying molecular mechanisms and clinical implications of CENPF in the context of bladder cancer.
Data availability
The data that support the findings of this study are included and will be available from the corresponding author upon reasonable request. The high-throughput sequencing datasets generated during the current study are available in the [Network Disk Sharing] repository. (https://pan.baidu.com/s/1Z-3hQZwSENI2JMKEBukJcQ? pwd=SR25)
References
Sanguedolce, F. et al. HER2 expression in bladder cancer: a focused view on its diagnostic, prognostic, and predictive role. Int. J. Mol. Sci. 24, 145. https://doi.org/10.3390/ijms24043720 (2023).
Li, J. et al. Construction of a novel mRNA-signature prediction model for prognosis of bladder cancer based on a statistical analysis. BMC Cancer. 21, 858. https://doi.org/10.1186/s12885-021-08611-z (2021).
Contieri, R. et al. Accuracy of the European association of urology (EAU) NMIBC 2021 scoring model in predicting progression in a large cohort of HG T1 NMIBC patients treated with BCG. Minerva Urol. Nephrol. 75, 180–187. https://doi.org/10.23736/s2724-6051.22.04953-9 (2023).
Babjuk, M. et al. European association of urology guidelines on Non-muscle-invasive bladder Cancer (Ta, T1, and carcinoma in Situ). Eur. Urol. 81, 75–94. https://doi.org/10.1016/j.eururo.2021.08.010 (2022).
Cathomas, R. et al. The 2021 updated European association of urology guidelines on metastatic urothelial carcinoma. Eur. Urol. 81, 95–103. https://doi.org/10.1016/j.eururo.2021.09.026 (2022).
Compérat, E. et al. Current best practice for bladder cancer: a narrative review of diagnostics and treatments. Lancet 400, 1712–1721. https://doi.org/10.1016/s0140-6736(22)01188-6 (2022).
Raspollini, M. R. et al. News in the classification of WHO 2022 bladder tumors. Pathologica 115, 32–40. https://doi.org/10.32074/1591-951x-838 (2022).
Zeng, S. et al. Transcriptome sequencing identifies ANLN as a promising prognostic biomarker in bladder urothelial carcinoma. Sci. Rep. 7, 3151. https://doi.org/10.1038/s41598-017-02990-9 (2017).
Ma, S. et al. Identification of PTK6, via RNA sequencing analysis, as a suppressor of esophageal squamous cell carcinoma. Gastroenterology 143, 675–686e612. https://doi.org/10.1053/j.gastro.2012.06.007 (2012).
Ma, L., Zhao, X. & Zhu, X. Mitosin/CENP-F in mitosis, transcriptional control, and differentiation. J. Biomed. Sci. 13, 205–213. https://doi.org/10.1007/s11373-005-9057-3 (2006).
Hussein, D. & Taylor, S. S. Farnesylation of Cenp-F is required for G2/M progression and degradation after mitosis. J. Cell. Sci. 115, 3403–3414. https://doi.org/10.1242/jcs.115.17.3403 (2002).
Göbel, C. et al. Upregulation of centromere protein F is linked to aggressive prostate cancers. Cancer Manag Res. 10, 5491–5504. https://doi.org/10.2147/cmar.S165630 (2018).
Huang, Y. et al. Centromere protein F (CENPF) serves as a potential prognostic biomarker and target for human hepatocellular carcinoma. J. Cancer. 12, 2933–2951. https://doi.org/10.7150/jca.52187 (2021).
Sun, J. et al. Overexpression of CENPF correlates with poor prognosis and tumor bone metastasis in breast cancer. Cancer Cell. Int. 19, 264. https://doi.org/10.1186/s12935-019-0986-8 (2019).
Li, M. X. et al. Overexpression of CENPF is associated with progression and poor prognosis of lung adenocarcinoma. Int. J. Med. Sci. 18, 494–504. https://doi.org/10.7150/ijms.49041 (2021).
Yu, B. et al. TOP2A and CENPF are synergistic master regulators activated in cervical cancer. BMC Med. Genomics. 13, 145. https://doi.org/10.1186/s12920-020-00800-2 (2020).
