
Abstract
To explore the role of Methyltransferase-like 1 (METTL1) in the prognosis, diagnosis, and immune infiltration of Breast invasive carcinoma (BRCA), transcriptome data of BRCA from The Cancer Genome Atlas (TCGA) database were analyzed. Then, the METTL1 expression in normal and cancer tissues was compared using the DESeq2 package. Next, the diagnostic and prognostic value of METTL1 was evaluated using receiver operating characteristic curves and survival analysis, respectively. Co-expressed genes and differentially expressed genes related to METTL1 were identified using Pearson correlation and Wilcoxon tests, with overlapping genes subjected to protein-protein interaction network construction to identify hub genes via degree algorithm. After that, immune infiltration analyses were performed using CIBERSORT and xCELL algorithms. METTL1 expression was significantly higher in BRCA tissues compared to normal tissues, with significant diagnostic value. Furthermore, BRCA patients with low METTL1 expression showed better 5-year overall survival and disease specific survival than those with high METTL1 expression. A total of 1829 differentially expressed genes and 236 co-expressed genes were identified, with five hub genes selected. These genes, along with METTL1, were positively associated with immunosuppressive cell types, such as regulatory T cells and follicular helper T cells, suggesting their role in shaping an immunosuppressive tumor microenvironment.
Similar content being viewed by others


Introduction
Breast cancer remains the most common malignant tumor in women, posing a serious threat to women’s health1,2. Despite significant advancements in improving breast cancer prognosis over recent decades, breast invasive carcinoma (BRCA) continues to be associated with a poor prognosis, which affects the overall survival of patients3,4. The prognosis of BRCA is related to molecular heterogeneity, and different subtypes may exhibit different histological features and treatment sensitivities5,6. Evidence at both the transcriptomic and genetic levels that BRCA progression and drug resistance have complex signaling7,8. Developing more effective biomarkers is an urgent priority to improve clinical treatment for BRCA.
RNA modifications affect the biological characteristics of tumors9. As an important m6A writer, methyltransferase-like 1 (METTL1) promotes cancer development when overexpressed10,11. In addition to m6A, there is evidence that N7-methylguanosine (m7G) tRNA modification is involved in tumor progression and drug resistance by selectively regulating the translation of oncogenic transcripts12,13. In terms of BRCA, tRNA modifications affect the metabolism and growth of breast cells and may accelerate tumor metastasis14,15. METTL1, an important paralogue of METTL3 located on chromosome 12, plays an essential role in normal mRNA translation and embryonic stem cell self-renewal and differentiation16. Alterations in this chromosomal region have been observed in various cancers17. It is reported that overexpression and amplification of METTL1 may contribute to poor prognosis in hepatocellular carcinoma18. In lung cancer, loss of METTL1 leads to impaired proliferation and invasion of lung cancer cells, thereby improving patient outcomes19. Mechanistically, recent studies have shown that METTL1 modifies m7G to influence chemotherapeutic drug sensitivity20,21. As reported by a previous study, METTL1 promotes the translation of EGFR/EFEMP1 via mediating m7G tRNA modification, thereby driving tumorigenesis22. By virtue of bioinformatics techniques, Zijie Gao et al. indicated that METTL1 might play a role in tumor progression-related pathways and contribute to tumor immune infiltration and stemness in pan-cancer23. Besides, higher METTL1 expression was correlated with better anti-PD-L1 treatment response and drugs sensitivity. However, despite these findings, the specific role of METTL1 in BRCA remains unclear.
In contrast to previous research that broadly investigates m7G modifications across multiple cancer types24, our study provides a more focused analysis of METTL1’s diagnostic and prognostic relevance specifically in BRCA using The Cancer Genome Atlas (TCGA) dataset. By examining the relationship between METTL1 expression and the clinical characteristics, prognosis and immune infiltration, our study aims to offer new insights into the role of METTL1 in BRCA and propose therapeutic targets for this malignancy.
Methods
Date sources
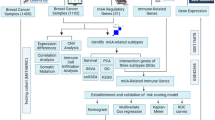
The TCGA database (https://portal.gdc.cancer.gov/repository) provided transcriptome profiling data (expressed in fragments per kilobase million, FPKM) and corresponding clinical information for 601 BRCA tumor samples and 64 normal breast tissue samples. Samples with incomplete clinical information or missing RNA-seq data were excluded to ensure data integrity. To correct for potential batch effects across samples, batch correction was performed using the sva package in R software. The overall study workflow was summarized in Fig. 1.

Flowchart of the study design.
METTL1 expression in breast invasive carcinoma
The METTL1 expression in BRCA was analyzed using RNA-seq transcriptome data from the TCGA-BRCA dataset. Differential expression analysis was performed using the DESeq2 package to compare METTL1 expression between BRCA tumor tissues and normal tissues25. For samples with matched tumor and adjacent normal tissues from the same patient, a paired analysis was performed using the Wilcoxon signed-rank test to account for inter-patient variability. To further validate the clinical relevance of METTL1, we analyzed its expression in subgroups of BRCA patients stratified by age (≦ 65 years vs. > 65 years) and tumor stage (Stage I-II vs. Stage III-IV). Besides, the GENT226 and CPTAC databases27 were used to analyze the mRNA and protein expression of METTL1 in breast cancer, respectively. A formal power calculation using the “pwr” package in R confirmed that the study had sufficient power (80%) at a 95% confidence level to detect significant differences in METTL1 expression across subgroups based on expected hazard ratios (HRs).
