
Abstract
Relative quantification of gene expression is a fundamental method used in the molecular biology field to analyse real-time PCR data to determine transcriptional differences between groups of samples (or technical replicates of a single sample). The main methods used for relative quantification of gene expression are delta Ct methods that allow to compare means of gene expression between groups of samples without preparing a standard curve. Despite the availability of several tools for delta Ct methods, it is difficult to select the best one that would be characterized by sufficient flexibility and comprehensive functionality. Therefore, we developed the RQdeltaCT R package, which was designed to analyse real-time PCR data for the relative quantification of gene expression using delta Ct methods (including 2−dCt and 2−ddCt methods), either to compare independent groups of samples or groups with paired samples. Furthermore, the package offers functions that cover other essential steps of analysis, including importing datasets, multistep quality control of data, numerous visualisations, and enrichment of the standard workflow with additional analyses (correlation analysis, Receiver Operating Characteristic analysis, linear and logistic regression). All obtained results can be conveniently saved as tables and publication-ready images. The RQdeltaCT package has been designed with the intention of being friendly to beginners in R programming. The package (version 1.3.2) is freely available on the GitHub (https://github.com/Donadelnal/RQdeltaCT) and CRAN (https://CRAN.R-project.org/package=RQdeltaCT) repositories.
Similar content being viewed by others
Introduction
Quantification of gene expression is a common subject in the field of molecular biology. One of the main purposes of such studies is to identify genes with increased or decreased expression levels related to the studied experimental condition. The traditional method used for gene expression analysis is real-time PCR, a simple, sensitive, and flexible technique that can be performed on a large number of samples and many different genes in the same experiment, depending on the format of used reaction plates (96-well plates, 384-well plates or 3072 through-holes in OpenArray plates).
Gene expression levels in real-time PCR experiments can be determined using in two approaches: absolute quantification and relative quantification. For absolute quantification, the precise amount of the template is used to draw the standard curve, and then samples are compared with the standard curve, and the result is extrapolated. In relative quantification, the absolute amount of template is not necessarily known because calculated means of expression levels are compared between groups of samples, and transcriptional changes are determined in the study group in relation to the control group by calculation of a fold change values. This approach does not require a standard curve; thus, a working load is significantly reduced. However, the additional assumption of amplification efficiencies of all primers are equal should be met. For the relative quantification of gene expression levels, delta Ct methods were developed1,2. The main idea is to normalise gene expression values using an endogenous control gene, transform gene expression values into linear form using the 2−(value) formula, and calculate differences in means of gene expression levels between groups of samples (or technical replicates of a single sample).
Two main delta Ct methods are used for relative quantification, depending on the study design and experimental setup: the 2−dCt and 2−ddCt methods 2. In 2−dCt method, the Ct values are normalised by endogenous control gene (often glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or beta-actin, or another) by subtracting the Ct value of the endogenous control in each sample from the Ct value of the gene of interest in the corresponding sample, obtaining delta Ct (dCt) values. Subsequently, the dCt values are transformed using the 2−dCt formula, summarised by means in the compared study groups, and a ratio of means (fold change) is calculated for a study group versus control group. This method is useful in scenarios where samples should be analysed as individual data points (according to the example no. 5 in2). In the case of endogenous control gene testing, unnormalized Ct values can be used (according to the example 3 in2).
Similarly to the 2−dCt method, the 2−ddCt method also includes normalisation of Ct values by endogenous control gene, but the obtained delta Ct (dCt) values are not exponentially transformed, but are summarised by means in the compared study groups, and the mean dCt in a control group is subtracted from the mean dCt in a study group, giving the delta delta Ct value (ddCt). Subsequently, ddCt values are transformed using the 2−ddCt formula to obtain the fold change value, also called the RQ value (according to the examples 1 and 2 in2).
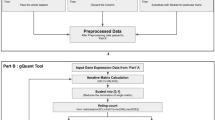
Because of the multitude of data processing procedures and factors that affect the final results 3,4 (Fig. 1), the optimal workflow for the analysis of real-time PCR data is not easy to establish and often requires the use of multiple tools to cover all the steps required to answer the research question. This means that, despite the fact that there several tools are available for the relative quantification analysis of real-time PCR data, identifying the best one that integrates comprehensiveness, flexibility, and user-friendliness remains challenging. Pabinger et al. in 2014 thoroughly summarized tools developed for the analysis of quantitative PCR data5, presenting a spectrum of either standalone, web-based, and R-based software. Several new tools have been developed, including qRAT6, qPCRtools7, tidyqpcr8, ddCt9, Auto-qPCR10 and Pcr11.

