
Abstract
Intervertebral disc degeneration (IDD), a common cause of chronic low back pain, strongly impacts daily life. Although previous studies have identified certain biomarkers indicating IDD, comprehensive analyses that integrate transcriptomic and proteomic data to elucidate age-related changes in IDD are lacking. We addressed this issue by integrating transcriptomic and proteomic analyses to identify key molecular signatures that may be potential therapeutic targets for improving the treatment of age-related IDD. We used transcriptomic and proteomic analyses to identify key regulatory genes associated with IDD. We performed RNA sequencing and mass spectrometry of 3 elderly patients with IDD and 3 younger patients with intervertebral disc lesions. Statistical analysis and GO and KEGG enrichment analyses were employed to interpret the transcriptomic and proteomic data. Validation was performed with external datasets and RT‒qPCR. Gene regulatory network and ceRNA network analyses revealed the factors associated with characteristic genes. Transcriptomic and proteomic analyses revealed 45 differentially expressed genes (DEGs) and 34 differentially expressed proteins (DEPs) associated with IDD. We identified CXCL14 as the sole molecule significantly upregulated in IDD at both the transcriptome (4.2-fold, p < 0.001) and proteome levels (3.8-fold, p = 0.003). RT‒qPCR confirmed CXCL14 overexpression in elderly IDD patients (|log2-fold change| =4.1, p < 0.001), consistent with external dataset analysis (GSE147383: |log2-fold change| =1.3, p = 0.008). Gene regulatory networks revealed that CXCL14 interacts with inflammatory mediators (IL-1β and TNF-α) and activates the NF-κB pathway, a key driver of extracellular matrix degradation and inflammation. ceRNA network analysis further identified hsa-miR-582-3p and hsa-miR-150-5p as potential upstream regulators of CXCL14. We analysed the expression profiles of elderly IDD patients and younger patients through transcriptomic and proteomic analyses, identifying unique molecular features associated with IDD. These findings lay a foundation for developing targeted treatments for elderly IDD patients and provide broader insights into potential therapeutic strategies for managing ageing-related IDD across different patient populations. CXCL14 is a potential therapeutic target for pain associated with age-related IDD and could inform the development of novel drug therapies and diagnostic tools, potentially improving clinical outcomes and providing a basis for personalized treatment approaches in managing chronic low back pain and IDD.
Similar content being viewed by others


Introduction
Intervertebral disc degeneration (IDD) is one of the leading causes of chronic low back pain and is particularly prevalent in the elderly population1. Studies have shown that approximately 84% of people worldwide experience back pain at some point in their lives. More than 90% of patients over 50 years old with intervertebral disc disorders suffer from degenerative intervertebral disc conditions2. The cost of treating low back pain caused by IDD is extremely high worldwide3. The incidence of low back pain continues to increase, placing increasing pressure on clinical resources. The financial strain associated with managing this condition is intensifying, as the increase in IDD spending surpasses that of overall healthcare spending4. IDD-related treatments are estimated to cost billions of dollars annually among global healthcare spending. In addition to these economic burdens, this condition also leads to a substantial loss of social resources, as individuals with chronic pain often experience reduced mobility, diminished quality of life, and increased reliance on healthcare services.
Currently, common clinical treatments for IDD mainly include conservative drug therapies, such as nonsteroidal anti-inflammatory drugs (NSAIDs) and opioids, as well as surgical interventions, such as spinal fusion and disc replacement. However, these treatments provide only temporary pain relief and do not address the underlying degenerative processes of the intervertebral discs5. Furthermore, surgical options carry risks of complications, and their long-term effectiveness remains uncertain. These issues highlight the urgent need for more targeted and sustainable therapies that can address the root causes of IDD and improve long-term patient outcomes.
IDD is a complex process influenced by multiple factors, and elucidation of the cellular components involved is crucial for determining its molecular mechanisms. Nucleus pulposus (NP) cells (NPCs) and annulus fibrosus (AF) cells (AFCs) are important components of the intervertebral disc. With increasing age, damage to NPCs and AFCs leads to gradual degeneration of the structure and function of the disc, manifested by pathological changes such as extracellular matrix (ECM) degradation, cell apoptosis, and inflammatory responses6. Although previous studies have explored the pathological mechanisms of IDD7,8including imbalanced metabolism of the ECM, the mediating role of inflammatory factors, and the process of cell apoptosis, the deeper mechanism by which IDD is regulated by ageing genes remains unclear and requires further research.
Transcriptomic and proteomic analyses, important tools in modern biological research, can reveal changes in gene expression and protein levels, as well as their dynamic interactions9. Transcriptomic and proteomic approaches have been applied to the study of IDD; however, previous studies have often focused on the roles of single molecules or proteins and have relied primarily on independent analyses of either transcriptomic or proteomic data. These issues limit the ability of these approaches to comprehensively reveal the correlations between transcriptomic and proteomic profiles and their potential impacts on IDD. Therefore, this research method is not sufficient to fully elucidate the complex mechanisms of this disease. Cherif H et al.10 reported the use of single-cell sequencing analysis to reveal cell phenotypes and biomarkers related to IDD, including C2orf40, CD44, and MT2A. Zhang Y et al.11 discovered new chondrocyte subtypes in the NP in the context of IDD through single-cell RNA sequencing (scRNA-seq), providing a theoretical basis for further understanding the pathological changes involved in IDD. Although single-omic studies have provided important clues about the mechanisms of disease, their limited coverage, inability to analyse multilevel regulation, and neglect of molecular network interactions have limited the comprehensiveness of the obtained results.