Role of METTL1 in prognosis
Patients were divided into high (above the median) and low (below the median) METTL1 expression groups based on the median expression level of METTL1 in the TCGA-BRCA dataset. Using the survival and survminer packages, we analyzed the effect of METTL1 expression level on overall survival (OS, defined as the time from diagnosis to death from any cause), disease specific survival (DSS, defined as the time from diagnosis to death specifically due to breast cancer), and disease free survival (DFS, defined as the time from initial treatment to cancer recurrence or death from any cause) in BRCA patients.
To assess the diagnostic value of METTL1 in differentiating BRCA tumor tissues from normal tissues, a receiver operating characteristic (ROC) curve was generated using the ROCR package28. The continuous METTL1 expression values were directly used as the predictor variable, and the binary response variable (BRCA vs. normal) was used as the classification outcome. The area under the ROC curve (AUC) was calculated to evaluate METTL1’s discriminatory power.
In addition, univariate and multivariate Cox regression analyses were performed based on METTL1 and clinicopathological features using the survival and rms packages in R software.
METTL1 co-expressed genes
A total of 236 METTL1 co-expressed genes (Table S1) were identified using RNA-seq expression data from the TCGA-BRCA cohort. The Pearson correlation analysis (p < 0.05 and |correlation coefficient| > 0.4) was performed to calculate the correlation between METTL1 expression and all other protein-coding genes in the dataset. The top 11 genes were illustrated in a circular diagram created with the corrplot and circlize packages29. Additionally, we plotted correlation scatter diagrams of the top 5 most significant METTL1 co-expressed genes. To further understand the biological functions of 236 METTL1 co-expressed genes, we carried out Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses using the clusterProfiler and enrichplot packages30,31,32, with false discovery rate (FDR)-adjusted p-values < 0.05 considered statistically significant.
Extraction of METTL1 differentially expressed genes
Wilcoxon test was used to screen METTL1 differentially expressed genes (DEGs) based on FDR threshold of < 0.05. The DEGs between normal tissue and BRCA tissue in TCGA were extracted according to FDR < 0.05 & |logFC| ≥ 1. These DEGs were plotted through volcano diagrams created with the ggplot2 package and heatmaps generated with the pheatmap package. After merging the two groups of DEGs, 1829 intersection genes were obtained using the VennDiagram package. In addition, we performed GO and KEGG enrichment analyses on the 1829 intersection genes, and considered results with FDR-adjusted p-value < 0.05 as statistically significant.
Identification of hub genes
Further, the METTL1 DEGs and co-expressed genes were intersected, resulting in the identification of 38 overlapping genes. Then, the protein-protein interaction network of the 38 genes was analyzed in the STRING database33 with an interaction score of 0.15. The top 5 hub genes were identified using the degree algorithm in the cytoHubba plugin of Cytoscape (3.9.1)34,35. The correlation between METTL1 and the five hub genes was validated in the bc-GenExMiner 4.5 database36. In addition, the association between the expression of the five hub genes and overall survival was further analyzed in both the TCGA-BRCA dataset and the Kaplan-Meier plotter database37.
Profiling tumor infiltrating immune cells
We analyzed the correlation between the METTL1-related gene signature and the immune infiltration of BRCA with CIBERSORT38 and xCELL39 algorithms. The immune cell types were quantified as continuous variables, and we categorized them into low and high expression groups based on the median expression value for each cell type. This binarization was applied uniformly across all immune cell types to assess their correlation with METTL1 expression levels.
Cell culture
Human normal breast cell line MCF10A (CL-0525) and breast cancer cell lines MCF7 (CL-0149) and MDA-MB-231 (CL-0150B) were obtained from Procell (Wuhan, China). These cells were cultured in a Dulbecco’s Modified Eagle Medium (FUSHENBIO, Shanghai, China) supplemented with 10% fetal bovine serum (FUSHENBIO, Shanghai, China) at 37 ℃ with 5% CO2.
Quantitative polymerase chain reaction (qPCR)
Total RNA was extracted from cells by TRIzol reagent (Invitrogen, USA), and cDNA was then synthesized using PrimeScript™ RT Master Mix (Perfect Real Time) (RR036A, TAKARA, Japan). Real-time quantification was performed with ChamQ Universal SYBR qPCR Master Mix (Q711-03, Vazyme China). The relative gene expression were normalized to GAPDH mRNA levels by the 2-ΔΔCt method. The primers were synthesized by Guangzhou all-perfect Biological Technology Co., Ltd. (Guangzhou, China), whose sequences are shown in Table S2.
Statistical analysis
All bioinformatics analyses were performed using R software (4.1.2). The mRNA expression of genes in cells were analyzed using GraphPad 9.0.0 and the data was expressed as mean ± standard deviation (SD). The statistical analysis was performed with t-test. P < 0.05 was considered to be statistically different. The Benjamini-Hochberg multiple testing correction method was used to calculate the adjusted p value.
Results
METTL1 expression in breast invasive carcinoma
We analyzed METTL1 expression in BRCA and normal tissues. The analysis results showed that METTL1 expression was significantly higher in BRCA tissues compared to normal tissues (p < 0.001) (Fig. 2A). To further validate this finding, we performed a paired analysis using samples with matched tumor and normal tissues from the same patients, which consistently demonstrated significantly elevated METTL1 expression in tumor tissues (p < 0.001) (Fig. 2B).

