
Abstract
SIRT3 knockout mice develop cardiac insufficiency due to decreased mitochondrial function. However, upregulation of the NAD+/NADH ratio can compensate for SIRT3 deficiency through the SIRT1/PGC-1α pathway, thereby improving mitochondrial function. We therefore hypothesized that upregulation of SIRT1 expression could improve cardiac function in SIRT3 knockout mice through its interactive compensatory effect. We first determined that SIRT3 knockout mice would develop cardiac insufficiency at 8 weeks of age by ultrasound cardiac function testing, and then we selected 6-week-old SIRT3 knockout mice with similar body weights of both sexes and performed intraperitoneal injections of NMN over a 14-day period to increase the content of NAD + in the myocardial tissue of the mice. The results showed that NMN injection effectively increased the NAD + content as well as the NAD+/NADH ratio within the myocardial tissue of SIRT3 knockout mice and stimulated the expression of SIRT1 protein. In addition, protein expression of PGC-1α and its downstream molecules as well as molecules related to subunits of the respiratory chain complex was increased in mouse myocardial mitochondria. Meanwhile, NMN injection improved the cardiomyocyte and mitochondrial structure of mice, ultimately ameliorating cardiac insufficiency in SIRT3 knockout mice. In conclusion, our results suggest that NMN can compensate for SIRT3 deficiency via the SIRT1/PGC-1α pathway and improve mitochondrial biosynthesis and oxidative respiration, thereby improving cardiac function in Sirt3−/− mice. This may provide new ideas for the clinical treatment of cardiac insufficiency.
Similar content being viewed by others


Introduction
Cardiac insufficiency is a clinical syndrome characterized by cardiac output failing to meet systemic tissues’ metabolic demands due to impaired pumping function. Its manifestations typically include weakness, dyspnea, and lower extremity edema1,2. The pathogenesis of cardiac insufficiency is mainly the reduction of myocardial systolic and diastolic function, and the underlying cause is the alteration of myocardial mitochondrial system function. The heart has a very high demand for energy and must constantly produce large amounts of ATP to maintain normal contractile function3,4 so the myocardial tissue has a highly developed mitochondrial system, which provides large amounts of energy and metabolites to maintain the normal function of the myocardium. The ATP required by the normal adult heart is mainly derived from mitochondrial oxidative phosphorylation5. When cardiac insufficiency occurs, mitochondria are damaged due to membrane rupture and matrix depletion6 which leads to a decrease in the activity of oxidative phosphorylation-related enzymes, resulting in a significant decrease in the mitochondria’s ability to synthesize ATP7,8. Also in the presence of impaired respiratory chain activity and ATP depletion, dysfunctional mitochondria can initiate cardiomyocyte apoptosis, causing irreversible damage to myocardial tissue9.
The sirtuins, a family of deacetylases in cells, are important molecules involved in maintaining mitochondrial function. Among them, SIRT3(Sirtuin 3) plays an important role in many reaction processes within mitochondria, such as promoting mitochondrial biosynthesis, counteracting oxidative stress, and facilitating the tricarboxylic acid cycle and electron transport10,11.Deficiency of SIRT3 reduces cardiac systolic function12 and decreased expression of SIRT3 interferes with oxidative phosphorylation processes, leading to an imbalance in myocardial energy metabolism13. There is an interactive compensatory mechanism for the role of Sirtuins, and it has been found that SIRT1(Sirtuin 1) expression compensates for SIRT3 deficiency, and the cofactor PGC-1α(Peroxisome proliferative activated receptor, gamma, coactivator 1α), which is biosynthesized by mitochondria, is the main molecule in which the two play an interactive role. In the absence of SIRT3, changes in intracellular NAD+( Nicotinamide adenine dinucleotide) levels can affect mitochondrial function through the SIRT1/PGC-1α pathway14. In addition, it has been demonstrated that up-regulation of the NAD+/NADH ratio can improve mitochondrial function via the SIRT1/PGC-1α pathway15 but the role of this pathway in the myocardium is unclear.