Overview of the key steps in the real-time PCR workflow with highlighting critical factors that influence accurate data analysis and interpretation.
Among the R-based tools developed to date for the relative quantification of gene expression using delta Ct methods, some high quality R packages are available. HTqPCR was published in 2009 and offers extensive functions for real-time PCR data import, quality control, visualization, and normalization12; however, the number of methods available for the stability assessment of reference genes is limited. In the same year, another package named ddCt was released 9, but it has limited methods of data visualisation and, similarly to HTqPCR, the number of stability parameters for reference gene selection is insufficient. In 2011, ReadqPCR and NormqPCR packages were developed to assist researchers read, quality check, and normalize Ct data, as well as to enable selection of the optimal reference gene and handle missing data13. However, data visualisation and statistical testing are outside the scope of these tools. The pcr package, published several years later, was enriched in functions for the calculation of amplification efficiency and linear regression; however, methods for missing data handling and data visualization are limited11.
Two R packages have been released in recent years, tidyqpcr8 and qPCRtools7, which provide functions that are helpful for setting experiments, calculating of efficiency, and applying of various methods for relative quantification. However, these tools do not provide sufficient methods for quality control and visualisation of data. Limited methods for reference gene stability assessment have been observed in the latest package rtpcr developed by Ghader Mirzaghaderi, published on CRAN in July 2024 (https://CRAN.R-project.org/package=rtpcr). In summary, each of the mentioned R-based packages contributes to a spectrum of tools for real-time PCR data analysis; however, the majority of them cover only a part of the analysis pipeline and have limited methods for quality control of data, insufficient number of data visualisations, or the lack of additional methods for differential expression assessment to complete the workflow (Table 1). Due to these issues, researchers have to use multiple tools specific for a particular part of the analysis, which involves additional data transformations and the risk of unnoticed errors.
The RQdeltaCT package (Relative Quantification using delta CT methods) was developed to address these issues and overcome the limitations of other tools. The presented package effectively balances comprehensive functionality, flexibility of programming, and ease of use for the analysis of real-time PCR data using delta Ct methods for relative quantification, according to the procedure described by Livak and Schmittgen1,2. The RQdeltaCT package implements the 2−dCt and 2−ddCt methods, enabling convenient import, preparation, and quality control of data. Its functions allow to create various publication-ready visualisations and complement standard pipeline by additional methods useful for differential gene expression analysis. Compared to standalone and web-based tools, the RQdeltaCT R package provides a prominently higher level of flexibility, enabling not only highly customizable analysis parameters, but also full access to the source code that can be freely modified and expanded to perfectly fit the specific requirements of each analysis.
Design and implementation
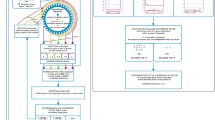
The RQdeltaCT package is implemented in the R programming environment and offers a set of 32 functions (version 1.3.2) that encompass delta Ct methods for relative quantification workflow, together with steps that include import, quality control, and filtering of data, as well as reference gene selection, data normalization, and multiple visualisation methods. To complement the standard pipeline, additional methods useful for differential analysis were implemented, including correlation analysis, simple linear and logistic regression, Receiver Operating Characteristic (ROC) analysis, and principal component analysis (PCA) with sample classification using the k-means method (Fig. 2).

Overview of the RQdeltaCT package workflow.
Tables with calculated results and all created plots can be conveniently exported to .txt and .tiff files, respectively. Most data processing steps and visualisations are powered by the tidyverse 2.0.0 package (https://www.tidyverse.org/).
Reading, processing, and quality check of data
The RQdeltaCT package provides functions that are useful for importing plain text files containing a table of Ct values. Given the variability in table formats produced by real-time PCR software, the package supports the import of both long- and wide-format data structures, offering a convenient and flexible approach for handling input files in standard .txt or .csv formats. The specific requirements for the file structure are stated in the package vignette (https://cran.r-project.org/web/packages/RQdeltaCT/vignettes/my-vignette.html).