METTL1 highly expresses in breast invasive carcinoma patients. (a–b) The unpaired (a) and paired (b) analysis were performed using DESeq2 package to compare the METTL1 expression in BRCA and normal tissues. (c) The METTL1 expression in BRCA patients with age ≦ 65 years or > 65 years. (d) The METTL1 expression in BRCA patients with Stage I-II or Stage III-IV. (e) The ROC curve of METTL1 in BRCA patients was performed using ROCR package. BRCA, breast invasive carcinoma; ROC, receiver operating characteristic.
We further analyzed METTL1 expression in relation to age and clinical stage in BRCA patients. In brief, METTL1 expression was significantly higher in patients aged > 65 years compared to those aged ≦ 65 years (p = 0.004) (Fig. 2C). Additionally, patients with stage III-IV showed slightly higher METTL1 expression than those with stage I-II, with the difference being marginally significant (p = 0.047) (Fig. 2D). However, the median expression levels between the two groups were similar, and overlapping data points indicated that METTL1 expression alone might not strongly differentiate clinical stages.
To explore the relationship between METTL1 expression and age further, we divided all samples into high and low METTL1 expression groups based on the median METTL1 expression level. The proportion of patients aged > 65 years was higher in the high METTL1 expression group (32.6%) compared to the low expression group (24.6%) (Table 1).
Overall, these findings indicated that higher METTL1 expression was associated with older age and advanced clinical stage in BRCA patients, although its ability to stratify clinical stages remained limited.
Clinical value of METTL1 in breast invasive carcinoma
The AUC for METTL1 was 0.817 (95% CI: 0.764–0.871), indicating that METTL1 had good diagnostic value for BRCA (Fig. 2E). Survival analysis showed that patients with low METTL1 expression had significantly better 5-year OS and DSS compared to those with high METTL1 expression (p < 0.05) (Fig. 3A–B). However, no significant difference was observed in DFS between the two groups (p = 0.068) (Fig. 3C).

Correlation of METTL1 expression with the prognosis of breast invasive carcinoma patients. (a–c) The correlation between overall survival (a), disease specific survival (b), and disease free survival (c) of BRCA patients and METTL1 expression were analyzed by survival and survminer packages. (d–e) The univariate (d) and multivariate (e) Cox regression analysis was performed to evaluate the predictive value of METTL1 expression and clinical variables (age, cancer stage, tumor size, metastasis, lymph node involvement) for overall survival in BRCA patients. BRCA, breast invasive carcinoma.
In addition, univariate and multivariate Cox regression analyses confirmed that METTL1 expression was an independent prognostic factor for BRCA (p = 0.013 and p = 0.044) (Fig. 3D–E). These results suggested that METTL1 expression was associated with BRCA prognosis, with low METTL1 expression indicating a more favorable prognosis.
Enrichment analysis of METTL1 co-expressed genes
The circle diagram showed the top 11 co-expressed genes of METTL1 (Fig. 4A). Notably, METTL1 expression showed significant positive correlations with BANF1 (R = 0.520, p < 0.001), NABP2 (R = 0.530, p < 0.001), and SHMT2 (R = 0.520, P < 0.001), and significant negative correlations with RORA (R = −0.520, p < 0.001) and ZC3H6 (R = −0.520, p < 0.001) (Fig. 4B–F).

The association between METTL1 and its co-expressed genes. (a) The top 11 co-expressed genes of METTL1 was screened. (b–f) The correlation analysis between METTL1 and top 5 co-expressed genes, including BANF1 (b), NABP2 (c), RORA (d), SHMT2 (e), and ZC3H6 (f).
Role of METTL1 differentially expressed genes
DEGs were identified by comparing high and low METTL1 expression groups (Fig. 5A) and by comparing BRCA tumor tissues with normal tissues from the TCGA dataset (Fig. 5B). The changes in the top 50 DEGs were shown in Figures S1 and S2, respectively. A total of 1829 overlapping DEGs were identified between the two comparisons (Fig. 5C).

Screening and identification of differentially expressed genes. (a) The volcano diagrams of DEGs between METTL1 high and low expression group. (b) The volcano diagrams of DEGs between tumor and normal tissues. (c) The 1829 overlapping DEGs was found. (d–e) GO (d) and KEGG (e) enrichment analysis for 1829 overlapped DEGs. DEGs, differentially expressed genes; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Functional enrichment analysis of these 1829 DEGs revealed their biological roles. GO enrichment analysis showed that the most significant enrichment functions of DEGs were extracellular matrix organization and regulation of cytosolic calcium ion concentration (Fig. 5D). KEGG enrichment analysis showed that the most significant enrichment pathways of DEGs were focal adhesion, PI3K−Akt signaling pathway, cell cycle, ECM-receptor interaction, and calcium signaling pathway (Fig. 5E, Table S3).
Screening of hub genes
To identify potential hub genes, we analyzed the 38 intersecting genes shared by the 1829 overlapping DEGs and 236 co-expressed genes (Fig. 6A). A protein-protein interaction network was constructed to explore their interactions (Fig. 6B), and the top 5 hub genes (NME1, ANAPC11, POP7, MRPL14, and MRPL55) were identified using a degree-based algorithm (Fig. 6C).