Multiomic analysis, which integrates data from various biological layers, such as genomics, transcriptomics, proteomics, and metabolomics, provides a more comprehensive and accurate understanding of the molecular mechanisms underlying diseases. By combining these different types of data, multiomics enables researchers to uncover complex interactions among genes, proteins, and metabolic pathways that individual omic studies may miss. This holistic approach has been successfully applied in various disease studies, offering deeper insights into the mechanisms of disease progression, the identification of novel biomarkers, and the discovery of new therapeutic targets. In the context of IDD, multiomic analysis has the potential to provide a more complete picture of the molecular changes involved, advancing our understanding and ultimately improving treatment strategies. Therefore, the use of multiomic technologies to study the pathogenesis of IDD and identify potential therapeutic targets can help us gain a deeper understanding of the IDD mechanism of ageing regulation.
Competing endogenous RNA (ceRNA) networks have emerged as important mechanisms in the regulation of gene expression. A ceRNA refers to any RNA molecule, including mRNAs, long noncoding RNAs (lncRNAs), and pseudogenes, that can act as a molecular sponge for microRNAs (miRNAs), thereby modulating the activity of miRNAs and influencing gene expression. In this model, miRNAs interact with various RNA molecules, creating a complex regulatory network that controls gene expression at the posttranscriptional level. Recent studies have highlighted the importance of ceRNA networks in various biological processes and diseases. Understanding these interactions is crucial for deciphering the molecular mechanisms underlying diseases such as IDD.
In this study, we aimed to systematically identify key genes and proteins that are regulated by ageing mechanisms associated with IDD, with a particular focus on the identification of CXCL14, a novel factor implicated in this disease. Using transcriptomic and proteomic data from the NP of the intervertebral disc, we hypothesize that CXCL14 regulates the biological processes underlying IDD through the NF-κB signalling pathway. Our goal was to uncover the potential pathogenic mechanisms underlying IDD that are related to ageing and provide new theoretical foundations and clinical references for the development of targeted interventions and treatments.
Methods
Participant inclusion criteria
Clinical and pathological data for the patients, including age, sex, and site of onset, were collected. The following criteria were used to exclude participants from the study: (1) the presence of other diseases affecting the intervertebral disc (such as infectious spondylitis, tuberculosis, tumours, and rheumatic diseases); (2) a history of previous spinal surgery (such as discectomy or spinal fusion); and (3) a history of severe spinal trauma, which may lead to acute injury rather than chronic degeneration. In addition, (4) patients with IDD classified as Pfirrmann grades12 1–2 with acute disc herniation were assigned to the young/normal group, whereas those with IDD classified as Pfirrmann grades 6–7 with disc herniation were assigned to the elderly/degenerative group. Patients whose IDD severity did not meet the study requirements or who fell outside the scope of the study design were excluded. Patients with (5) systemic comorbidities or chronic pain conditions that could influence intervertebral disc biology, including but not limited to diabetes mellitus, autoimmune diseases, chronic inflammatory disorders, fibromyalgia, and other chronic widespread pain syndromes were also excluded. All participants underwent a comprehensive clinical history review and physical examination to ensure compliance with these criteria.
Clinical sample collection and storage
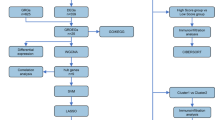
This study was approved by the Ethics Review Committee of the Affiliated Hospital of Hubei University of Chinese Medicine (Approval No. HBZY2022-C03-02). Written informed consent was obtained from each patient before any research procedures were performed. The samples used in this study were collected in accordance with the ethical guidelines established by the Declaration of Helsinki. From June 2022 to December 2023, 208 patients who underwent lumbar surgery at the Department of Orthopaedics, Affiliated Hospital of Hubei University of Traditional Chinese Medicine, were assessed. Among these patients, 6 met the eligibility criteria: 3 in the elderly group and 3 in the young group. Therefore, valid NP samples were collected from these 6 patients. The samples were collected immediately after surgery and rapidly frozen in liquid nitrogen for further processing. Transcriptomic and proteomic analyses were performed by Shenzhen BGI Technology Co., Ltd. (Wuhan). The workflow adopted by the research institute for studying the transcriptomes and proteomes of IDD patients is shown in Figs. 1 and 2.

Transcriptomic analysis workflow diagram. (A) Experimental Flowchart. (B) Bioinformatics Pipeline.

Proteomic analysis workflow diagram. (A) Sample Experimental Flow. (B) Bioinformatics Pipeline.
RNA extraction and quality assessment
Total RNA was extracted from intervertebral disc tissues using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. Briefly, tissue samples were ground in liquid nitrogen and processed using a chloroform/isopropanol extraction protocol. RNA was precipitated with isopropanol and washed with 75% ethanol before being resuspended in RNase-free water. RNA quality and quantity metrics for each sample are summarized in Table 1. RNA concentration was measured using the Qubit RNA HS Assay Kit (Thermo Fisher Scientific) and is reported in nanograms per microliter (ng/µL). RNA integrity was assessed using the Agilent Bioanalyzer 2100 system, which provides the RNA Integrity Number (RIN) as an indicator of degradation. Additionally, the 28 S/18S rRNA ratio is included to provide further insight into RNA integrity, although it is known to be sensitive to sample type and experimental conditions. These values were used to screen for high-quality RNA suitable for library construction and downstream sequencing analysis. Although some samples exhibited 28 S/18S ratios slightly above the typical threshold (e.g., > 2.2), all six selected RNA samples passed internal library quality control checks and were successfully used in sequencing.