It should be noted that the RQdeltaCT workflow does not include preprocessing steps for raw real-time PCR data, such as noise reduction, calculation of amplification efficiency, quality control of amplification curves, and final Ct calculation. These steps are typically covered by software coupled with PCR devices and used to generate raw data files during real-time PCR experiments; therefore, it is not necessary to include these functionalities in the presented package. Tables with final raw Ct values exported from such software (in .txt or .csv format) can be directly passed to the RQdeltaCT workflow.
The RQdeltaCT package allows for the comprehensive quality control of Ct data, which is an essential step required to assess the suitability of data for analysis, ensuring the reliability of the obtained results. This procedure can be performed based on reliability criteria established for Ct values, such as maximum Ct value and flag information used to label undetermined Ct values. The package enables the assessment of the number of Ct values that meet or fail predefined reliability criteria, facilitating the identification and filtering of samples and genes with a high proportion of low-quality Ct values. Additionally, it supports the exploration of replicate number across samples using heatmap, which is particularly critical in situations where genes are analysed across varying numbers of samples or where samples themselves differ in the number of replicates. It significantly aids in elucidating the underlying data structure, which is essential for proper data processing. The RQdeltaCT workflow encompasses also steps such as the collapsing of technical replicates and the imputation of missing values using group-wise mean substitution. In general, a majority of functions in the RQdeltaCT package can deal with missing data; however, some used methods (e.g. PCA) are sensitive to missing data (see the package vignette).
Data normalization
Data normalization is necessary for minimizing the influence of technical and sampling variability. In this step, Ct values of the reference gene (or mean of the Ct values of reference genes, if more than one reference gene is used) are subtracted from Ct values of the gene of interest across all samples, diving delta Ct (dCt) values. A critical aspect of data normalization in qPCR analysis is the selection of an appropriate reference gene. The reference gene must fulfill several essential criteria, including stable expression across all experimental conditions, moderate expression levels (avoiding extreme high or low abundance), consistency across different sample types, and the absence of co-regulation with the target genes. The RQdeltaCT package is helpful in selecting the best reference gene by calculating several stability parameters, including descriptive statistics such as minimum, maximum, standard deviation, and variance, as well as stability scores calculated using the NormFinder14 and geNorm15 algorithms. NormFinder scores are computed using the code adapted from the original NormFinder code (https://www.moma.dk/software/normfinder). For the geNorm score, the geNorm function of the ctrlGene 1.0.1 package (https://cran.r-project.org/web/packages/ctrlGene/index.html) is used. An ideal reference gene is expected to exhibit low standard deviation, variance, and stability scores, reflecting minimal expression variability across samples. However, universally accepted threshold values for these parameters are lacking, and the selection of an appropriate reference gene ultimately relies on the researcher’s judgment.
Homogeneity assessment of data
Before proceeding to the relative quantification analysis, the prepared data should undergo homogeneity assessment to identify outlier samples or errors that could introduce significant bias into results. The RQdeltaCT tool allows to perform this assessment by generating boxplots presenting data distribution and using more advanced methods, such as hierarchical clustering and PCA. All of these functions highlight the variability within gene expression data and facilitate the identification of samples exhibiting distinct expression values that deviate from the overall sample distribution. These methods could be applied not only for samples, but also for genes to evaluate similarities and differences between expression of analysed genes.
However, interpretation of the resulting plots requires caution due to inherent methodological limitations. PCA plots generated by RQdeltaCT package are based on the first two principal components, which may not capture all relevant variance, as can be associated with the third or higher-order components. In turn, hierarchical clustering is highly sensitive to the choice of distance metrics and linkage algorithms, potentially leading to inconsistent or unreliable outlier detection. The classification of a sample data point as an outlier remains, to some extent, subjective and dependent on the researcher’s judgment. Despite these limitations, the methods implemented in the package provide valuable insights into data variability and homogeneity, which are critical for ensuring the validity of downstream analyses.