Identification of hub genes. (a) The 39 genes was found to belong to both DEGs and co-expressed genes of METTL1. (b) The protein-protein interaction network analysis for 39 genes was performed using STRING database. (c) The top 5 hub genes were selected using degree algorithm in cytoHubba plugin of Cytoscape. (d–f) The association between the overall survival of BRCA patients and the expression of ANAPC11 (d), NME1 (e), and POP7 (f) were analyzed. BRCA, breast invasive carcinoma.
Next, we evaluated the impact of hub gene expression on the survival probability of BRCA. The evaluation results showed that high expression levels of NME1 (p = 0.022) and ANAPC11 (p = 0.043), along with low expression of POP7 (p = 0.021), were significantly associated with improved survival outcomes (Fig. 6D–F). However, MRPL14 (P = 0.194) and MRPL55 (P = 0.085) did not exhibit significant prognostic value (Figure S3).
We also compared the mRNA and protein expression levels of these six genes in tumor and normal tissues. Consistently, the mRNA expression of METTL1, NME1, ANAPC11, POP7, MRPL14, and MRPL55 was significantly higher in tumor tissues compared to normal tissues (p < 0.001) (Figure S4). The CPTAC database analysis showed significant upregulation of METTL1 and MRPL55 protein expression in cancer tissues (p < 0.05), whereas ANAPC11 and POP7 were found to be downregulated (p < 0.05) (Figure S5). This discrepancy might be explained by potential post-transcriptional regulation or protein degradation mechanisms, which could result in reduced protein levels in tumor tissues.
Additionally, correlation analysis revealed strong interrelationships among these six genes (Figure S6). Validation using the Kaplan-Meier plotter database supported the prognostic relevance of these genes, as high expression of all six genes was associated with poor overall survival (Figure S7).
Effects of METTL1-related gene signature on immune infiltration
To investigate the potential effects of METTL1 and its related gene signature on immune infiltration, we analyzed their correlation with tumor-infiltrating immune cells using CIBERSORT and xCell algorithms. As shown in Fig. 7, high METTL1 expression was significantly associated with increased fractions of follicular helper T cells (p < 0.001) and regulatory T cells (Tregs) (p < 0.01), suggesting its potential contribution to an immunosuppressive tumor microenvironment. Similarly, all five hub genes demonstrated positive correlations with follicular helper T cells and Tregs, further supporting their involvement in modulating immune responses. Additionally, these genes also exhibited associations with other immune cell types, such as macrophages and CD8 + T cells, indicating a broader impact on immune regulation. Notably, METTL1 and NME1 exhibited no statistically significant correlation with CD8 + T cells (p > 0.05), suggesting that their role in immune modulation might not directly influence CD8 + T cell infiltration.

Effects of hub genes on the immune infiltration of breast invasive carcinoma patients. (a–f) CIBERSORT algorithm was adopted to analyze the correlation between immune infiltration and METTL1 (a), ANAPC11 (b), MRPL14 (c), MRPL55 (d), NME1 (e), POP7 (f).
As demonstrated in Fig. 8, METTL1 and its hub genes displayed positive correlations with stromal-related scores and immunosuppressive cell types, further supporting their role in establishing an immunosuppressive tumor microenvironment. Interestingly, METTL1 and MRPL55 exhibited a weak negative correlation with CD8 + T cells, highlighting their potential role in suppressing anti-tumor immunity, which may contribute to poor prognosis in BRCA.

Correlation analysis between METTL1-related gene and immune infiltration. xCELL algorithm was used to analyze the association between immune infiltration and METTL1 and hub genes.
Collectively, these findings suggested that METTL1 and its related genes correlated with BRCA immune infiltration by enhancing immunosuppressive cell recruitment and dampening cytotoxic immune responses, ultimately influencing the tumor microenvironment and prognosis.
Expression of METTL1-related gene signature
To validate the expression of METTL1-related gene signature, qPCR was performed to analyze breast cancer cell lines and normal breast epithelial cells. The analysis results showed that the mRNA expression of METTL1, ANAPC11, MRPL14, MRPL55, and NME1 was significantly higher in MDA-MB-231 and MCF7 cells compared to MCF10A cells, suggesting their upregulation in breast cancer (Fig. 9A–E). However, there was no significant difference in POP7 expression among MDA-MB-231, MCF7, and MCF10A cells (Fig. 9F). Hence, POP7 expression might not be cell-line dependent or regulated differently in breast cancer cells. These findings corroborated the RNA-seq results and further validated the potential involvement of these genes in BRCA progression.

The expression of METTL1 and hub genes in breast invasive carcinoma cells. (a–f) The METTL1 (a), ANAPC11 (b), MRPL14 (c), MRPL55 (d), NME1 (e), and POP7 (f) in MDA-MB-231, MCF7, and MCF10A cells were detected by qPCR. qPCR, quantitative polymerase chain reaction.
Discussion
In this study, we identified significantly increased METTL1 expression in BRCA tissues compared to normal tissues, with strong associations with age, disease severity, and immune infiltration. Importantly, high METTL1 expression correlated with poor prognosis in BRCA patients. Through co-expression and differential gene analyses, five METTL1-related hub genes (ANAPC11, MRPL14, MRPL55, NME1, and POP7) were identified. These genes demonstrated significant associations with BRCA prognosis, immune infiltration, and tumor microenvironment modulation.