Analysis of differentially expressed genes (DEGs)/differentially expressed proteins (DEPs)
DEG analysis was performed using Bowtie2 (v2.3.4.3). Gene expression quantification was conducted with RSEM (v1.3.1), and expression clustering heatmaps of different samples were generated using the R package pheatmap (v1.0.8). DEGs were identified using DESeq2 (v1.4.5), which applies the Benjamini-Hochberg procedure to adjust for multiple testing. Genes with an adjusted Q value ≤ 0.05 were considered significantly differentially expressed. For differentially expressed protein (DEP) analysis, the fold change in protein expression in each comparison group (elderly vs. young) was calculated, and Welch’s t test was used to assess statistical significance. Prior to testing, the protein abundance values were log2-transformed to improve normality and stabilize variance across samples. Significant differences were identified using |log2-fold change| > 1.5 and P value < 0.05 as screening thresholds. Due to the limited number of differentially expressed proteins, multiple testing correction was not applied to proteomic p-values; however, stringent thresholds were set to minimize false positives. Finally, enrichment analysis was performed on the DEPs.
Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) enrichment analyses
GO (http://www.geneontology.org/) and KEGG13,14,15 (https://www.kegg.jp/) enrichment analyses of the DEGs were conducted using hypergeometric tests with the phyper function. A Q value ≤ 0.05 was set as the threshold, and terms meeting this criterion were considered significantly enriched among the candidate genes.
Validation with external datasets
DEGs were identified from the GSE147383 gene expression dataset, a publicly available resource titled “Gene Expression Data of Human Intervertebral Discs”, using the “limma” package (v 3.52.2) in R software (v 4.3.3). This dataset, which was released on June 30, 2020, includes 8 samples from 4 individuals, representing AF and NP tissues from each individual. This study aimed to analyse the transcriptomes of AF and NP tissues from young individuals without degenerative disc disease (n = 2) and elderly individuals with degenerative disc disease (n = 2) to explore the molecular dynamics of the intervertebral disc during human ageing. The criteria for DEG screening were a |log2-fold change| > 0.5 and a Benjamini-Hochberg adjusted P value < 0.05. This more relaxed cutoff (fold change > 1.4) was chosen due to the limited sample size and higher variability of the external dataset, allowing the inclusion of biologically relevant genes that may exhibit modest expression changes. To ensure data quality and alignment with research objectives, we performed preprocessing to remove genes with low expression and samples with substantial missing values. Heatmaps were generated using the “ggplot2” package (v 3.4.4) to visualize expression patterns. Volcano plots were generated using the R software (v 4.2.1) with the “clusterProfiler” package (v 4.4.4) to verify the significance of the DEGs. Boxplots were created using R (v 4.2.1) to illustrate the intergroup expression levels of the target genes.
Construction of ceRNA and Gene-Gene interaction networks
The interactions and functions of the CXCL14 gene were analysed via GeneMANIA (http://genemania.org)16. The predicted miRNAs and lncRNAs for the DEG CXCL14 were obtained using the starBase (v 2.0) platform. These predicted miRNAs and lncRNAs were then imported into Cytoscape software to construct the ceRNA regulatory network.
RT‒qPCR validation
NP tissues were extracted from patients and homogenized on ice using a grinder for 10‒15 cycles to prepare tissue homogenates. Total RNA was then isolated from the NP tissues via TRIzol reagent (Vazyme, Nanjing, China) and reverse-transcribed into cDNA. RT‒qPCR was performed using the AceQ qPCR SYBR Green Master Mix (Q111‒02, Vazyme, China) according to the manufacturer’s instructions on a 7500 real-time PCR system (Applied Biosystems, Inc., Carlsbad, CA, USA). The sequences of the qPCR primers are provided in the supplementary files (Table 2). GAPDH was used as the internal reference gene, and the log(2^-(ΔΔCt)) method was applied for statistical analysis.
Statistical information
All the statistical analyses were performed using R software. Data are presented as the means with standard deviations (SDs) for continuous variables. The Wilcoxon test was used for comparisons between two groups. For the clinical validation results, an unpaired Student’s t test was used to determine the statistical significance of differences between the two groups. A P value < 0.05 was considered to indicate statistical significance. Due to the small sample size, potential confounding factors such as sex and BMI were not included as covariates in the statistical models, and efforts were made to minimize their effects through matched sample selection. Nevertheless, we acknowledge that residual confounding may exist and may influence the magnitude or direction of the observed molecular differences. This limitation should be considered when interpreting the findings and underscores the need for larger, more stratified future studies.
Detailed experimental materials and methods
Detailed descriptions of the experimental procedures and statistical analyses used in this study are provided in the Supplementary Materials and Methods.
