
Abstract
Cancer cells exhibit metabolic reprogramming to fulfill their increased demands for abnormal growth and proliferation. We studied metabolic profiles of squamous cell lung carcinoma (LUSC).
Elevated arginine levels and reduced acylcarnitine C18:2, along with decreased phosphatidylcholine (PC) with acyl-alkyl residues C38:0 were associated with the diagnosis and prognosis of LUSC. Most of the PCs demonstrated a decrease, while lysophosphatidylcholines (LPC) exhibited an increase in LUSC patients. Network analysis unveiled that LPCs mediated PC and amino acids subgroup in LUSC compared to the control group. Analysis of public LUSC data confirmed associations between the expression levels of genes encoding enzymes involved in the biosynthesis pathways of arginine, proline (ASL, OTC, PYCR2), PC (CEPT1, CHPT1, LPCAT1) and LPC (LCAT, PLA2G16, PLB1) with a 5-yr survival outcome. The observed metabolic reprogramming in LUSC patients suggested the potential utility of metabolites as a supportive biomarkers for LUSC diagnosis.
Similar content being viewed by others


Introduction
Lung carcinoma ranks as the second most prevalent malignancy, trailing behind breast cancer and followed by colorectal, prostate and stomach cancers in global statistics1. However, lung cancer registers the highest mortality rate, surpassing breast, colorectal, and liver cancers in 20201. Among non-small cell lung carcinoma cases, approximately 30% are attributed to squamous cell type (LUSC), while non-squamous cell types constitute about 65%, with the remainder classified as other types2,3. As therapeutic options, particularly targeted therapies, are relatively limited for LUSC in comparison to the lung adenocarcinoma, and considering the unfavorable prognosis associated with LUSC, early diagnosis of LUSC could be a measure that might lead to better outcomes.
The proliferation of cancerous cells necessitates diverse building blocks and metabolites to support their abnormal growth and rapid proliferation4,5. Metabolomics, intertwined with genomics and proteomics, plays a pivotal role in unraveling the mechanisms underlying the growth and proliferation of these cancerous cells. The metabolic profiles of these cancerous cells reflect pathological alterations in the supply and demand of metabolites specific to the state or stage of cancer. Targeted analysis of acylcarnitines, amino acids, biogenic amines, phosphatidylcholine (PC), lysophosphatidylcholine (LPC), and hexoses can be conducted using tissue, blood, or other body fluids6,7. Arginine, categorized as a semi-essential amino acid, can be acquired through synthesis, protein turnover, and extracellular reservoir by diet. Arginine serves as a precursor for various compounds, including nitric oxide, creatine, polyamines and other amino acids. Furthermore, downstream metabolites of proline, such as hydroxyproline, have implications for tumor aggressiveness8,9.
PCs and LPCs are precursors in the formation of membrane lipids associated with plasma membrane remodeling in cancerous cells. PCs are synthesized from precursors of diacylglycerol and CDP-choline precursors, derived from phosphocholine, choline, and acetylcholine. Enzymatic reactions by phospholipase or lecithin-cholesterol acyltransferase (LCAT) lead to the separation of acyl or alkyl moieties from PC, resulting in the production of LPC9,10,11. Conversely, cancer cells utilize LPC as substrate for LPC acyltransferase 1 (LPCAT1), converting LPC into various PCs. These PCs are crucial for plasma membrane remodeling, and detectable in tissue or peripheral blood samples from cancer patients.
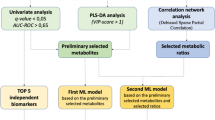
To investigate metabolomic characteristics specific to LUSC using peripheral blood samples, both healthy control and LUSC patient samples were analyzed. As metabolites are correlated in high degree, a feature selection method followed by a dimension reduction method was utilized to construct a diagnostic model. Additionally, to interpret the intricate nature of metabolites, network analysis was conducted for both healthy controls and LUSC patients, further stratified into early (EA) stages I and II, and advanced (ADV) stages III and IV for the LUSC patient groups.