Dynamic RNA modifications, such as m7G modifications mediated by METTL1 in complex with WDR4, are known to regulate gene expression, protein stability, and translation40,41,42. Dysregulation of tRNA modifications, including m7G, has been linked to tumor prognosis43. METTL1 has been reported to exhibit abnormal expression in multiple cancers, including digestive tract tumors, lung cancer, and breast cancer, with links to tumor progression, treatment resistance, and immune suppression18,19,20,44. For example, elevated METTL1 has been reported to promote immunosuppressive environments by inducing myeloid-derived suppressor cells and inhibiting CD8 + T cell infiltration45.
In our study, high METTL1 expression in BRCA was associated with poor prognosis, clinicopathological features, and immune infiltration. Notably, Tregs, which inhibit antitumor immunity and promote tumor progression27, and effector immune cells were significantly enriched in the high METTL1 expression group, suggesting a potential association with an immunosuppressive tumor microenvironment. However, we emphasize that these associations are based on transcriptomic correlation analyses and do not imply direct causality. Further studies are needed to determine whether METTL1 directly influences immune cell infiltration or function in BRCA.
Notably, while METTL1 expression correlates with poor prognosis in BRCA, survival differences, particularly in DFS, were not statistically significant, raising questions about its clinical utility. Further validation in larger cohorts and integration into multi-biomarker models are needed to confirm its predictive value for BRCA prognosis.
In contrast to established clinical biomarkers such as Ki-67, HER2, and hormone receptor status (ER/PR), which are critical components in the prognostic assessment and therapeutic stratification of breast cancer46, METTL1 offers unique mechanistic insights into tumor biology through m7G tRNA modification and mRNA translation. Our study demonstrated a promising diagnostic AUC of 0.817 for METTL1, suggesting its potential in distinguishing BRCA tumor tissues from normal tissues. Moreover, METTL1 may offer added value in understanding immune modulation and drug resistance. Still, these implications require functional confirmation, as the associations presented here are observational. Future studies should evaluate the combined performance of METTL1 and existing biomarkers to enhance the accuracy and predictive value of BRCA diagnosis and prognosis.
Biological pathways potentially regulated by METTL1 include cytokine signaling, antigen presentation, and immune checkpoint regulation, all of which may be influenced by METTL1’s m7G modification activity47. For instance, METTL1 may alter the translation of immune-related genes, such as PD-L1, by m7G modification, thereby influencing the immune response in BRCA. This modulation could help tumor cells evade immune detection and establish an immunosuppressive microenvironment. Nevertheless, these possibilities remain hypothetical and are based on indirect transcriptomic evidence. Further experimental studies are required to validate these hypotheses and confirm METTL1’s direct involvement in immune regulation.
Several hub genes identified in this study were found to contribute to tumor progression, prognosis, and immune-related processes. Among them, ANAPC11 plays a critical role in cell cycle regulation and ubiquitin-mediated degradation, processes essential for tumor cell proliferation and survival36,48. In our analysis, high ANAPC11 expression was associated with better survival outcomes in BRCA patients, potentially reflecting its role in maintaining genomic stability or facilitating tumor-specific metabolic adaptations. Similarly, NME1, a well-known tumor suppressor, was correlated with poor prognosis and increased aggressiveness in breast cancer when expressed at low levels33,49. Consistent with this, our survival analysis showed that high NME1 expression was associated with better prognosis. In contrast, POP7, which has been implicated in promoting breast cancer progression50,51, was associated with a poorer prognosis in this study. However, POP7 mRNA expression did not significantly differ between tumor and normal cells, possibly due to cell-line specific regulation or post-transcriptional modifications. This suggests that POP7’s regulation may involve alternative splicing, mRNA stability, or other post-transcriptional processes affecting protein expression without altering mRNA levels. Further experiments, including protein analysis and alternative splicing studies, are needed to better understand POP7’s role in BRCA. Additionally, it is important to note that the discrepancies in survival analysis for hub genes across databases may reflect differences in tumor subtypes, patient cohorts, and methodological factors. These context-dependent effects underline the need for further research to clarify their roles and reconcile these inconsistencies.
Furthermore, MRPL14 and MRPL55, both involved in mitochondrial translation, were also linked to BRCA prognosis and immune infiltration38. MRP14 is thought to promote mitochondrial translation in coordination with C7orf30, and its deletion could lead to mitochondrial translation defects29. MRPL55, known for its association with ovarian cancer prognosis, may have a similar role in BRCA by affecting mitochondrial function and contributing to immune cell dynamics in the tumor microenvironment38,52,53. Interestingly, we found that POP7, ANAPC11, MRP14, and MRPL55 were associated with increased infiltration of both tumor-killing immune cells (CD8 + T cells and activated natural killer cells) and immunosuppressive cells (Tregs). This dual role may contributes to the variability in their prognostic significance observed across databases. However, these associations are descriptive and require mechanistic studies to elucidate causality.
Notably, the correlation between METTL1 expression and immune cells is intriguing, but further investigation is required. While these associations suggest that METTL1 may influence immune responses, it is important to recognize that correlation does not imply causation. The observed relationships could arise from confounding tumor-intrinsic or microenvironmental factors. Future experimental validation, such as knockdown or overexpression studies, are necessary to determine whether METTL1 directly regulates immune infiltration or if the observed correlations reflect broader tumor microenvironment changes.