Results
Patient and sample characteristics
To determine the key molecular factors involved in age-related IDD, we first aimed to identify differential gene and protein expression by analysing NP samples from patients with different degrees of disc degeneration. A total of 6 patients were included in this study, and valid NP samples were collected from these patients and stored in a sample repository until use. After preliminary screening of the samples, 3 young patients with typical intervertebral disc NP tissues classified as modified Pfirrmann grades 1–2 were selected as the normal control group, and 3 elderly patients with degenerated intervertebral disc NP tissues classified as modified Pfirrmann grades 6–7 were selected as the ageing-related degeneration group on the basis of the modified Pfirrmann classification. Patient information, including sex, age, surgical segment, and modified Pfirrmann grade, is summarized in Table 1. The specific experimental workflow is shown in Fig. 3.

Study flowchart.
Transcriptomic analysis
To identify the molecular differences related to IDD in young and elderly patients, we performed RNA-seq analysis of NPC transcripts. A total of 17,389 genes were identified in the database (Fig. 4A). After quality control of all the samples, high sequencing accuracy and consistency were observed. Clean reads were then aligned to the reference genome (Homo sapiens GRCh38) using HISAT2 and Bowtie2, with an average alignment rate of 98.05%, indicating ideal genomic alignment of the samples. Among the identified genes, 16,268 genes were shared between the young and elderly groups, with 593 DEGs in the young group and 528 in the elderly group. Differential expression analysis revealed 45 genes that were significantly differentially expressed between the young and elderly groups (Fig. 4B) according to the screening criteria of a |log2-fold change| > 1.5 and a P value < 0.05. This cutoff was selected as a fold change greater than 2.8 to enhance result robustness given the small sample size and high-quality dataset. The gene expression correlation heatmap (Fig. 4C) showed strong within-group consistency among the young samples, with Pearson correlation coefficients ranging from 0.717 to 0.967. In contrast, the elderly samples exhibited greater variability, with coefficients ranging from 0.167 to 0.806. This heterogeneity among elderly samples may reflect individual differences in the severity or molecular features of age-related IDD. Notably, among the elderly samples, E1 showed a relatively higher correlation with the younger group in Fig. 4C, which may suggest that E1 represents an earlier or less advanced stage of degeneration at the molecular level, despite being classified as elderly by clinical criteria. These results underscore the importance of stratified analysis and support the overall quality of the transcriptomic data. Principal component analysis (PCA) revealed a general grouping trend between young and elderly samples (Fig. 4D), with partial within-group clustering. Some variation was observed among biological replicates, particularly in the elderly group, which may reflect underlying molecular heterogeneity. The first two principal components explained 92.5% of the total variance, supporting the robustness of the transcriptomic dataset.
Moreover, among these 45 DEGs, key genes related to apoptosis, the inflammatory response, and ECM degradation were included. GO annotation of the DEGs (Fig. 5A, B) revealed that these genes were enriched primarily in biological processes related to ECM organization, apoptosis, and the immune response. KEGG pathway analysis (Fig. 5C, D) indicated that the DEGs were significantly enriched in metabolic pathways as well as cell signalling pathways associated with ageing.

(A) Gene Expression Venn Diagram. (B) Bar Chart of Differential Genes. (C) Sample Correlation Heatmap. (D) Principal Component Analysis.

(A) GO Cellular component Enrichment Bubble Chart. (B) GO Molecular function Enrichment Bubble Chart. (C) GO Biological process Enrichment Bubble Chart. (D) KEGG Pathway Enrichment Bubble Chart.
Proteomic analysis
To further investigate the protein-level changes associated with IDD, we performed proteomic analysis and identified 2,301 proteins using TMT technology. For quantitative comparisons, 35 DEPs were selected on the basis of the criteria of |log2-fold change| > 1.5 and P < 0.05 (Fig. 6A). The gene expression correlation heatmap revealed high Pearson correlation coefficients between samples (Fig. 6B) within each group, further supporting the reliability of the experimental data. Similarly, in the proteomic PCA plot (Fig. 6C), young and elderly samples showed partial separation, though with some overlap. This observation highlights both the biological diversity of the samples and the complexity of IDD.
In the comparison between the young and elderly groups, the DEPs were primarily involved in biological processes such as ECM remodelling, the inflammatory response, the cellular stress response, and cell migration. GO annotation of the DEPs (Fig. 7A, B) revealed significant enrichment of pathways related to collagen biosynthesis, the inflammatory response, and ECM degradation, further supporting the gene changes observed in the transcriptomic analysis. KEGG pathway analysis (Fig. 7C, D) further revealed that the DEPs associated with IDD were predominantly enriched in cell signalling and inflammation-related pathways.

(A) Bar Chart of Differential Protein. (B) Heat map of sample correlation analysis. (C) PCA result.

(A) GO Cellular component Enrichment Bubble Chart. (B) GO Molecular function Enrichment Bubble Chart. (C) GO Biological process Enrichment Bubble Chart. (D) KEGG Pathway Enrichment Bubble Chart.