Results
Metabolite concentrations
Out of 188 initially measurable metabolites, 123 metabolites underwent analysis after exclusion of molecules that exhibited values either below the limit of detection, below the limit of quantitation, or exceeding the upper limit of quantification in 70% of the samples. Notably, a statistically significant difference in mean age was observed between the control group (66.7 year) and LUSC group (72.4 year). Additionally, sex-based disparity was noted in LUSC cohort, with a higher prevalence of male patients (87.7%) (Supplementary Table S1).
The mean and standard deviation values for these metabolites in the healthy control group (n = 98) and LUSC group (n = 73) are provided (Supplementary Table S3, Supplementary Figure S1-S4). Concentrations of metabolites from early state (EA, n = 32) and advanced stage (Adv, n = 41) LUSC are listed and plotted (Supplementary Table S4, Supplementary Figure S5). Comparative analyses among normal, EA LUSC, and advanced (Adv) LUSC groups revealed statistically significant differences in the concentrations of glutamate, phenylalanine, serotonin, PC.aa.C34.4, PC.aa.C36.2, PC.aa.C36.6, PC.ae.C36.2, PC.ae.C38.0, PC.ae.C44.6, SM.OH.C22.1, and SM.C24.1.
MetaboanalystR
Further analysis was conducted using MetaboAnalystR, an R package synchronized with its website. Among the various functions in MetaboAnalystR, statistical analysis was performed. There are more than a dozen statistical functions, and among them, we selected the ones that best represent this study. A t-test with the false discovery rate (FDR) correction was applied to compare the concentrations of metabolites between normal controls and LUSC patients. A volcano plot wasused to summarize the metabolites. The Volcano plot shows the fold change of metabolites, comparing normal and LUSC groups, along with the p-value of fold changes. Enrichment analysis results are plotted to demonstrate the status of involved metabolic pathways.
T-tests were employed and represented in a plot based on P-value (Fig. 1A). Notably, metabolites such as C18:2, C18:1, serotonin, Arg, C10.1, C2, C12.1, PC.aa.C38.5, and PC.aa.C38.3 demonstrated lower P-values. The volcano plot displaying the fold change of metabolites between normal control and LUSC group revealed statistically significant increases in serotonin, Arg, glutamic acid (Glu), aspartic acid (Asp) and PC.aa.C38.6 in LUSC, while C18.2, C10.1, and C14.2showed decreases in LUSC (Fig. 1B).

Analysis results of metabolites by T-test (A), volcano plot (B) and pathway enrichment analysis (C).
Enrichment analysis focusing on amino acids was depicted in a bar graph (Fig. 1C), revealing significant pathways, including methylhistidine, urea cycle, ammonia recycle, Arg and Pro metabolism, glycine and serine metabolism, aspartate metabolism and spermidine, spermine biosynthesis. PCA analysis was performed based on metabolite concentration dataset including normal control and LUSC groups. PCA, an unsupervised dimension reduction method, indicated that component 1 and 2 explained 33.7% and 16.8% of the total dataset variance, respectively (Supplementary Figure S6). Additionally, PLS-DA analysis, a supervised dimension reduction method, explained 51.5% of the variance between control and LUSC groups, effectively distinguishing between them (Supplementary Figure S6). Clustering analysis showed that the majority of the normal and LUSC samples formed distinct clusters (Supplementary Figure S7).
Feature selection
Due to multicollinearity observed among the measured metabolites, a feature selection process employing seven distinct algorithms was utilized to identify variables (Fig. 2). Among them, Arg, C18.1, C18.2, hexoses, and serotonin were selected by all seven algorithms. Additionally, PC.ae.C38.0 was selected by six algorithms, with the exception of XGboost. Moreover, C10.1, PC.aa.C38.3, and PC aa C38.5 were selected by five algorithms, excluding LASSO and Random Forest. Further, C14, glutamic acid (Glu) and PC.aa.C38.4 were selected by four different methods. To validate these selections, dimension reduction method utilizing LASSO and subsequent LASSO analyses was implemented for variable selection (Table 1). The results demonstrated the selection of Arg, C18.2, Serotonin, PC.aa.C38.6, and PC.ae.C38.0, which corroborated the variables identified in the prior feature selection analysis.