During the preliminary work of this experiment, we found that SIRT3 knockout (Sirt3−/−) mice develop cardiac insufficiency from 8 weeks of age onwards, and cardiac insufficiency in Sirt3−/− mice was improved by administration of the precursor of NAD+, NMN. We hypothesized that NMN could improve cardiac function in Sirt3−/− mice by upregulating SIRT1 expression to compensate for SIRT3 deficiency. In our experiments, we found that the protein expression of mitochondrial biosynthesis-related molecules PGC-1α, NRF1(Nuclear respiratory factor 1), NRF2 Nuclear respiratory factor , and TFAM Mitochondrial transcription factor A was decreased in myocardial tissues of 8-week-old Sirt3−/− mice, as well as the protein expression of respiratory chain complex subunits MTND1(mitochondrially encoded NADH dehydrogenase 1), SDHB(succinate dehydrogenase complex iron sulfur subunit B), MTCO2(mitochondrially encoded cytochrome c oxidase II), and ATP5A(ATP synthase F1 subunit alpha). After the administration of NMN, the expression of all the above proteins was increased in myocardial tissues of Sirt3−/− mice, and the expression of SIRT1 protein was also elevated. Therefore, we conclude that NMN can improve cardiac function in Sirt3−/− mice by increasing mitochondrial biosynthesis capacity through the SIRT1/PGC-1α pathway.
Materials and methods
Animals
Wild-type C57BL/6J mice were purchased from Beijing Viton Lihua Laboratory Animal Technology Co. ltd.SIRT3 knockout C57BL/6J mice were originally purchased from JAX in the United States, and then bred in the barrier facility of the School of Basic Medical Sciences of Jilin University. The animal experiment has been approved by the Animal Ethics Committee. All experiments were performed in accordance with relevant named guidelines and regulations. The authors complied with the ARRIVE guidelines.
Drugs and antibodies
Drugs: β-nicotinamide mononucleotide (NMN) (Shanghai Yuanye Biotechnology Co., Ltd., China).
Antibodies: SIRT1 (FNab10371, Fine Test, China), SIRT3 (10099-1-AP, Proteintech, USA), PGC-1α (66369-1-Ig, Proteintech, USA), NRF1 (66832-1-Ig. Proteintech, USA), NRF2 (66504-1-Ig, Proteintech, USA), TFAM (22586-1-AP, Proteintech, USA), MTND1 (A17967, ABclonal, China), OXPHOS (45-8199,ThermoFisher, USA), BNP(13299-1-AP, Proteintech, USA).
Experimental grouping and treatment
Genetically characterized 6-week-old WT mice and Sirt3−/− mice were randomly divided into saline (Control, Sirt3−/−) and NMN groups (WT + NMN, Sirt3−/−+NMN), respectively, with six mice in each group. The four groups of mice received different treatments under the condition of maintaining normal life: NMN solution at a concentration of 500/mg/kg/d for 14 days and each mouse in the saline group was injected with saline according to the same dosage for 14 days, and the relevant assays were performed starting on the 15th day.
Western blot
An appropriate amount of left ventricular zone tissues were rapidly removed from mice after they were humanely euthanized by overdose of carbon dioxide anesthesia, After adding RIPA, the cells were lysed by grinding with an electric grinder, and then the protein content was determined using Bradford’s reagent (Beyotime Biotechnology, Shanghai, China). The samples were then assayed using Western Blot, and proteins were separated using 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis at 80 V for 2 h, followed by 100 V for 1 h. The proteins were transferred to PVDF membranes. 5% skim milk powder was used to block at room temperature for 2 h. The samples were incubated with the corresponding primary antibodies at 4 °C overnight, and the secondary antibodies were incubated at room temperature for 2 h on the following day. After that, the samples were analyzed using ECL reagents (YEASEN, Shanghai, China) and a gel imager (Syntyos, Cambridge, MA, USA), automatic exposure.
Determination of tissue ATP content
An appropriate amount of left ventricular zone tissues were rapidly removed from mice after they were humanely euthanized, washed with ice-cold saline, and then processed according to the instructions of the enhanced ATP assay kit (Beyotime Biotechnology, Shanghai, China).