Integrating the transcriptomic and proteomic analyses
To explore the correlation between mRNA and protein expression, we performed integrative transcriptomic and proteomic analyses. A Venn diagram (Fig. 8A) revealed that 44 genes were differentially expressed at the transcriptomic level, 34 proteins were differentially expressed at the proteome level, and only one factor was differentially expressed at both the gene and protein levels. This common molecule was CXCL14, which was found to be significantly upregulated at both the mRNA and protein levels. The consistent change in expression observed for this factor suggests that this gene may be regulated at multiple levels and may play an important role in specific biological processes. Boxplots were used to visualize gene expression distributions between the correlated and noncorrelated protein groups (Fig. 8B). While no formal statistical tests were applied, visual inspection of medians, quartiles, and outliers suggested potential differences between the two groups. Each point on the scatter plot (Fig. 8C) represents the expression level of a gene in the young and elderly groups. The correlations between the expression levels of all quantified proteins and genes can be identified by analysing their expression associations (Fig. 8D). Further GO enrichment analysis of the DEGs and DEPs (Fig. 9A, B) revealed that ageing-induced IDD is associated with multiple key biological processes, molecular functions, and cellular components. Specifically, GO terms such as “inflammatory response” (GO:0006954), “positive regulation of cytokine production” (GO:0001819), and “neutrophil chemotaxis” (GO:0030593) highlighted the involvement of inflammatory pathways in the IDD group. Likewise, terms like “extracellular matrix organization” (GO:0030198) and “regulation of apoptotic signaling pathway” (GO:1902175) suggested significant remodelling of the disc microenvironment and increased cell turnover. KEGG pathway enrichment analysis revealed multiple signalling pathways associated with ageing and IDD. As shown in Fig. 9C, the top significantly enriched pathways were related to the immune response, cell death, and matrix regulation processes. Figure 9D presents a four-dimensional bubble plot that integrates enrichment scores, gene counts, and p-values, further highlighting the complexity and diversity of biological pathways involved. Together, these visualizations underscore the multifactorial nature of age-related IDD at the pathway level. The findings suggest that CXCL14, which is regulated at both the gene and protein levels, may be a central factor in the molecular mechanisms driving ageing-related IDD.

(A) Venn diagram of the correlation number between differential proteome and transcriptome at the quantitative level. (B) Boxplot of correlated and uncorrelated gene expressions. (C) Scatterplots of correlated and uncorrelated gene expression levels. (D) Expression correlation diagram of all quantitative proteins and genes.

(A) Expression correlation diagram of all quantitative proteins and genes. (B) Four-dimension bubble diagram of GO enrichment correlation analysis. (C) Pathway enrichment correlation analysis significance distribution diagram. (D) Four-dimension bubble diagram of pathway enrichment correlation analysis.
Verification with an external dataset
Differential expression analysis was performed using the GSE147383 gene dataset, identifying a total of 326 DEGs, with 151 genes upregulated and 175 genes downregulated in IDD samples compared with controls. Among these genes, CXCL14 was significantly upregulated in IDD, with a fold change of 4.61 and a P value of 7.08 × 10−4, indicating a strong association with IDD. The volcano plot (Fig. 10A) confirmed the differential expression of CXCL14, revealing its prominence among the upregulated genes. A boxplot (Fig. 10B) further illustrated the significant upregulation of CXCL14 in IDD samples, with clear differences between the elderly and young groups, suggesting its potential role in the inflammatory processes driving IDD progression.
Construction of CeRNA and Gene-Gene interaction networks
To further elucidate the functional roles of CXCL14-related genes in IDD, we employed extensive transcriptomic and proteomic data17 to construct a gene interaction network using the GeneMANIA database. This database was utilized to validate the functions of CXCL14-related genes, and the resulting gene interaction network (Fig. 10C) confirmed their involvement in the aforementioned biological functions. The network portrays diverse types of gene interactions, encompassing physical interactions, coexpression, predicted associations, colocalization, genetic interactions, signalling pathways, and shared protein domains. Functional enrichment analysis revealed that these genes are involved mainly in crucial biological processes, such as cytokine activity, chemokine receptor binding, the cellular response to chemokines, granulocyte migration, the humoral immune response, the regulation of macrophage migration, and the response to IL-1. After constructing the ceRNA regulatory network (Fig. 10D), we identified four miRNAs, namely, hsa-miR-582-3p, hsa-miR-2116-3p, hsa-miR-532-3p, and hsa-miR-150-5p, which may play a role in the regulatory mechanisms of IDD via their interactions with CXCL14 and its associated genes. These findings suggest potential posttranscriptional regulation of CXCL14 by these miRNAs, which could influence the molecular pathways involved in IDD progression.
Verification by rt‒qpcr
To investigate the expression of CXCL14 in different age groups, we performed RT‒qPCR on NP tissues from young and elderly patients. The results (Fig. 10E) revealed a significant difference in CXCL14 gene expression between the young and elderly groups. Specifically, CXCL14 expression was significantly upregulated in the elderly group, whereas it was downregulated in the young group. The bar graph in the figure shows the statistically significant difference between the two groups, as indicated by the P value. The asterisk (*) indicates that the P value is less than 0.05, indicating that the result is unlikely to be due to random factors. Therefore, the upregulation of CXCL14 gene expression in the elderly group can be considered biologically significant.

(A) Volcano plot showing the differential expression of genes in the GSE147383 dataset. (B) Boxplot of CXCL14 expression levels in the control and IDD groups based on the GSE147383 dataset. (C) GeneMANIA Gene Interaction Network related to CXCL14. (D) CXCL14-related mRNA-miRNA-lncRNA regulatory networks in ENCORI. (E) RT-qPCR was used to detect the differential expression of feature genes in young and elderly groups.