This study innovatively explored the role of METTL1 and its related genes in BRCA through multi-dimensional approach, combining transcriptomic analysis, survival studies, immune infiltration profiling, and qPCR validation. However, the direct regulatory relationship between these key hub genes with METTL1 remains unclear. It is essential to investigate whether METTL1 regulates these genes transcriptionally or post-transcriptionally, particularly through m7G modification, which may influence protein synthesis and thereby affect tumor progression and immune regulation. Future studies should focus on confirming these interactions and regulatory mechanisms using functional assays like RNA immunoprecipitation (RIP) or m7G pull-down assays.
However, several limitations should be noted. First, while we established associations between METTL1, its hub genes, and BRCA aggressiveness, the underlying molecular mechanisms remain speculative and require further experimental validation. Functional studies, such as gene knockdown or overexpression in cell and animal models, Western blot, IHC, and immunofluorescence to assess METTL1 protein levels and localization in BRCA tissues are needed to confirm causative relationships and confirm the effects of METTL1 on tumor behavior. Additionally, while METTL1 was linked to pathways such as focal adhesion and PI3K-AKT signaling, this study did not include direct experiments to investigate these pathways. Future work should explore whether METTL1-mediated m7G modifications affect key transcripts involved in these pathways, potentially contributing to tumor migration, invasion, and immune evasion. Advanced technologies, such as high-plex protein and transcriptome co-mapping with spatial CITE-seq and in vivo CRISPR screens via perturb-DBiT, could provide greater precision in uncovering these mechanisms54,55. Lastly, while our analysis used publicly available datasets, their limitations in sample selection and data quality highlight the need for independent validation using larger, more diverse cohorts (e.g., METABRIC) to confirm the robustness and generalizability of these findings.
Despite these limitations, this study provides new insights into the oncogenic role of METTL1 in BRCA and highlights its potential as a diagnostic and therapeutic target. We emphasize the need for future experimental validation and independent cohort validation to enhance the confidence in our conclusions and further establish METTL1 as a key player in BRCA progression.
Conclusion
This study comprehensively investigated the role of METTL1 in BRCA, revealing its significant diagnostic, prognostic, and immunomodulatory potential. On the one hand, METTL1 was highly expressed in BRCA tissues and significantly associated with patient age, disease stage, and poor survival outcomes. On the other hand, five hub genes closely related to METTL1 were identified, and their expression influenced the immune microenvironment by promoting immunosuppressive cell types. Future studies should focus on exploring the molecular mechanisms underlying METTL1-mediated immune regulation and its potential as a therapeutic target in BRCA.
Data availability
All data relevant to the study are included in the article. Additional information, including analysis scripts and detailed filtering steps used in the bioinformatics workflow, is available from the corresponding author upon reasonable request.
References
Ghoncheh, M., Pournamdar, Z. & Salehiniya, H. Incidence and mortality and epidemiology of breast Cancer in the world. Asian Pac. J. Cancer Prev. 17, 43–46. https://doi.org/10.7314/apjcp.2016.17.s3.43 (2016).
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 72, 7–33. https://doi.org/10.3322/caac.21708 (2022).
DeSantis, C. E. et al. Breast cancer statistics, 2019. CA Cancer J. Clin. 69, 438–451. https://doi.org/10.3322/caac.21583 (2019).
Jacobson, G. et al. Palliative radiation therapy for symptomatic advance breast cancer. Sci. Rep. 11, 5282. https://doi.org/10.1038/s41598-021-84872-9 (2021).
Pedrosa, R., Mustafa, D. A., Soffietti, R. & Kros, J. M. Breast cancer brain metastasis: molecular mechanisms and directions for treatment. Neuro Oncol. 20, 1439–1449. https://doi.org/10.1093/neuonc/noy044 (2018).
Nagini, S. Breast cancer: current molecular therapeutic targets and new players. Anticancer Agents Med. Chem. 17, 152–163. https://doi.org/10.2174/1871520616666160502122724 (2017).
Chen, Y., Lin, Y., Shu, Y., He, J. & Gao, W. Interaction between N(6)-methyladenosine (m(6)A) modification and noncoding RNAs in cancer. Mol. Cancer. 19, 94. https://doi.org/10.1186/s12943-020-01207-4 (2020).
Bianchini, G., Balko, J. M., Mayer, I. A., Sanders, M. E. & Gianni, L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 13, 674–690. https://doi.org/10.1038/nrclinonc.2016.66 (2016).
Barbieri, I. & Kouzarides, T. Role of RNA modifications in cancer. Nat. Rev. Cancer. 20, 303–322. https://doi.org/10.1038/s41568-020-0253-2 (2020).
Kumari, R., Michel, A. M. & Baranov, P. V. PausePred and rfeet: webtools for inferring ribosome pauses and visualizing footprint density from ribosome profiling data. RNA 24, 1297–1304. https://doi.org/10.1261/rna.065235.117 (2018).
Vu, L. P. et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 23, 1369–1376. https://doi.org/10.1038/nm.4416 (2017).