Measurement of NAD content in mouse myocardial tissue
After euthanizing the mice, weigh an appropriate amount of myocardial tissue (approximately 20 mg), wash it with ice-cold physiological saline, place it in a 1.5 ml centrifuge tube, add NAD+/NADH extraction buffer, and homogenize thoroughly. Centrifuge at 13,000×g for 10 min at 4 °C. Transfer the supernatant while keeping the samples on ice throughout the procedure. Take a new 1.5 ml centrifuge tube, pipette approximately 100 µL of the supernatant, and incubate it in a 60 °C water bath for 30 min. Detect the total NAD and NADH content of the resulting samples according to the kit instructions (Beyotime Biotechnology, Shanghai, China, S0175).
Mouse echocardiography
Anesthetize wild-type and Sirt3-/- mice after 14 days of group-specific treatment with avertin, depilate them, fix them in a supine position on a flat plate, and perform small-animal echocardiography(Esaote sigma VET system).
HE staining and electron microscopy
After 14 days of group-specific treatment, wild-type and Sirt3-/- mice were euthanized, rapidly excise left ventricular tissue and fix it in corresponding fixatives. Subsequent slide preparation, staining, and imaging were performed by the Pathology Laboratory of the School of Basic Medicine, Jilin University.
Statistical analysis
All data in the manuscript are presented as the mean ± standard deviation. Statistical analyses were conducted using SPSS version XX (SPSS Inc., Chicago, IL, USA). For intergroup comparisons, independent two - sample t - tests were used for comparing two groups, and one - way analysis of variance (one - way ANOVA) was applied for comparing more than two groups. After one - way ANOVA indicated significant differences among groups, We further performed post - hoc pairwise comparisons between groups using Tukey’s HSD (Honestly Significant Difference) test. The quantitative values of Western Blot bands were expressed as the ratio of grayscale values calculated by the Image J software to those of a certain reference sample.
Results
Sirt3−/− mice develop cardiac insufficiency
To verify the role of SIRT3 in maintaining cardiac function, we used a small animal ultrasound system to examine the cardiac function of 4-week-old, 8-week-old, and 12-week-old WT and Sirt3−/− mice, respectively (Fig. 1.supplement Fig. 1). The results showed that compared with the WT mice, the Sirt3−/− mice would exhibit cardiac insufficiency, with a decrease in cardiac output and stroke volume starting from 8 weeks of age.

Ultrasound examination of cardiac function in WT mice and Sirt3−/− mice at different weekly ages(n = 6). (a ) Stroke Volume of mice; (b) Cardiac Output of mice (*P < 0.05 vs. WT).
Cardiac insufficiency in Sirt3−/− mice is associated with decreased mitochondrial biosynthesis capacity
Subsequently, we observed the microstructure of myocardial tissues in mice, and the results of HE staining (Fig. 2a, b Black arrow: hypertrophic myocardial cells) showed that at 4 weeks of age, there was no significant difference in the arrangement and morphology of cardiomyocytes between WT and Sirt3−/− mice, but at 8 weeks of age, cardiomyocytes of WT mice were more neatly arranged, with dense nuclei, clear cytoplasmic structure, and homogeneous staining, whereas the arrangement of cardiomyocytes of Sirt3−/− mice were more disorganized, with nuclei and local cytoplasm showing cavitation-induced alterations, and hypertrophic cardiomyocytes could be observed. In contrast, Sirt3−/− mice had disorganized cardiomyocytes with vacuolated nuclei and localized cytoplasmic changes, and deeply stained hypertrophic cardiomyocytes, which may be due to compensatory changes of cardiomyocyte hypertrophy in 8-week-old Sirt3−/− mice due to insufficient myocardial contractile and diastolic function to maintain normal myocardial ejection. Electron microscopic observation of cardiomyocyte ultrastructure (Fig. 2c Yellow arrow: disorganized mitochondria; white arrow: lipid droplets) showed that at 4 weeks of age, the myonodular bright and dark bands of cardiomyocytes from WT mice and Sirt3−/− mice were clearly structured, the myofilaments were neatly arranged, and the mitochondrial bilayer membrane and cristae were well defined; however, at 8 weeks of age, the myonodular bright and dark bands of cardiomyocytes were not clearly structured, the size of mitochondria had become larger, the bilayer membrane and cristae were not well defined, and the intermitochondrial structure was disordered, with mitochondria being larger in size. The mitochondrial volume was enlarged, the bilayer membrane structure and cristae were unclear, and more lipid droplets were seen among mitochondria. The above results indicate that mitochondrial morphology and dysfunction have been observed in 8-week-old Sirt3−/− mice cardiomyocytes. Western blot analysis of BNP expression in cardiomyocytes revealed that 8-week-old Sirt3−/− mice exhibited significantly higher BNP levels compared to age-matched wild-type controls (Fig. 2d, e).