Discussion
This study aimed to explore the effects of ageing on IDD and the associated regulatory mechanisms, with a particular focus on the key role of CXCL14 in IDD. By integrating transcriptomic and proteomic analyses, we explored in detail how ageing contributes to IDD and identified relevant molecular processes. GO enrichment analysis of the DEGs and DEPs identified in this study revealed that ageing-induced IDD involves multiple key biological processes, molecular functions, and cellular components. In terms of biological processes, the DEGs and DEPs were enriched mainly in ECM organization, apoptosis, and the immune response, suggesting that one of the key mechanisms of IDD is degradation of the ECM and programmed cell death, which may exacerbate disc degeneration, especially in the context of ageing. In terms of molecular functions, the DEGs and DEPs were enriched primarily in metalloprotease activity, receptor binding, and cell‒cell adhesion, further indicating that IDD is regulated by complex signalling pathways. In terms of cellular components, the DEPs were enriched mainly in the ECM, endoplasmic reticulum, and plasma membrane structures, and changes in these cellular components may be closely related to cell damage and ECM metabolic imbalance in IDD. Specifically, changes in endoplasmic reticulum stress and cell membranes can affect intervertebral disc cell survival and function, further driving disc degeneration.
Consistent with the findings of previous studies, our findings also support the importance of inflammation, ECM degradation, and abnormal regulation of signalling pathways in the progression of IDD18. Notably, CXCL14, identified in this study, may contribute to the inflammatory response and ECM degradation observed in IDD. Proinflammatory factors such as IL-1β and TNF-α, which were previously shown to promote immune cell recruitment and activate inflammatory signalling, are consistent with our findings that identified CXCL14 as a key mediator in these processes. Furthermore, our results align with the involvement of various signalling pathways, including the NF-κB, MAPK, and Wnt/β-catenin pathways, in IDD development. Specifically, CXCL14 can act through these pathways to regulate proinflammatory gene expression and influence cell survival and apoptosis, thereby exacerbating disc degeneration. These insights not only reinforce the relevance of these established mechanisms but also introduce CXCL14 as a novel player in IDD pathogenesis, offering potential for targeted therapeutic strategies.
This study revealed that CXCL14 is significantly upregulated in ageing-related IDD and that its expression changes are evident not only at the mRNA level but also at the protein level, suggesting that CXCL14 may regulate downstream signalling pathways through both transcriptional and translational regulation19.
CXCL14 is a chemokine that mediates inflammation and ECM remodeling, two hallmarks of IDD. While it has been studied in oncological contexts, its upregulation in degenerative NP tissue indicates a localized, non-neoplastic role. Specifically, CXCL14 may function as a chemotactic signal that enhances immune cell infiltration and protease activation within intervertebral discs, thereby exacerbating the catabolic and inflammatory microenvironment characteristic of IDD20,21,22,23,24. These findings broaden the understanding of CXCL14 beyond its role in systemic disease and support its potential relevance in spine pathology.
Prior studies have shown that CXCL14 can activate the NF-κB signalling pathway via receptors such as ACKR2 and SDC125,26. Given the known relevance of NF-κB in IDD progression, we hypothesize that CXCL14 contributes to IDD pathogenesis by promoting proinflammatory signalling, enhancing ECM degradation, and disrupting NP cell homeostasis. These mechanisms align with established features of degenerative disc disease and warrant further functional studies.
In this study, we further explored the functional implications of CXCL14 in IDD through integrative analyses. CXCL14 may exacerbate disc inflammation by attracting immune cells27 and may contribute to matrix degradation through upregulation of proteases such as MMPs and ADAMTS28. These effects, together with elevated IL-1β and TNF-α, may form a feed-forward loop promoting disc degeneration29. Additionally, ceRNA network analysis revealed regulatory interactions with inflammation-associated miRNAs, reinforcing the position of CXCL14 in the degenerative molecular network.
Therapeutic strategies targeting CXCL14, including the inhibition of its expression or blockade of its downstream signalling pathways, offer promising avenues for delaying disease progression and optimizing clinical outcomes in IDD patients. By directly addressing the molecular drivers of IDD, these approaches have the potential to improve upon current treatments, offering more targeted and effective therapies.
To validate the role of CXCL14 in IDD, we analysed an external dataset (GSE147383), which confirmed the significant upregulation of CXCL14 in the elderly group. Differential expression analysis revealed a statistically significant P value, supporting the hypothesis that CXCL14 plays an important role in disease progression. This finding was further validated by boxplots and RT‒qPCR analysis, which confirmed the increased expression of CXCL14 in IDD patients, consistent with our earlier findings. Additionally, gene regulatory network and ceRNA network analyses revealed strong associations between CXCL14 and several inflammation-related factors, such as hsa-miR-150-5p and NEAT1. These findings suggest that CXCL14 may contribute to the development and progression of IDD by regulating key inflammatory and molecular pathways, which is consistent with its known role in inflammatory diseases and tissue remodelling.
Notably, our study has several limitations that warrant acknowledgement. First, owing to ethical constraints and the difficulty in obtaining healthy human intervertebral disc samples, we utilized the NP tissues of young patients with mild IDD and acute protrusion as substitutes for the control group. While this approach enabled us to conduct omic analyses, it is possible that the data did not fully capture the distinct cell populations, as the tissue samples from the acute herniation group may not accurately reflect the molecular characteristics of healthy discs. Another limitation of this study is the use of mildly degenerated discs as control samples. While these tissues did not show overt radiological or histopathological signs of severe degeneration, they may still harbour early molecular alterations that distinguish them from truly healthy intervertebral discs. Such baseline deviations may decrease the contrast between groups, potentially underestimating the extent of degeneration-related transcriptomic and proteomic changes. This limitation reflects a broader challenge in IDD research, where ethical access to non-degenerated discs—particularly from aged, asymptomatic individuals—is often restricted. Additionally, this study included only ten NP samples (3 from the elderly group and 3 from the young group). Although we rigorously screened for confounding factors, the small sample size may limit the statistical power and generalizability of our findings. However, despite these limitations, the consistent molecular patterns observed between the two groups increase the reliability of our conclusions. Furthermore, our findings provide valuable insights that merit further investigation in larger, more diverse cohorts. Future studies with larger sample sizes and healthy control tissues are necessary to validate these results and elucidate the precise molecular mechanisms of CXCL14 in IDD.