Dai, Z. et al. N(7)-Methylguanosine tRNA modification enhances oncogenic mRNA translation and promotes intrahepatic cholangiocarcinoma progression. Mol Cell 81, 3339–3355 e3338, (2021). https://doi.org/10.1016/j.molcel.2021.07.003
Xia, P. et al. MYC-targeted WDR4 promotes proliferation, metastasis, and Sorafenib resistance by inducing CCNB1 translation in hepatocellular carcinoma. Cell. Death Dis. 12, 691. https://doi.org/10.1038/s41419-021-03973-5 (2021).
Pavon-Eternod, M. et al. tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res. 37, 7268–7280. https://doi.org/10.1093/nar/gkp787 (2009).
Kwon, N. H., Lee, J. Y. & Kim, S. Role of tRNAs in breast Cancer regulation. Adv. Exp. Med. Biol. 1187, 121–145. https://doi.org/10.1007/978-981-32-9620-6_6 (2021).
Lin, S. et al. Mettl1/Wdr4-Mediated m(7)G tRNA methylome is required for normal mRNA translation and embryonic stem cell Self-Renewal and differentiation. Mol. Cell. 71 (e245), 244–255. https://doi.org/10.1016/j.molcel.2018.06.001 (2018).
Wikman, H. et al. CDK4 is a probable target gene in a novel amplicon at 12q13.3-q14.1 in lung cancer. Genes Chromosomes Cancer. 42, 193–199. https://doi.org/10.1002/gcc.20122 (2005).
Tian, Q. H. et al. METTL1 overexpression is correlated with poor prognosis and promotes hepatocellular carcinoma via PTEN. J. Mol. Med. (Berl). 97, 1535–1545. https://doi.org/10.1007/s00109-019-01830-9 (2019).
Ma, J. et al. METTL1/WDR4-mediated m(7)G tRNA modifications and m(7)G codon usage promote mRNA translation and lung cancer progression. Mol. Therapy: J. Am. Soc. Gene Therapy. 29, 3422–3435. https://doi.org/10.1016/j.ymthe.2021.08.005 (2021).
Liu, Y. et al. Overexpressed methyltransferase-like 1 (METTL1) increased chemosensitivity of colon cancer cells to cisplatin by regulating miR-149-3p/S100A4/p53 axis. Aging 11, 12328–12344. https://doi.org/10.18632/aging.102575 (2019).
Chen, B. et al. N(7)-methylguanosine tRNA modification promotes tumorigenesis and chemoresistance through WNT/beta-catenin pathway in nasopharyngeal carcinoma. Oncogene 41, 2239–2253. https://doi.org/10.1038/s41388-022-02250-9 (2022).
Ying, X. et al. METTL1-m(7) G-EGFR/EFEMP1 axis promotes the bladder cancer development. Clin. Transl Med. 11, e675. https://doi.org/10.1002/ctm2.675 (2021).
Gao, Z. et al. A comprehensive analysis of METTL1 to immunity and stemness in Pan-Cancer. Front. Immunol. 13, 795240. https://doi.org/10.3389/fimmu.2022.795240 (2022).
Li, A. Y. et al. Prognostic and immune implications of a novel 7-methylguanosine-related MicroRNA signature in breast invasive carcinoma: from exploration to validation. J. Cancer Res. Clin. Oncol. 149, 9105–9128. https://doi.org/10.1007/s00432-023-04849-1 (2023).
Love, M. I., Huber, W. & Anders, S. Moderated Estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. https://doi.org/10.1186/s13059-014-0550-8 (2014).
Zeng, X. et al. Eliminating METTL1-mediated accumulation of PMN-MDSCs prevents hepatocellular carcinoma recurrence after radiofrequency ablation. Hepatology 77, 1122–1138. https://doi.org/10.1002/hep.32585 (2023).
Ohue, Y., Nishikawa, H. & Regulatory, T. Treg) cells in cancer: can Treg cells be a new therapeutic target? Cancer Sci. 110, 2080–2089. https://doi.org/10.1111/cas.14069 (2019).
Sing, T., Sander, O., Beerenwinkel, N. & Lengauer, T. ROCR: visualizing classifier performance in R. Bioinformatics 21, 3940–3941. https://doi.org/10.1093/bioinformatics/bti623 (2005).
Fung, S., Nishimura, T., Sasarman, F. & Shoubridge, E. A. The conserved interaction of C7orf30 with MRPL14 promotes biogenesis of the mitochondrial large ribosomal subunit and mitochondrial translation. Mol. Biol. Cell. 24, 184–193. https://doi.org/10.1091/mbc.E12-09-0651 (2013).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. ClusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–462. https://doi.org/10.1093/nar/gkv1070 (2016).
Antar, S., Mokhtar, N., Elghaffar, A., Seleem, A. K. & M. A. & Association of polymorphisms in metastasis suppressor genes NME1 and KISS1 with breast cancer development and metastasis. J. Egypt. Natl. Canc Inst. 32 https://doi.org/10.1186/s43046-020-00037-1 (2020).
Chin, C. H. et al. CytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8 (Suppl 4). https://doi.org/10.1186/1752-0509-8-S4-S11 (2014).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. https://doi.org/10.1101/gr.1239303 (2003).
Shi, Y. J. & Huo, K. K. Knockdown expression of Apc11 leads to cell-cycle distribution reduction in G2/M phase. Genet. Mol. Res. 11, 2814–2822. https://doi.org/10.4238/2012.August.24.6 (2012).
Lanczky, A. & Gyorffy, B. Web-Based survival analysis tool tailored for medical research (KMplot): development and implementation. J. Med. Internet Res. 23, e27633. https://doi.org/10.2196/27633 (2021).