Mitochondrial function assay of myocardial tissue from WT mice and Sirt3−/− mice(n = 6). (a) HE staining (×400) of mouse myocardial tissues (hypertrophied cardiomyocytes indicated by arrows, scale bar is 50 μm); (b)Quantification of cardiomyocyte cross-sectional area; (c) morphology of mouse cardiomyocytes and mitochondria under electron microscopy (scale bar is 0.5 μm); (d) BNP expression level; (e) Quantification graph of BNP expression level (*P < 0.05 vs. WT; **P < 0.01 vs. WT).
We then further examined the protein expression of mitochondrial biosynthesis-related molecules in myocardial tissues of 8-week-old WT mice and Sirt3−/− mice (Fig. 3a,b), and the results showed that cardiomyocytes of Sirt3−/− mice showed decreased protein expression of PGC-1α, NRF1, NRF2, and TFAM, which suggests that the biosynthesis capacity of mitochondria in the cardiac muscle tissues of Sirt3−/− mice was reduced under the condition of SIRT3 deficiency. The protein expression of mitochondrial respiratory chain complex subunits in myocardial tissues of 8-week-old mice was examined (Fig. 3c, d), and the results showed that the protein expression of MTND1, SDHB, MTCO2, and ATP5A was decreased, that the respiratory capacity of mitochondria in the cardiac tissue of Sirt3−/− mice was decreased.

(a) protein expression of molecules related to mitochondrial biosynthesis in mouse myocardial tissues; (b) quantitative analysis of protein expression in the c plot; (c) protein expression of subunits of the respiratory chain complex in mouse myocardial tissue; (d) quantitative analysis of protein expression in the e plot (*P < 0.05 vs. WT; **P < 0.01 vs. WT). original blots are presented in original data Fig. 3a, c.
NMN administration improves cardiac function in Sirt3−/− mice by increasing mitochondrial oxidative respiratory capacity
After 14 days of administration of saline or NMN, we performed ultrasound cardiac function tests on four groups of mice (Fig. 4a, b, supplement Fig. 2), and the results showed that mice in the Sirt3−/− group showed obvious signs of cardiac insufficiency compared with mice in the Control group, whereas mice in the Sirt3 − /− + NMN group, although still showing signs of cardiac insufficiency, demonstrated that NMN was effective in improving the cardiac function of Sirt3 − /− mice. This demonstrates that NMN can effectively improve cardiac insufficiency in Sirt3−/− mice. Immediately after that, we observed the myocardial tissue structure of mice, and the HE staining results (Fig. 5a,b) showed that compared with the Control group mice, the cardiomyocytes of the Sirt3−/− group mice were more disorganized, and deeply stained hypertrophic cardiomyocytes were seen, whereas the cardiomyocytes of the Sirt3−/−+NMN group mice were more regularly arranged, and staining was more homogeneous, with occasional hypertrophic cells in the field of view. It was demonstrated that the occurrence of compensatory hypertrophy of cardiomyocytes in Sirt3−/− mice was improved after administration of NMN. Electron microscopy results (Fig. 5c, Yellow arrow: disorganized mitochondria; white arrow: lipid droplets) showed that compared with Control group mice, Sirt3−/− group mice had uneven size of myocardial mitochondria, partial loss of bilayer membrane structure, and unclear cristae structure, whereas Sirt3−/−+NMN group mice had more uniform size of myocardial mitochondria, clear structure of bilayer membrane, and clearer cristae structure. It was demonstrated that the myocardial mitochondrial structure of Sirt3−/− mice was improved after administration of NMN.