Xu, P. et al. N6-methyladenosine modification of CENPF mRNA facilitates gastric cancer metastasis via regulating FAK nuclear export. Cancer Commun. (Lond). 43, 685–705. https://doi.org/10.1002/cac2.12443 (2023).
Wang, D. et al. CENPF knockdown inhibits adriamycin chemoresistance in triple-negative breast cancer via the Rb-E2F1 axis. Sci. Rep. 13, 1803. https://doi.org/10.1038/s41598-023-28355-z (2023).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kotolloshi, R., Hölzer, M., Gajda, M., Grimm, M. O. & Steinbach, D. SLC35F2, a transporter sporadically mutated in the untranslated region, promotes growth, migration, and invasion of bladder Cancer cells. Cells 10, 36. https://doi.org/10.3390/cells10010080 (2021).
Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507, 315–322, doi:https://doi.org/10.1038/nature12965 (2014).
Shahid, M. et al. Downregulation of CENPF remodels prostate Cancer cells and alters cellular metabolism. Proteomics 19, 752. https://doi.org/10.1002/pmic.201900038 (2019).
Shahid, M. et al. Downregulation of CENPF remodels prostate Cancer cells and alters cellular metabolism. Proteomics 19, e1900038. https://doi.org/10.1002/pmic.201900038 (2019).
Aytes, A. et al. Cross-Species regulatory network analysis identifies a synergistic interaction between and that drives prostate Cancer malignancy. Cancer Cell. 25, 638–651. https://doi.org/10.1016/j.ccr.2014.03.017 (2014).
Du, J., Li, Y. & Zhu, X. Involvement of CENP-F in histone methylation. Acta Biochim. Biophys. Sin. 42, 173–176. https://doi.org/10.1093/abbs/gmq001 (2010).
Rattner, J. B., Rao, A., Fritzler, M. J., Valencia, D. W. & Yen, T. J. CENP-F is a.ca 400 kda kinetochore protein that exhibits a cell-cycle dependent localization. Cell. Motil. Cytoskeleton. 26, 214–226. https://doi.org/10.1002/cm.970260305 (1993).
Dai, Y. et al. Characterization of the oncogenic function of centromere protein F in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 436, 711–718. https://doi.org/10.1016/j.bbrc.2013.06.021 (2013).
Pabón-Peña, L. M., Goodwin, R. L., Cise, L. J. & Bader, D. Analysis of CMF1 reveals a bone morphogenetic Protein-independent component of the cardiomyogenic pathway. J. Biol. Chem. 275, 21453–21459. https://doi.org/10.1074/jbc.M000518200 (2000).
Yang, Z. et al. Silencing Mitosin induces misaligned chromosomes, premature chromosome decondensation before anaphase onset, and mitotic cell death. Mol. Cell. Biol. 25, 4062–4074. https://doi.org/10.1128/mcb.25.10.4062-4074.2005 (2005).
Shi, J., Zhang, P., Liu, L., Min, X. & Xiao, Y. Weighted gene coexpression network analysis identifies a new biomarker of CENPF for prediction disease prognosis and progression in nonmuscle invasive bladder cancer. Mol. Genet. Genom. Med. 7, 96. https://doi.org/10.1002/mgg3.982 (2019).
Bomont, P., Maddox, P., Shah, J. V., Desai, A. B. & Cleveland, D. W. Unstable microtubule capture at kinetochores depleted of the centromere-associated protein CENP-F. EMBO J. 24, 3927–3939. https://doi.org/10.1038/sj.emboj.7600848 (2005).
Mokhosoev, I. M., Astakhov, D. V., Terentiev, A. A. & Moldogazieva, N. T. Human cytochrome P450 Cancer-Related metabolic activities and gene polymorphisms: a review. Cells 13, 123. https://doi.org/10.3390/cells13231958 (2024).
Liu, Y. et al. Analysis of gene expression in bladder cancer: possible involvement of mitosis and complement and coagulation cascades signaling pathway. J. Comput. Biol. 27, 987–998. https://doi.org/10.1089/cmb.2019.0237 (2020).