Relative quantification
Data that passed the quality control criteria can be subjected to relative quantification using the 2−dCt method implemented in the RQ_dCt function or 2−ddCt method implemented in the RQ_ddCt function; each method can be used for comparison of independent groups of samples or groups containing paired samples. Statistical testing of differences in means of gene expression levels between compared groups is done using parametric two-sided the Student’s t test and nonparametric the Mann–Whitney U test, together with the correction of P values for multiple testing (the Benjamini–Hochberg method is used by default). The implementation of both parametric and nonparametric statistical tests makes the presented package suitable for analyzing data regardless of whether they follow a normal distribution. Normality is assessed using the Shapiro–Wilk test integrated within the tool. To enhance the comprehensiveness of the analysis, the package additionally computes descriptive statistics, including group-wise means and standard deviations of expression data, along with both raw and log10-transformed fold change values. All these results are returned in one collective table and can be used to select significantly differentially expressed genes based on fold change values and P values appropriate for the normality of the data.
Visualisations
The RQdeltaCT package provides a comprehensive set of visualizations designed to effectively present the obtained results. The arrangement of genes based on fold change values and P values can be illustrated on a volcano plot, while fold change values can be visualised on the barplot, where bars of significant genes are distinguished by colors and/or significance labels. Statistical significance labels are added to the plots using the ggsignif 0.6.4 package (https://cran.r-project.org/web/packages/ggsignif/index.html) and can be fully customised by the user (for example, ‘not sig.’, ‘*’ or ’p = 0.03’).
Two subsequent types of visualisations allow to compare data distribution values of specific genes between compared groups. These are barplot that illustrates mean and standard deviation values of the data for all or selected genes and boxplot that illustrates the distribution of the data for the genes. The presented package allows the selection of genes for presentation, faceting of plot, and addition of fully customised statistical significance labels.
Another type of visualisation offered by the RQdeltaCT package is a heatmap powered by the pheatmap 1.0.12 package (https://cran.r-project.org/web/packages/pheatmap/index.html). Heatmaps are advantageous in gene expression analysis as they provide an intuitive visual representation of expression patterns across samples and conditions. They facilitate quick comparison of expression levels, making it easier to identify clusters of co-expressed genes and underlying biological trends. To minimize granularity of the heatmap, rows and columns are aggregated based on hierarchical clustering that can be carried out with various methods of distance calculation and agglomeration. The colours used for the heatmap and sample annotation can be freely customized by the user.
In pairwise analysis, the parallel_plot function can be used to illustrate pairwise changes in gene expression. This type of visualisation enables the simultaneous comparison of expression patterns across multiple genes and conditions, facilitating the identification of coordinated expression trends and potential regulatory relationships among genes.
Further analyses
The RQdeltaCT package greatly expands the scope of standard analysis by enabling additional methods that are useful for further analysis of gene expression levels and differences between groups:
-
1.
Principal Component Analysis (PCA) coupled with k-means clustering (pca_kmeans function) provides an unsupervised approach to assess sample clustering based on the gene expression data. The integration of these methods represents an effective strategy for dimensionality reduction and pattern recognition in high-dimensional gene expression datasets. In this approach, sample variability captured by the first two principal components of the PCA is analyzed through k-means clustering to identify distinct groupings, that are independent of predefined group assignments. Unlike supervised methods, this unsupervised approach facilitates the identification of previously unrecognized sample clusters, which may arise from underlying biological variation or technical bias. Results are presented in a scatter plot, and a confusion matrix of sample classification is returned. Although the confusion matrix is conventionally utilized in the context of supervised learning, we applied it to the results of the implemented unsupervised methods as a quantitative measure to assess the correspondence between the derived clusters and the known sample classifications.
-
2.
Correlation analysis that is used to generate and visualise the correlation matrix of samples (corr_sample function) or genes (corr_gene function) using functionalities inherited from the corrplot 0.92 package (https://cran.r-project.org/web/packages/corrplot/index.html). This analysis helps identify relationships between gene expression levels, both across genes (revealing potential co-regulation or functional associations among genes) and samples (allowing for the detection of similarities or differences in gene expression between samples).
-
3.
Simple linear regression that is used to analyse and visualise relationships between pairs of samples (single_pair_sample function) or genes (single_pair_gene function). Regression results such as regression equation, coefficient of determination, F value, or P value can optionally be added to the plot.
-
4.
The Receiver Operating Characteristic (ROC) analysis (implemented in the ROCh function) is useful for assessing the discriminatory power of individual genes between distinct different groups. The ROCh function allows drawing ROC curves together with the calculation of parameters such as the area under curve (AUC), specificity, sensitivity, accuracy, and positive and negative predictive values by implementation of pROC 1.18.5 package16 (https://cran.r-project.org/web/packages/pROC/index.html). By assessing these metrics, it facilitates the identification of candidate biomarkers with high diagnostic or predictive accuracy.