Variable selection using seven feature selection method and concentration of metabolites. Color of the bar indicates feature selection method and arginine was selected by 7 methods (A) and concentration of metabolites between control and LUSC are plotted (B).
Cox regression analysis of the selected variables showed that Arg was the only variable that showed statistical significance (hazard ratio of 0.8981, p alue 0.0355).
As in survival analysis, high Arg concentration showed better survival.
Survival analysis
A 5- year overall survival analysis was conducted for all metabolites included in the feature selection process (Supplementary Figure S8). Conversely, metabolites not incorporated were also plotted separately (Supplementary Figure S9). Higher concentrations of amino acids and LysoPCs exhibited a trend towards favorable survival outcomes compared to lower concentrations, while elevated concentrations of PCs tended to correlate with unfavorable prognoses.
AUROC and Cox regression analysis
Among the 5 selected metabolites, Arg, C18.2, and PC.ae.C38.0 resulted in an AUROC (area under the receiver operator characteristics) (95% CI) of 0.911 (0.859–0.963), 0.997 (0.992-1.000), and 0.858 (0.802–0.912) (Supplementary Figure S10, Supplementary Table 5). Hazard ratios (95% CI) from the Cox regression analysis for these three metabolites were as follows: 1.060 (1.040–1.090); 0.860 (0.800–0.930); and 0.160 (0.090–0.280), respectively.
Network analysis
Construction of networks based on Spearman’s correlation matrix between control and LUSC groups, as well as between EA LUSC and ADV LUSC was performed (Supplementary Figure S11, S12). The network analysis revealed that the healthy normal group demonstrated higher network density, heterogeneity, centralization and average degree, whereas the LUSC group showed higher shortest path, assortativity and modularity (Table 2). For EA LUSC and ADV LUSC, most parameters was increased in ADV LUSC, except for the number of links, heterogeneity, shortest path, and diameter. The network topology of LUSC resulted in increased interactions between subgroups or modules, especially between amino acids and LPC. Interestingly, LPC mediated interactions between amino acids and PC (Fig. 3). For ADV LUSC, amino acids and LPC revealed an isolated amino acid subgroup compard to EA LUSC (Supplementary Figure S13).

Network construction based on correlation matrix of metabolites of healthy control (A) and LUSC (B) group.
Public data analysis
From the Xena browser, a dataset comprising 765 RNA-seq gene expression data from GDC TCGA LUSC was downloaded. Among these, 537 samples with associated survival data were selected for Kaplan-Meier analysis (Supplementary Figure S14, S15). Metabolite concentrations were interpolated within pathways. Notably, Arg, ornithine, glutamate, Pro, and 4-hydroxyproline were found to be elevated in this study, indicated by blue arrows (Fig. 4A). The gene expression status associated with Arg (ASL gene), ornithine (OTC gene) and Pro (PYCR2 gene) demonstrated prognostic significance (Fig. 4B, Supplementary Figure S13). Most PCs was decreased, while LPC levels were increased(Fig. 4C). Furthermore, gene expression related to PCs (CEPT1, CHPT1, LPCAT1) and LPC (LCAT, PLA2G16, PLB1) was associated with prognostic outcomes (Fig. 4D, Supplementary Figure S14). Cox Regression analysis for genes related to amino acid and lipid metabolism was performed (Supplementary Table S16). Multivariable analysis showed that the ASL gene, related with amino acid metabolism, and LPCAT1 gene, related to lipid metabolism, showed statistical significance.

Biosynthesis pathway and GDC TCGA LUSC data with overlaid study data. Arginine, proline biosynthesis pathway (A), survival data from GDC TCGA LUSC RNA-seq (B, D), phospholipid (PC) biosynthesis pathway (C).
Discussion
The analysis of the highly inter-correlated metabolome matrix necessitated feature extraction or feature selection methods and subsequent dimension reduction techniques12. Feature extraction encompassed PCA and PSLDA, merging features into new informative sets (Fig. 1)13. Feature selection methods aimed to retain informative features while discarding irrelevant ones14,15,16,17,18.