To further verify whether mitochondrial function was improved in Sirt3−/− mice, we first examined the ATP content in myocardial tissues of mice (Fig. 5d), found that the ATP content in myocardial tissues of mice in the Sirt3−/− group was decreased compared with the Control group, whereas the ATP content in myocardial tissues of mice in the Sirt3−/−+NMN group was increased compared with the Sirt3− /− group. Western blot analysis showed that BNP expression was increased in Sirt3−/− mice, but decreased after NMN administration. (Figure 5e,f)
Afterwards, we examined the protein expression of mitochondrial respiratory chain complex subunits in the myocardial tissues of mice (Fig. 5g,h), which showed that the protein expression of MTND1, SDHB, MTCO2, and ATP5A was decreased in the Sirt3−/− group of mice compared with that in the Control group, whereas the expression of the above proteins was elevated by the administration of NMN.
In summary, NMN improves cardiac insufficiency by increasing mitochondrial oxidative respiratory capacity and promoting ATP production in Sirt3−/− mouse cardiomyocytes.

Cardiac function in four groups of mice after different treatments(n = 6). (a) Mouse stroke volume ; (b) mouse cardiac output (*P < 0.05 vs. Control);

(a) HE staining of mouse myocardial tissues (×400) (arrowheads point to hypertrophied cardiomyocytes, scale bar is 50 μm); (b)Quantification of cardiomyocyte cross-sectional area (c) Morphology of mouse cardiomyocytes and mitochondria under electron microscopy (scale bar is 1 μm, Yellow arrow: disorganized mitochondria; white arrow: lipid droplets); (d) ATP content of mouse cardiac tissues (*P < 0.05 vs. Control; **P < 0.01 vs. Control; #P < 0.05 vs. Sirt3−/−); (e)BNP expression level (f) Quantification graph of BNP expression level; (g) Protein expression of subunits of respiratory chain complex in mouse cardiac tissues; (h) Quantitative analysis of protein expression in Figure g (*P < 0.05 vs. Control; **P < 0.01 vs. Control; ##P < 0.01 vs. Sirt3−/−). original blots are presented in original data Fig. 5g.
NMN improves mitochondrial biosynthesis in Sirt3−/− mice via the SIRT1/PGC-1α pathway
To verify whether NMN improves the mitochondrial biosynthetic capacity of Sirt3−/− mice via the SIRT1/PGC-1α pathway, we first examined the contents of NAD + and NADH as well as the NAD+/NADH ratios in the myocardial tissues of the two types of mice before and after the administration of NMN, as seen in Fig. 6a: Although the levels of NAD + and NADH in the myocardial tissues of the Sirt3 − /− mice were elevated, the NAD+/NADH ratio was significantly decreased. After the administration of NMN, it can be observed that the NAD+, NADH content and NAD+/NADH ratio were increased in the myocardial tissues of mice in the WT + NMN group, and NAD + and NAD+/NADH ratio were increased in the myocardial tissues of mice in the Sirt3−/−+NMN group, while the NADH content was decreased. These results proved that the administration of NMN effectively increased the NAD + content and improved the metabolism of NAD in myocardial tissues of Sirt3−/− mice.