-
5.
Simple logistic regression analysis (log_reg function) that allows to investigate the impact of the analysed variable on the odds of the occurrence of the studied experimental condition. This function is based on the functionalities of the oddsratio 2.0.1 package (https://cran.r-project.org/web/packages/oddsratio/index.html) and calculates the chances (odds ratio, OR) of being included in the study group when gene expression level increases by one unit (suitable for non-transformed data) or by mean of expression levels (more suitable for transformed data).
While the RQdeltaCT package, as a tool developed in the R programming language, offers significant flexibility and transparency, its command-line-driven access and absence of a graphical user interface may pose challenges for users unfamiliar with R programming. Therefore, the functions developed within the RQdeltaCT package are designed with the intention of being maximally easy to use, even for beginner users of R. The parameters of functions were prepared to sufficiently range all essential tasks and options, and no additional, extensive coding steps are necessary in the standard workflow. In addition, rich documentation that includes detailed descriptions of all built-in functions, as well as vignette providing detailed step-by-step instructions on how to perform the entire analysis, were incorporated and made openly available. The included documentation provides great support for users, allowing people with no programming experience to work effectively with the presented package.
Results
To illustrate the use of the RQdeltaCT package and familiarise the user with all its functionalities, a real dataset from the previously published article17 was included in this package. These data contain raw Ct values for 20 genes quantified using real-time PCR in peripheral blood mononuclear cells samples derived from the group of 40 patients with abdominal aortic aneurysm (AAA group) and 24 subjects without this disease (Control group). These data can be easily loaded into the R environment using a data function (the entire code used for this article is available in the Supplementary File 3). For presentation purposes, ten randomly selected samples from the AAA group and ten randomly selected samples from the Control group were selected for analysis to save the space needed for images and avoid duplication of data previously shown in the package vignette.
The quality check of the loaded Ct data was performed using the control_Ct_barplot_sample and the control_Ct_barplot_gene functions to calculate and visualise the numbers of undetermined or unreliable Ct values (flagged as ‘Undetermined’ or those with a value above 35) across genes and samples (Fig. 3A,B, respectively). Corresponding tables with the results are available in the Supplementary File 2 (Tables S1 and S2).

Example visualisations useful for the quality check of data. Comparison of the numbers of reliable and unreliable Ct values across genes (A) and samples (B). Data homogeneity check based on comparison of gene expression distributions between samples (C), variance analysis using PCA (D), and hierarchical clustering based on averages of Euclidean distances calculated for each sample (E).
Visual inspection of the returned plots provides a clear and comprehensive image of the data quality and structure. Gene FGF23 was analysed exclusively in the Control group, whereas FGF23 and GAPDH exhibited two times more Ct values in the Control group compared to the other genes. This observation suggests that these two genes have a greater number of technical replicates across samples than the other genes. The variability in the number of sample replicates within the dataset was confirmed on a heatmap illustrating the distribution of replicates for each gene across samples, generated using the control_heatmap function (Figure S1 in Supplementary File 1). The presented example shows that the analysed data can have complex structure that can be difficult to recognize; therefore, the presented tool provides great support for users to familiarize themselves with dataset and avoid serious issues that are consequences of data complexity.
Unreliable Ct values, together with samples and genes with more than half of unreliable data, were conveniently filtered out using the filter_Ct function. Retained data were subjected to collapsing of technical replicates and imputation of missing data by means within groups using the make_Ct_ready function.
In the next step, the expression data were normalized using the delta_Ct function and the GAPDH gene as an endogenous control, which exhibited high stability of Ct values in the samples assessed using algorithms implemented in the find_ref_gene function (Table S3 in Supplementary File 2). The homogeneity of the normalized data was assessed by visualization of the data distribution across samples, PCA, and hierarchical clustering (Fig. 3C–E, respectively). None of these methods showed outlier samples.
In this example, the most popular 2−ddCt method was used for relative quantification; thus, the RQ_ddCt function was used for this purpose. Due to the large number of columns in the obtained table, the most important results were selected and are presented in Table 2. The entire table with the results is provided as Table S4 in the Supplementary File 2.