Decision tree-based algorithms (Random Forest, Gradient Boosting) and penalized regularization models (LASSO) ranked important features. This study utilized Adaboost, GAMboost, XGboost, Boruta, LASSO, Random Forest, SDWD algorithms for feature selection (Fig. 2). Features most frequently selected by these methods were initially considered potential informative variables. Additionally, to validate these features, LASSO was followed by additional LASSO for dimension reduction, yielding five variables: Arg, C18.2, PC.ae.C38.0, PC.aa.C38.6, and serotonin (Fig. 2). Among these variables, Arg, C18.2, and PC.ae.C38.0 were selected based on their concentration, accuracy, sensitivity, and specificity. Consequently, Arg, C18.2, and PC.ae.C38.0 could potentially be applied for the diagnosis of LUSC, although further studies involving various pathologic subtypes of lung cancer and large samples sizes are warranted.
MetaboAnalystR offers feature extraction methods along with a volcano plot displaying scattered data points with fold change and p-value (Fig. 1)13. Enrichment analysis focusing on proteins indicated statistically significant pathways such as the Argand Pro pathway, spermine, spermidine pathway and methylhistidine metabolism. Among metabolites in the Argand Pro pathway, 1-pyrroline-5-carboxylic acid is recognized for its association with tumor aggressiveness. Activation of the Pro biosynthesis pathway, triggered by hypoxia, a common factor, in the tumor microenvironment, may contribute to metabolic reprogramming the characteristics of cancer generation and progression19. These distinct characteristics were observed in LUSC patients compared to healthy individuals, evident through differences in metabolite concentrations and topological variations in network analysis20,21.
Concentrations of metabolites such as Arg, glutamate, glycine, phenylalanine, serine were found to be associated with tumor aggressiveness (Supplementary Figure S5). Glutamate and glycine are involved in glutathione synthesis, which is crucial for detoxifying reactive oxygen species (ROS)22. Cancer cells, due to their rapid proliferation, generate a significant amount of ROS, which is balanced by anti-ROS systems within the cells. The increased levels of glutamate and glycine may contribute to maintaining intracellular redox balance within LUSC cells. Moreover, higher levels of hexoses, including glucose, were observed in ADV LUSC. Reprogramming of glucose metabolism is also important in anabolic processes of cancer cells. Hexoses serve as energy sources and building blocks for cancer cells. These alterations in glycolysis pathways can affect methylation, glutathione and purine synthesis6. The increased levels of PC.aa.C32.2, PC.aa.C34.4, PC.aa.C36.2 and PC.aa.C36.6 in ADV LUSC warrant further studies.
Network analysis is applicable to complex systems, offering a global perspective on variable interrelatedness23,24. In contrast to feature extraction, selection, or dimension reduction methods, network analysis conserves data for topological analysis. By forming a correlation matrix and constructing a network based on these matrices (Supplementary Figure S11, S12), the network parameter analysis elucidated decreased network density, heterogeneity, average degree, and centrality in LUSC and ADV LUSC networks (Fig. 3, Supplementary Figure S13). Interestingly, in the LUSC group compared to the control group, LPC mediated amino acid and PC subgroups. These topological characteristics imply that LPC might play an important role in abnormal utilization of amino acids in LUSC. Indeed, concentrations of the LPCs were increased in the LUSC group compared to the control group. The role of LPC in mediating amino acids among LUSC requires further studies.
Reduced average degree and density may suggest a lack of coordination of metabolic pathways and a shift in the metabolome. Decreased metabolite heterogeneity implies a diminished diversity of metabolite functions, potentially serving as a source of energy or anabolic materials (Table 2). Additionally, a reduced shortest path in the LUSC and ADV LUSC network might indicate increased metabolic function, warranting further validation through additional studies25,26,27.
The TCGA data represent valuable resources for researchers, serving as crucial references or beacons for comparative analysis. The attached survival data addclinical significance and potential functionality to the obtained results28,29. The UCSC Xena browser offers web-based interfaces for downloading data. The integration of our study findings into these publicly available datasets indicates an association between the Arg, Pro, glutamate, and glutamine pathways and cancer prognosis. Furthermore, various enzymes related to the anabolism and catabolism of PC and LPC are also associated with cancer prognosis. However, precise functional roles of diverse metabolites necessitate further studies.