Conclusion
The mechanisms underlying the role of CXCL14 in IDD are multifaceted and include processes such as inflammation, ECM degradation, cell apoptosis, and immune regulation. This study aimed to elucidate these mechanisms through integrated transcriptomic and proteomic analyses, thereby providing novel insights into the pathological processes associated with IDD. Our findings indicate that CXCL14 plays a pivotal role in IDD development and may serve as a potential therapeutic target for mitigating inflammatory responses and excessive ECM degradation. Future research should further investigate the specific roles of CXCL14 in IDD and refine these therapeutic strategies to optimize patient prognosis and quality of life.
Data availability
All raw transcriptomic data for this study are available at the NCBI Sequence Read Archive (SRA) under the accession number [PRJNA1280549], the mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the iProX partner repository30,31 with the dataset identifier PXD065307. Further inquiries can be directed to the corresponding author.
Abbreviations
- IDD:
-
intervertebral disc degeneration
- DEGs:
-
differentially expressed genes
- DEPs:
-
differentially expressed proteins
- NSAIDs:
-
nonsteroidal anti-inflammatory drugs
- NP:
-
nucleus pulposus
- NPCs:
-
nucleus pulposus cells
- AF:
-
annulus fibrosus
- AFCs:
-
annulus fibrosus cells
- ECM:
-
extracellular matrix
- scRNA-seq:
-
single-cell RNA sequencing
- ceRNA:
-
competing endogenous RNA
- lncRNAs:
-
long noncoding RNAs
- miRNAs:
-
microRNAs
- RIN:
-
RNA integrity number
- GO:
-
gene ontology
- KEGG:
-
kyoto encyclopedia of genes and genomes
- SDs:
-
standard deviations
- PCA:
-
principal component analysis
- TIL:
-
tumour-infiltrating lymphocyte
- NSCLC:
-
non-small cell lung cancer
- EMT:
-
epithelial‒mesenchymal transition
- ALD:
-
alcoholic liver disease
- MMPs:
-
matrix metalloproteinases
References
Hoy, D. et al. The global burden of low back pain: estimates from the global burden of disease 2010 study. Ann. Rheum. Dis. 73, 968–974. https://doi.org/10.1136/annrheumdis-2013-204428 (2014).
Song, C. et al. An in-depth analysis of the Immunomodulatory mechanisms of intervertebral disc degeneration. JOR Spine. 5, e1233. https://doi.org/10.1002/jsp2.1233 (2022).
Dagenais, S., Caro, J. & Haldeman, S. A systematic review of low back pain cost of illness studies in the united States and internationally. Spine J. 8 https://doi.org/10.1016/j.spinee.2007.10.005 (2008).
Martin, B. I. et al. Expenditures and health status among adults with back and neck problems. JAMA 299, 656–664. https://doi.org/10.1001/jama.299.6.656 (2008).
Saeidian, A. H., Youssefian, L., Vahidnezhad, H. & Uitto, J. Research techniques made simple: Whole-Transcriptome sequencing by RNA-Seq for diagnosis of Monogenic disorders. J. Invest. Dermatol. 140 https://doi.org/10.1016/j.jid.2020.02.032 (2020).
Romaniyanto, F. N. U. et al. Adipose-Derived stem cells (ASCs) for regeneration of intervertebral disc degeneration: review Article. Stem Cells Cloning. 15, 67–76. https://doi.org/10.2147/SCCAA.S379714 (2022).
Li, W., Xu, Y. & Chen, W. Bone mesenchymal stem cells deliver exogenous LncRNA CAHM via exosomes to regulate macrophage polarization and ameliorate intervertebral disc degeneration. Exp. Cell. Res. 421, 113408. https://doi.org/10.1016/j.yexcr.2022.113408 (2022).
Zhang, J. et al. The influence of intervertebral disc microenvironment on the biological behavior of engrafted mesenchymal stem cells. Stem Cells Int. 2022 (8671482). https://doi.org/10.1155/2022/8671482 (2022).
Ben-Chetrit, N. et al. Integration of whole transcriptome Spatial profiling with protein markers. Nat. Biotechnol. 41, 788–793. https://doi.org/10.1038/s41587-022-01536-3 (2023).
Cherif, H. et al. Single-Cell RNA-Seq analysis of cells from degenerating and Non-Degenerating intervertebral discs from the same individual reveals new biomarkers for intervertebral disc degeneration. Int. J. Mol. Sci. 23 https://doi.org/10.3390/ijms23073993 (2022).
Zhang, Y. et al. Single-cell RNA-seq analysis identifies unique chondrocyte subsets and reveals involvement of ferroptosis in human intervertebral disc degeneration. Osteoarthr. Cartil. 29, 1324–1334. https://doi.org/10.1016/j.joca.2021.06.010 (2021).