Genes with P values below 0.05 and at least 1.5-fold change in expression (FCh ≥ 1.5 or ≤ 0.667) were selected as significantly differentially expressed. Only one gene, VEGFB, satisfied the predefined selection criteria. However, to ensure a comprehensive and coherent demonstration of the package’s functionalities, a minimum of two genes was necessary for inclusion in downstream analyses. Consequently, the TGFB gene was also selected, as it exhibited the lowest adjusted P value among the remaining genes, and also met the fold change threshold.
The obtained differential expression results were presented on barplot (Fig. 4A) and volcano plot (Fig. 4C) generated using the FCh_plot and results_volcano functions, respectively. The differential expression levels of two selected genes, TGFB and VEGFB, were visualized on the boxplot, which illustrates the distribution of the data in the compared groups (Fig. 4B), as well as on the barplot that shows the mean and standard deviation values of these genes in the compared groups (Fig. 4D). The expression patterns of these genes are presented in the heatmap (Fig. 4E).

Example visualisations of results obtained from relative quantification. (A) Barplot of fold changes obtained for each gene, with visually highlighted genes considered significant. Data distributions and means of expression for two genes selected as significant were compared on boxplot (B) and barplot (D), respectively. (C) Volcano plot presenting the arrangement of P values and fold change values calculated for analysed genes. (E) Heatmap illustrating the expression values of the two genes selected as significant in the studied samples. *—P < 0.05, ns.—not significant.
The differences in TGFB and VEGFB expression between the compared groups were further analysed using unsupervised PCA and k-means methods implemented in the pca_kmeans function (Fig. 5A). When two cluster centers were applied, 80% overall accuracy was achieved (Table S5 in Supplementary File 2). The results obtained from the ROC analysis implemented in the ROCh function confirmed that both genes have a good classification accuracy (Fig. 5B, Table S6). The logistic regression method implemented in the log_reg function showed that a two-fold decrease in VEGFB expression is associated with a 17.4-fold increase in the probability of belonging to the AAA group (Fig. 5C and Table S7 in Supplementary File 2). Correlation analysis performed using the corr_gene function demonstrated that the most correlated genes were TGFB and VEGFB (Fig. 5D and Table S8 in Supplementary File 2). The linear relationship between these genes was investigated separately for the AAA and Control groups using the linear regression method implemented in the single_pair_gene function (Fig. 5E).

Example visualisations of results generated by additional methods implemented in the RQdeltaCT package. (A) PCA and k-means clustering with two centres. (B) ROC curves for two genes selected as significant. (C) Odds ratios with 95% confidence interval and P values calculated using logistic regression. (D) Matrix of Pearson’s correlation coefficients calculated for analysed genes. (E) Trend lines and linear regression results obtained for two genes selected as significant.
Discussion
The presented example of workflow shows that RQdeltaCT provides a useful solution for the relative quantification of gene expression using delta Ct methods that balances comprehensive functionality, flexibility of programming, and ease of use. This tool is designed to enable real-time PCR data processing, analysis, and preparation of publication-ready visualisations using well-established and state-of-the-art statistical and graphical techniques, even by researchers with only basic programming skills. Compared to other tools, this package distinguishes itself through its capability to import data with diverse structural formats, comprehensive data quality control features, support for both the 2−dCt and 2−ddCt methods, and the integration of additional methodologies for differential expression assessment absent in comparable tools. The RQdeltaCT package is powered in the R environment, which allows the data to be processed efficiently and reproducibly. The average computation time for the complete workflow described in the manuscript was approximately 3.5 s when applied to the used dataset (20 samples × 20 genes), and was increased slightly to approximately 4.1 s for the full dataset (64 samples × 22 genes). This assessment was conducted on a workstation equipped with an AMD Ryzen 9 PRO 3900 (3.10 GHz), 32 GB RAM, running Windows 10 Pro.
The documentation provided with the package ensures a transparent and error-free workflow for data handling and analysis. Additionally, a discussion on GitHub in issue #1 to the RQdeltaCT package (https://github.com/Donadelnal/RQdeltaCT/issues/1) showed that this package has been successfully implemented in another independent laboratory.
Although the main work on the RQdeltaCT package has been completed, further improvements will be implemented based on the users feedback and the authors’ further inventions. Among functionalities that can be added to the RQdeltaCT package in the future are application of the linear regression method to the imputation of missing data, and extension of the relative quantification functions to include analysis of more than two groups. All errors and weaknesses reported by the users and noticed by the authors will be fixed without delay on an ongoing basis.