The limitation of this study is the relatively small sample size for subsequent machine learning analyses. Clinical data were unavailable, as the samples were de-identified. An observed age difference may have acted as a potential confounding factor. A sex difference was also noted in the LUSC group, with a predominance of male patients (87.7%). However, this difference might reflect real- world LUSC incidence or increased prevalence within the smoking population (Supplementary Table S1).
Conclusion
The metabolic reprogramming was evident in LUSC patients, characterized by elevated levels of Arg and reduced level of acylcarnitine C18.2 and phosphatidylcholine (PC) with acyl alkyl residues C38:0 (PC.ae.C38.0), showing associations with both the diagnosis and prognosis of LUSC. A decreased network density, heterogeneity, average degree and centrality was observed in the LUSC and ADV LUSC networks. Further studies are required by recruiting additional metabolites to elucidate other metabolic pathways underlying LUSC.
Methods
Patients
This study received approval from the Institutional Review Board of Seoul St. Mary’s Hospital and Eunpyeong St. Mary’s Hospital. Informed consent for researchers was waived, given that the samples were obtained after gaining informed consent from participants followed by anonymization procedures. The pool of 98 samples from healthy controls and 73 samples from LUSC patients was provided by the Sample Bank of Seoul St. Mary’s Hospital and Eunpyeong St. Mary’s Hospital, The Catholic University of Korea, Seoul, Korea, adhering to relevant regulations. All LUSC patients underwent surgical intervention, and pathology confirmation was conducted by the pathologists as a part of routine diagnostic protocols. Tissue samples from LUSC patients were categorized following the guidelines outlined in the World Health Organization of the American Joint Committee on Cancer Staging Manual30. The TNM (Tumor, Node, Metastasis) stages of the LUSC patients are described (Supplementary Table S1). Blood sample were obtained on the day of surgery from the LUSC patients after more than 8 hours of fasting, while healthy control samples were collected from individuals undergoing routine health checkups after more than 8 hours of fasting.
Targeted metabolomic measurement
Metabolite measurement was conducted using the AbsoluteIDQ p180 kit (Biocrates Life Science AG, Innsbruck, Austria), analyzed via mass spectrometry, employing isotope-labeled standards for quantification31,32. The kit facilitated the measurement of 188 metabolites, encompassing 21 amino acids and 21 biogenic amines, via liquid chromatography (LC)-mass spectrometry (MS) mode. Flow injection analysis was applied to 40 acylcarnitines, 76 phosphatidyl cholines (PC), 14 lysophosphatidyl cholines (LPC), 15 sphingomyelins (SM), and the sum of hexoses, respectively (Supplementary Table S2).
In brief, 10 µL of internal standard (amino acids, biogenic amines) followed by 10 µL of samples (calibrator, QC material, and patient samples) were added to the filter insert. This insert contained several internal standards related to glycerophospholipids, sphingomyelins, acylcarnitine and hexoses. This process was performed according to manufacturer’s instruction. The mixture was then dried with a nitrogen stream for 30 minutes. Subsequently, 50 µL of 5% phenyl isothiocyanate was added for derivatization of amino acids and biogenic amines. Following a 1 hour incubation, the molecules were extracted using 5mM ammonium acetate in 300 µL methanol. Half of the eluate was analyzed by LC-MS/MS, while the other half was analyzed by flow injection analysis MS/MS. For amino acids and biogenic amines, a Zorbax Eclipse XDBC 18 column (3 mm x 100 mm, 3.5 µM, Agilent Technologies, USA) was employed at a flow rate of 0.5 mL/min using a water and acetonitrile with 0.2% formic acid in the LC gradient conditions. The analysis of PCs, SM, LPCs, acylcarnitines and hexoses utilized a flow rate of 0.03 mL/min with an elution solvent in flow injection analysis gradient. Multiple reaction monitoring modes of the MS (API 4000, AB Sciex, Darmstadt, Germany) in positive electrospray ionization mode were used for quantification, while only hexoses were analyzed in negative mode20,21.