Touyz, R. M. et al. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 114, 529–539. https://doi.org/10.1093/cvr/cvy023 (2018).
Rosanò, L. & Bagnato, A. New insights into the regulation of the actin cytoskeleton dynamics by GPCR/β-arrestin in cancer invasion and metastasis. Int. Rev. Cell. Mol. Biol. 346, 129–155. https://doi.org/10.1016/bs.ircmb.2019.03.002 (2019).
Paluch, E. K., Aspalter, I. M. & Sixt, M. Focal Adhesion-Independent cell migration. Annu. Rev. Cell. Dev. Biol. 32, 469–490. https://doi.org/10.1146/annurev-cellbio-111315-125341 (2016).
Sanchez-Pulido, L. & Ponting, C. P. TMEM132: an ancient architecture of cohesin and Immunoglobulin domains define a new family of neural adhesion molecules. Bioinformatics 34, 721–724. https://doi.org/10.1093/bioinformatics/btx689 (2018).
Zhang, M. et al. Multidimensional analysis of TMEM132A in pan-cancer: unveiling its potential as a biomarker for treatment response prediction. J. Cancer. 15, 4386–4405. https://doi.org/10.7150/jca.96396 (2024).
Wang, Y. et al. Study of the role of E2F1 and TMEM132A in prostate Cancer development. Front. Biosci. (Landmark Ed). 29, 360. https://doi.org/10.31083/j.fbl2910360 (2024).
Roldán, F. L. et al. Prognostic gene Expression-Based signature in Clear-Cell renal cell carcinoma. Cancers (Basel). 14, 253. https://doi.org/10.3390/cancers14153754 (2022).
Li, B. & Niswander, L. A. TMEM132A, a novel Wnt signaling pathway regulator through Wntless (WLS) interaction. Front. Cell. Dev. Biol. 8, 599890. https://doi.org/10.3389/fcell.2020.599890 (2020).
Liu, R. et al. Hsa-miR-1 suppresses breast cancer development by down-regulating K-ras and long non-coding RNA MALAT1. Int. J. Biol. Macromol. 81, 491–497. https://doi.org/10.1016/j.ijbiomac.2015.08.016 (2015).
Huang, Z. G. et al. MiRNA-145-5p expression and prospective molecular mechanisms in the metastasis of prostate cancer. IET Syst. Biol. 15, 1–13. https://doi.org/10.1049/syb2.12011 (2021).
Xu, D., Jiang, J., He, G., Zhou, H. & Ji, C. miR-143-3p represses leukemia cell proliferation by inhibiting KAT6A expression. Anticancer Drugs 33, e662–e669. https://doi.org/10.1097/cad.0000000000001231 (2022).
Acknowledgements
This work was supported by National Key Research and Development Program of China (2022YFB3804504), the National Natural Science Foundation of China, Youth Project (Nos. 82101838, 82203367, 82203505, and 82203403), and the Experimental Animal Fund of Shanghai Science and Technology Commission (No. 22140903800).
Author information
Authors and Affiliations
Contributions
R.S., X.C., Z.Z. and X.Y. designed the study. R.S. and X.C. performed the experiments. R.S. and Z.S. drafted this manuscript. Z.Z. and X.Y. critically revised the manuscript. H.W. and J.X. coordinated the experiments, interpreted the data and prepared the manuscript. S.Z. performed the statistical analysis. W.Z. collected and analyzed the clinical samples. Z.S., X.C. and R.S designed the high‑throughput transcriptome sequencing experiments and functional enrichment analyses.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
All methods were performed in accordance with the relevant guidelines and regulations. This paper does not engage in animal experimentation or involve any animal ethics.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Song, R., Chen, X., Zhang, Z. et al. CENPF overexpression in bladder cancer cells enhances proliferation, migration, invasion, and apoptosis. Sci Rep 15, 25398 (2025). https://doi.org/10.1038/s41598-025-10677-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-10677-9