Summary
The RQdeltaCT package constitutes a unique, specialized collection of well-established methods and algorithms dedicated to the relative quantification of gene expression using delta Ct methods. This package has been developed to facilitate the processing, analysis and visualization of real-time PCR data using cutting-edge statistical and graphical methods. The presented tool distinguishes itself from existing solutions by offering diverse methods for data import, quality assessment, and visualization. Furthermore, it substantially enhances the standard workflow by integrating additional methodologies relevant to differential expression analysis. The RQdeltaCT package is designed with the intention of being maximally easy to use and includes a comprehensive vignette that provides step-by-step guidance on executing the complete analysis process.
Data availability
The RQdeltaCT package (version 1.3.2), together with the source code and vignette, is available under a GPL-3 licence and is publicly accessible in the GitHub (https://github.com/Donadelnal/RQdeltaCT) and CRAN (https://CRAN.R-project.org/package=RQdeltaCT) repositories. All package functions are free of charge and do not require login or registration. The data underlying this article are available as an integral part of the RQdeltaCT package. The R code used is available in Supplementary File 3.
References
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3, 1101–1108 (2008).
Bustin, S. A. Improving the quality of quantitative polymerase chain reaction experiments: 15 years of MIQE. Mol. Aspects Med. 96, 101249 (2024).
Taylor, S. C. et al. The ultimate qPCR experiment: Producing publication quality, reproducible data the first time. Trends Biotechnol. 37, 761–774 (2019).
Pabinger, S., Rödiger, S., Kriegner, A., Vierlinger, K. & Weinhäusel, A. A survey of tools for the analysis of quantitative PCR (qPCR) data. Biomol. Detect. Quant. 1, 23–33 (2014).
Flatschacher, D., Speckbacher, V. & Zeilinger, S. qRAT: An R-based stand-alone application for relative expression analysis of RT-qPCR data. BMC Bioinform. 23, 286 (2022).
Li, X. et al. qPCRtools: An R package for qPCR data processing and visualization. Front. Genet. 13, 1002704 (2022).
Wallace, E. W. J. & Haynes, S. J. tidyqpcr: Quantitative PCR analysis in the tidyverse. J. Open Source Softw. 7, 4507 (2022).
Zhang, J. D., Biczok, R. & Ruschhaupt, M. ddCt: The ddCt algorithm for the analysis of quantitative real-time PCR (qRT-PCR). (2023) https://doi.org/10.18129/B9.bioc.ddCt.
Maussion, G. et al. Auto-qPCR; a python-based web app for automated and reproducible analysis of qPCR data. Sci. Rep. 11, 21293 (2021).
Ahmed, M. & Kim, D. R. pcr: An R package for quality assessment, analysis and testing of qPCR data. PeerJ 6, e4473 (2018).
Dvinge, H. & Bertone, P. HTqPCR: High-throughput analysis and visualization of quantitative real-time PCR data in R. Bioinformatics 25, 3325–3326 (2009).
Perkins, J. R. et al. ReadqPCR and NormqPCR: R packages for the reading, quality checking and normalisation of RT-qPCR quantification cycle (Cq) data. BMC Genomics 13, 296 (2012).
Andersen, C. L., Jensen, J. L. & Ørntoft, T. F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Can. Res. 64, 5245–5250 (2004).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, research0034.1 (2002).
Robin, X. et al. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 12, 77 (2011).
Zalewski, D. et al. Dysregulations of key regulators of angiogenesis and inflammation in abdominal aortic aneurysm. Int. J. Mol. Sci. 24, 12087 (2023).
Funding
This work was supported by statutory funds of the Medical University of Lublin (DS43 A.B.-K.).
Author information
Authors and Affiliations
Contributions
D.Z.: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft. A.B-K.: Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zalewski, D., Bogucka-Kocka, A. RQdeltaCT: an open-source R package for relative quantification of gene expression using delta Ct methods. Sci Rep 15, 29762 (2025). https://doi.org/10.1038/s41598-025-11822-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-11822-0
Keywords
This article is cited by
-
Sub-inhibitory tobramycin concentration suppresses ToxA and LipA in single- and dual-species biofilms
Brazilian Journal of Microbiology (2026)