Data analysis
Quantification involved the utilization of internal standards in conjunction with calibration curves. Following peak integration and concentration calculation using Analyst 1.5.1 software (AB Sciex, Darmstadt, Germany), the processed data were imported into the Biocrates MetIDQ software31. Derivative metabolites, which included amino acids and biogenic amines, were quantitated employing seven-point-calibration curves integrated with internal standards. The semi-quantitative assessment of PCs, LPCs, SMs, acylcarnitines and hexoses relied on the specific stable isotope internal standard intensities. Subsequently, the determination of final metabolite concentrations was conducted, employing inter- and intra-plate normalization methods. This normalization was based on median values derived from three replicate injections of QC2 samples from each plate, aligned with the target values31.
Network analysis
Network parameters were established in accordance with previously documented methodologies23,24. Specifically, each metabolite was represented as a Node (N), while correlations between nodes was denoted as a Link (L). The average clustering coefficient (< C> ) denoted the average link among adjacent nodes. Network density indicated the ratio of actual links to the maximum potential links within the network. Network heterogeneity reflected the variation in the links connected to a node, divided by the mean link value. Connected compounds represented a connected subgraph not included in a larger giant component. The shortest path defined the minimum number of links between two nodes. The average degree (< k> ) depicted the total number of links divided by the total number of nodes. Network diameter signified the longest shortest path between nodes within the network. Assortativity implies a node’s inclination to connect with another node sharing similar features, typically defined by the number of a node’s links. Modularity is the preference of a node to form a cluster or subgraph with dense interconnection within the subgraph25,26,27. The hub node designated the node with the highest connected links. Calculation of these parameters was performed utilizing NetAnalyzer and Cytoscape applications 27,33,34. The formulas utilized in this study is described in the supplementary materials (Supplementary S17).
Public data analysis
For verification of the quantified metabolites, the LUSC (n = 765) dataset from The Genomic Data Commons (GDC) The Cancer Genome Atlas (TCGA), provided by the National Cancer Institute, was acquired through the UCSC Xena browser (www.xenabrowser.net)28,29. A subset comprising primary tumors with available survival data (n = 537) was selected for further analysis. Genes of interest were analyzed that was extracted from RNA-seq count data, and these genes were specifically associated with proteins or phosphatidylcholines pathways measured in this study. A comprehensive 5 year survival analysis was conducted for these pertinent genes.
To illustrate the arginine (Arg), proline (Pro) metabolism pathway (hsa T30003) and the PC and LPC pathway, graphical representations were generated based on modifications derived from the KEGG database35,36. These pathways were adjusted to integrate the measured metabolites obtained from our study, thus facilitating an interpolation of the identified metabolites into these pathways.
Statistical analysis
Due to the observed correlation and multicollinearity among the measured variables, feature selection was performed using following algorithms available in the caret R package and others, including Adaboost, Boruta, gradient boosting for additive models in mboost (GAMboost), least absolute shrinkage and selection operator (LASSO), Random Forest, sparse distance weighted discrimination (SDWD), and extreme gradient boosting (XGBoost)12,14,15,16,17,18. Independently, LASSO which is a penalized regression model, was performed and among the selected variables. As selected variables still had multicollinearity, additional analysis by LASSO was employed using the hdm package37,38. The variables selected by both LASSO and feature selection were compared. Differences in measured metabolites between healthy controls and individuals diagnosed with LUSC were assessed using Student’s t-test for variables with normal distribution and a non-parametric test for those with the non-normal distribution (Supplementary Table S18). For comparison between early and advanced LUSC, Kruskal-Wallis test followed by Wilcoxon signed rank test was performed using the R package moonBook39. Subsequent metabolite analyses involved the utilization of the MetaboAnalystR package, an R package synchronized with its website. Among various functions in MetaboAnalystR, statistical analysis was performed. There are more than dozen statistical functions and among them, we selected the ones that best represents this study. A t-test using the false discovery rate (FDR) option was applied to compare the concentrations of metabolites between normal control and LUSC. A volcano plot was applied to summarize the concentrations of metabolites. The Volcano plot shows the fold change of metabolites, which compares normal and healthy control groups and the p-value of compared fold changes. Enrichment analysis results are plotted to demonstrate the status of involved metabolic pathways. In addition, principal component analysis (PCA) and partial least squares discriminant analysis (PLS-DA)was performed.