Griffith, J. F. et al. Modified Pfirrmann grading system for lumbar intervertebral disc degeneration. Spine (Phila Pa. 1976). 32, E708–E712 (2007).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677. https://doi.org/10.1093/nar/gkae909 (2025).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Franz, M. et al. GeneMANIA update 2018. Nucleic Acids Res. 46, W60–W64. https://doi.org/10.1093/nar/gky311 (2018).
Warde-Farley, D. et al. The genemania prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 38, W214–W220. https://doi.org/10.1093/nar/gkq537 (2010).
Wang, B. et al. miR-31 from Mesenchymal Stem Cell-Derived Extracellular Vesicles Alleviates Intervertebral Disc Degeneration by Inhibiting NFAT5 and Upregulating the Wnt/β-Catenin Pathway. Stem Cells Int 2164057, (2022). https://doi.org/10.1155/2022/2164057 (2022).
Yu, Y. et al. Human embryonic Stem-Cell-Derived exosomes repress NLRP3 inflammasome to alleviate pyroptosis in nucleus pulposus cells by transmitting miR-302c. Int. J. Mol. Sci. 24 https://doi.org/10.3390/ijms24087664 (2023).
Hou, Y., Shi, J., Guo, Y. & Shi, G. DNMT1 regulates polarization of macrophage-induced intervertebral disc degeneration by modulating SIRT6 expression and promoting pyroptosis in vivo. Aging (Albany NY). 15, 4288–4303. https://doi.org/10.18632/aging.204729 (2023).
Gowhari Shabgah, A. et al. Chemokine CXCL14; a double-edged sword in cancer development. Int. Immunopharmacol. 97, 107681. https://doi.org/10.1016/j.intimp.2021.107681 (2021).
Parikh, A. et al. Malignant cell-specific CXCL14 promotes tumor lymphocyte infiltration in oral cavity squamous cell carcinoma. J. Immunother Cancer. 8 https://doi.org/10.1136/jitc-2020-001048 (2020).
Chang, T. M. et al. CXCL14 promotes metastasis of non-small cell lung cancer through ACKR2-depended signaling pathway. Int. J. Biol. Sci. 19, 1455–1470. https://doi.org/10.7150/ijbs.79438 (2023).
Li, N. et al. Targetable Brg1-CXCL14 axis contributes to alcoholic liver injury by driving neutrophil trafficking. EMBO Mol. Med. 15, e16592. https://doi.org/10.15252/emmm.202216592 (2023).
Tian, H. Y., Liang, Q., Shi, Z. & Zhao, H. Exosomal CXCL14 Contributes to M2 Macrophage Polarization through NF-κB Signaling in Prostate Cancer. Oxidative Medicine and Cellular Longevity 7616696, (2022). https://doi.org/10.1155/2022/7616696 (2022).
Ma, R. et al. PAX6/CXCL14 regulatory axis promotes the repair of corneal injury by enhancing corneal epithelial cell proliferation. J. Transl Med. 22, 458. https://doi.org/10.1186/s12967-024-05270-z (2024).
Lu, J., Chatterjee, M., Schmid, H., Beck, S. & Gawaz, M. CXCL14 as an emerging immune and inflammatory modulator. J. Inflamm. (Lond). 13, 1. https://doi.org/10.1186/s12950-015-0109-9 (2016).
Zhang, L. et al. Astragaloside IV relieves IL-1β-induced human nucleus pulposus cells degeneration through modulating PI3K/Akt signaling pathway. Med. (Baltim). 102, e34815. https://doi.org/10.1097/MD.0000000000034815 (2023).
Chu, L. W. et al. Loganin prevents chronic constriction injury-provoked neuropathic pain by reducing TNF-α/IL-1β-mediated NF-κB activation and Schwann cell demyelination. Phytomedicine 67, 153166. https://doi.org/10.1016/j.phymed.2019.153166 (2020).
Ma, J. et al. iProX: an integrated proteome resource. Nucleic Acids Res. 47, D1211–D1217. https://doi.org/10.1093/nar/gky869 (2019).
Chen, T. et al. iProX in 2021: connecting proteomics data sharing with big data. Nucleic Acids Res. 50, D1522–D1527. https://doi.org/10.1093/nar/gkab1081 (2022).
Funding
This work was supported by the 2023–2024 Chinese Medicine Research Project of Hubei Provincial Administration of Traditional Chinese Medicine (ZY2023F006), and Hubei Provincial Natural Science Foundation Joint Fund Project (2023AFD126).
Author information
Authors and Affiliations
Contributions
All authors participated in data acquisition. TLZ and WW contributed to the conception and design of the study. TLZ, ZWZ, YH, ZJC and WW did the data collection and analysis. TLZ, THZ and QWH performed experiments. TLZ and THZ contributed to the drafing and revision of the manuscript. All authors read and approved the fnal manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethical approval
Our research received approval from the Ethics Department of the Hubei Provincial Hospital of Traditional Chinese Medicine. Even though it was an experimental study based on clinical samples, we adhered to the Declaration of Helsinki and relevant policies in China.
Consent for publication
Patients signed informed consent regarding publishing their data and photographs.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, T., Zhang, T., He, Q. et al. CXCL14 drives age-related intervertebral disc degeneration via NF-κB pathway activation in a multiomic study. Sci Rep 15, 25307 (2025). https://doi.org/10.1038/s41598-025-10998-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-10998-9