Zahra, A., Hall, M., Chatterjee, J., Sisu, C. & Karteris, E. In Silico study to predict the structural and functional consequences of SNPs on biomarkers of ovarian Cancer (OC) and BPA Exposure-Associated OC. Int. J. Mol. Sci. 23 https://doi.org/10.3390/ijms23031725 (2022).
Aran, D., Hu, Z. & Butte, A. J. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220. https://doi.org/10.1186/s13059-017-1349-1 (2017).
Chou, H. J., Donnard, E., Gustafsson, H. T., Garber, M. & Rando, O. J. Transcriptome-wide analysis of roles for tRNA modifications in translational regulation. Mol. Cell. 68 (e974), 978–992. https://doi.org/10.1016/j.molcel.2017.11.002 (2017).
Liu, F. et al. ALKBH1-Mediated tRNA demethylation regulates translation. Cell 167, 1897. https://doi.org/10.1016/j.cell.2016.11.045 (2016).
Boccaletto, P. et al. MODOMICS: a database of RNA modification pathways. 2021 update. Nucleic Acids Res. 50, D231–D235. https://doi.org/10.1093/nar/gkab1083 (2022).
Torres, A. G., Batlle, E. & de Ribas, L. Role of tRNA modifications in human diseases. Trends Mol. Med. 20, 306–314. https://doi.org/10.1016/j.molmed.2014.01.008 (2014).
Pandolfini, L. et al. METTL1 Promotes let-7 MicroRNA Processing via m7G Methylation. Mol Cell 74, 1278–1290 e1279, (2019). https://doi.org/10.1016/j.molcel.2019.03.040
Zeng, X. et al. Eliminating METTL1-mediated accumulation of PMN-MDSCs prevents hepatocellular carcinoma recurrence after radiofrequency ablation. Hepatology https://doi.org/10.1002/hep.32585 (2022).
Prakash, D. P. Dr. Rashmi gudur. The prognostic role of Ki-67 in invasive breast cancer: correlation with ER, PR, and HER2 receptor status. Front. Health Inf. 13, 3. https://doi.org/10.52783/fhi.vi.556 (2024).
Wang, F. et al. METTL1 mediates PKM m7G modification to regulate CD155 expression and promote immune evasion in colorectal cancer. J. Transl Med. 22, 1161. https://doi.org/10.1186/s12967-024-05991-1 (2024).
Drouet, Y. et al. Integrated analysis highlights APC11 protein expression as a likely new independent predictive marker for colorectal cancer. Sci. Rep. 8, 7386. https://doi.org/10.1038/s41598-018-25631-1 (2018).
Palmieri, D., Horak, C. E., Lee, J. H., Halverson, D. O. & Steeg, P. S. Translational approaches using metastasis suppressor genes. J. Bioenerg Biomembr. 38, 151–161. https://doi.org/10.1007/s10863-006-9039-9 (2006).
Perederina, A., Esakova, O., Koc, H., Schmitt, M. E. & Krasilnikov, A. S. Specific binding of a Pop6/Pop7 heterodimer to the P3 stem of the yeast RNase MRP and RNase P RNAs. RNA 13, 1648–1655. https://doi.org/10.1261/rna.654407 (2007).
Huang, Y. et al. RNA binding protein POP7 regulates ILF3 mRNA stability and expression to promote breast cancer progression. Cancer Sci. https://doi.org/10.1111/cas.15430 (2022).
Zou, W. Immune regulation in the tumor microenvironment and its relevance in cancer therapy. Cell. Mol. Immunol. 19, 1–2. https://doi.org/10.1038/s41423-021-00738-0 (2022).
Li, J. et al. Epigenetic and transcriptional control of the epidermal growth factor receptor regulates the tumor immune microenvironment in pancreatic Cancer. Cancer Discov. 11, 736–753. https://doi.org/10.1158/2159-8290.CD-20-0519 (2021).
Liu, Y. et al. High-plex protein and whole transcriptome co-mapping at cellular resolution with Spatial CITE-seq. Nat. Biotechnol. 41, 1405–1409. https://doi.org/10.1038/s41587-023-01676-0 (2023).
Baysoy, A. et al. Spatially resolved in vivo CRISPR screen sequencing via Perturb-DBiT. BioRxiv https://doi.org/10.1101/2024.11.18.624106 (2024).
Acknowledgements
Not applicable.
Funding
This project is supported by the doctoral start-up fund of Guangzhou Women and Children’s Medical Center (1600042-04), National Natural Science Foundation of China (82103420) and Guangzhou Municipal Science and Technology Project (202102020182).
Author information
Authors and Affiliations
Contributions
Xiaobin Lin and Yuzhi Yao collected clinical data and wrote the manuscript. Xiaobin Lin and Hongmin Ma was involved in the patient’s medical care and surgery. Hongmin Ma and Yuhong Pan initiated and supervised the study, and wrote and revised the manuscript. All authors have given final approval of the version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent of participate
Not applicable.
Consent of publication
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lin, Xb., Yao, Yz., Ma, Hm. et al. Integrative bioinformatics analysis identifies METTL1 as a regulator of immune infiltration and prognosis in breast cancer. Sci Rep 15, 26297 (2025). https://doi.org/10.1038/s41598-025-11391-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-11391-2