Survival analysis encompassing 5 year overall survival rates, where death from any cause was considered an event, was carried out utilizing the survival and survminer packages40. Receiver operating characteristic (ROC) curve analysis was performed to identify metabolites potentially useful for diagnosing LUSC, employing the ROC R package41. All the statistical analyses were conducted in R program, and data were processed using packages such as dplyr, while graphical representations were plotted using ggplot2.
Data availability
All the data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- LUSC:
-
Squamous cell lung carcinoma
- Arg:
-
arginine
- C18.2:
-
Acylcarnitine C18:2, C18.2
- PC:
-
Phosphatidylcholine
- PC.ae.C38.6:
-
PC with acyl alkyl residues C38:6
- LPC:
-
Lysophosphatidylcholines
- EA:
-
Early stage cancer
- ADV:
-
Advanced stage cancer
- TNM:
-
Tumor, Node, Metastasis stages
- LC:
-
Liquid chromatography
- MS:
-
Mass spectrometry
- TCGA:
-
The Cancer Genome Atlas
- Pro:
-
Proline
- GAMboost:
-
Gradient boosting for additive models in mboost
- LASSO:
-
Least absolute shrinkage and selection operator
- SDWD:
-
Sparse distance weighted discrimination
- XGboost:
-
Extreme gradient boosting
- PCA:
-
Principal component analysis
- PLS-DA:
-
Partial least squares discriminant analysis
- ROC:
-
Receiver operating characteristic
- Glu:
-
Glutamic acid
- ASL :
-
Argininosuccinate lyase
- OTC :
-
Ornithine transcarbamylase
- PYCR2 :
-
Pyrroline-5-carboylate reductase 2
- CEPT1 :
-
Choline/Ethanolamine Phosphotransferase 1
- CHPT1 :
-
Choline phosphotransferse 1
- LPCAT1 :
-
1-acyl-sn-glycoerol-3-phosphate acyltrnsferase
- LCAT :
-
Lecithin-cholesterol acyltransferase
- PLA2G16 :
-
Phospholipase A and Acyltransferase 3
- PLB1 :
-
Phospholipase B1
References
Sung, H. et al. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Gridelli, C. et al. Non-small-cell lung cancer. Nat. Rev. Dis. Primers. 1, 15009 (2015).
Collisson, E. A. et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014).
Alvarado, A. & Arce, I. Metabolic functions of the lung, disorders and associated pathologies. J. Clin. Med. Res. 8, 689–700 (2016).
DeBerardinis, R. J. & Chandel, N. S. Fundamentals of cancer metabolism. Sci. Adv. 2, e1600200 (2016).
Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer. 16, 635–649 (2016).
Lesko, J. et al. Phospholipid dynamics in ex vivo lung cancer and normal lung explants. Exp. Mol. Med. 53, 81–90 (2021).
Lieu, E. L., Nguyen, T., Rhyne, S. & Kim, J. Amino acids in cancer. Exp. Mol. Med. 52, 15–30 (2020).
Wei, Z., Liu, X., Cheng, C., Yu, W. & Yi, P. Metabolism of amino acids in Cancer. Front Cell. Dev. Biol 8 (2021).
Xiao, M. et al. Functional significance of cholesterol metabolism in cancer: From threat to treatment. Exp. Mol. Med. 55, 1982–1995 (2023).
Singh, A. et al. Serum metabolic disturbances in lung Cancer investigated through an elaborative NMR-Based serum metabolomics approach. ACS Omega. 7, 5510–5520 (2022).
Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. 28, 1–26 (2008).
Chong, J. & Xia, J. MetaboAnalystR: an R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 34, 4313–4314 (2018).
Kursa, M. B. & Rudnicki, W. R. Feature selection with the Boruta package. J. Stat. Softw. 36, 1–13 (2010).
Lantz, B. PACKT,. Machine learning with R. (2019).
Terry Therneau, E. J. A. & Foundation, M. rpart: Recursive Partitioning and Regression Trees (2022). https://github.com/bethatkinson/rpart
Chen, T. C. G. XGBoost: A Scalable Tree Boosting System. arXiv (2016).
Wang, B. & Zou, H. Sparse distance weighted discrimination. J. Comput. Graph Stat. 25, 826–838 (2016).
Kuo, M. T., Chen, H. H. W., Feun, L. G. & Savaraj, N. Targeting the Proline-Glutamine-Asparagine-Arginine metabolic Axis in amino acid starvation Cancer therapy. Pharmaceuticals 14 (2021).
Shestakova, K. M. et al. Targeted metabolomic profiling as a tool for diagnostics of patients with non-small-cell lung cancer. Scic Rep. 13, 11072 (2023).
Saito, R. F., Andrade, L. N. S., Bustos, S. O. & Chammas, R. Phosphatidylcholine-Derived lipid mediators: The crosstalk between Cancer cells and immune cells. Front. Immunol. 13, 768606 (2022).
Barabási, A. L. & Oltvai, Z. N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 5, 101–113 (2004).
Newman, M. Networks: an Introduction (Oxford University Press, 2010).
Kim, K. S. et al. Immune gene expression networks in sepsis: A network biology approach. PLoS One. 16, e0247669 (2021).
Kim, K. S. et al. Immune gene networks form lung cancer patients treated with immune checkpoint inhibitors. Biomedicines 12, 628 (2024).
Pavlopoulos, G. A. et al. Using graph theory to analyze biological networks. BioData Min. 4, 10 (2011).
Doncheva, N. T., Assenov, Y., Domingues, F. S. & Albrecht, M. Topological analysis and interactive visualization of biological networks and protein structures. Nat. Protoc. 7, 670–685 (2012).
Hammerman, P. S. et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 (2012).
Goldman, M. J. et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 38, 675–678 (2020).
Edge, S. B. & Compton, C. C. The American joint committee on cancer: The 7th edition of the AJCC Cancer staging manual and the future of TNM. Ann. Surg. Oncol. 17, 1471–1474 (2010).
Kim, K. S. et al. Metabolic profiles of lung adenocarcinoma via peripheral blood and diagnostic model construction. Sci. Rep. 13, 7304 (2023).
Qi, S. et al. High-resolution metabolomic biomarkers for lung cancer diagnosis and prognosis. Sci. Rep. 11, 11805 (2021).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Saito, R. et al. A travel guide to cytoscape plugins. Nat. Methods. 9, 1069–1076 (2012).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG:biologicl systems database as a model of the real world. Nucleic Acids Res. 53, D672–677 (2025).
Chernozhukov, V. H. & Spindler, C. High-Dimensional metrics. R J. 8, 14 (2016).
Hastie, T. The Element of Statistical Learning (Springer, 2009).
Moon, K. W. R Statistics and Graphs for Medical Papers (Hannarae, 2015).
Alboukadel Kassambara, M. K. & Biecek, P. Scheipl Fabian. survminer: Drawing Survival Curves using ggplot2 (2021). https://cran.r-project.org/web/packages/survminer/index. html.
Robin, X. et al. pROC: An open-source package for R and S + to analyze and compare ROC curves. BMC Bioinform. 12, 77 (2011).
Acknowledgements
We are grateful to the patients and healthy normal individuals for the banking of their samples. The specimens for this study were provided by Sample Bank from Seoul St. Mary’s Hospital and Eunpyeong St. Mary’s Hospital, The Catholic University of Korea, Seoul, Korea.
Author information
Authors and Affiliations
Contributions
D.W.J. takes full responsibility of the content, data and the analysis result. D.W.J.: conceptualization, validation, investigation, data analysis, writing original draft, writing review and editing. K.K.S.: conceptualization, data analysis, writing original draft, writing review and editing, fund acquisition. S.W.M.: resources, supervision; M.H.M.: conceptualization, resources, data analysis; S.J.K.: resources, supervision; K.Y.K.: methodology, data curation, supervision.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, K.S., Moon, S.W., Moon, M.H. et al. Metabolic profiles of squamous cell lung carcinoma and diagnostic model construction. Sci Rep 15, 34015 (2025). https://doi.org/10.1038/s41598-025-12412-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-12412-w