After that we examined the protein expression of mitochondrial biosynthesis-related molecules in myocardial tissues of mice (Fig. 6b and c), and the results showed that the protein expression of PGC-1α, NRF1, NRF2, and TFAM in myocardial tissues of Sirt3 − /− mice was lower than that in WT mice, but the expression of the above proteins increased after the administration of NMN. (Figure 6d and e) By examining the protein expression of SIRT1 and SIRT3 in myocardial tissues of mice, we found that the protein expression of SIRT1 and SIRT3 in myocardial tissues of WT mice was elevated after administration of NMN, and that the expression of SIRT1 in myocardial tissues of Sirt3−/− mice was elevated even though they did not express SIRT3. It was demonstrated that the elevated NAD+/NADH ratio stimulated SIRT1 protein expression.
We conclude that NMN can increase the NAD+/NADH ratio, stimulate the expression of SIRT1, and improve the mitochondrial biosynthesis capacity of Sirt3−/− mice via the SIRT1/PGC-1α pathway.

Mitochondrial biosynthesis capacity assay in four groups of mice after different treatments(n = 6). (a) Changes of NAD+, NADH content and NAD+/NADH ratio in mouse myocardial tissues after different treatments (**P < 0.01 vs. Control; ##P < 0.01 vs. Sirt3−/−); (b) Protein expression of molecules related to mitochondrial biosynthesis in mouse myocardial tissues; (c) Quantitative analysis of the protein expression in b-plot (**P < 0.01 vs. Control; ##P < 0.01 vs. Sirt3−/−); (d) Protein expression of SIRT3 and SIRT1 in mouse myocardial tissues; (e) quantitative analysis of protein expression in d graph (**P < 0.01 vs. Control; ##P < 0.01 vs. Sirt3−/−). original blots are presented in original data Fig. 6b,d.
Conclusion
Sirt3−/− mice develop cardiac insufficiency at 8 weeks of age due to the reduction in mitochondrial biosynthesis capacity and oxidative respiratory capacity, but the administration of NMN improves cardiac function in Sirt3−/− mice by enhancing mitochondrial function through the SIRT1/PGC-1α pathway, taking advantage of the compensatory effect of SIRT1.
Discussion
Cardiac insufficiency is a common clinical syndrome that can develop from a variety of diseases, and although the etiologies leading to cardiac insufficiency are not identical, the main mechanism is mitochondrial damage and dysfunction, which leads to insufficient energy production and ultimately manifests itself as a decrease in the heart’s ejection capacity16. Myocardial tissue has a high demand for energy, so cardiomyocytes contain a large number of mitochondria, and mitochondria serve as the hub of energy metabolism within cardiomyocytes, the morphological and functional stability of mitochondria is a prerequisite for maintaining normal cardiac function. Among them, SIRT3 is an indispensable molecule to maintain mitochondrial function.SIRT3 is an important deacetylase, which is localized in mitochondria and is involved in the maintenance of cellular metabolic homeostasis, mitochondrial quality control, and mitochondrial unfolded protein response10.SIRT3 can deacetylate mitochondrial TCA and fatty acid oxidation-related enzymes, regulate mitochondrial OXPHOS, and ensure energy supply11thereby protecting cardiac function. On the contrary, SIRT3 deficiency decreases the contractile function of the heart12 while decreased SIRT3 expression interferes with OXPHOS, leading to an imbalance in myocardial energy metabolism13. In addition, it has been found that Sirt3−/− mice are more likely to exhibit cardiac microvessel thinning and functional hypoxia than WT mice, and Sirt3−/− mice have enhanced expression of markers of myocardial fibrosis and develop significant mitochondrial dysfunction17,18. Therefore, it can be said that SIRT3 is an indispensable molecule for maintaining normal mitochondrial morphology and functional stability.
In the early stage of this experiment, we found that Sirt3−/− mice would develop cardiac insufficiency at 8 weeks of age, which was manifested as a decrease in cardiac output, while HE staining revealed that myocardial tissues showed compensatory changes of cellular hypertrophy in morphology. Afterwards, we observed the morphology of cardiomyocytes and mitochondria in mice by transmission electron microscopy, and found that the bilayer membrane structure and cristae structure of mitochondria were unclear, and more lipid droplets were visible, which indicated that mitochondria were altered, resulting in oxidative respiration impairment. Cardiomyocytes mainly rely on ATP produced by oxidative respiration to maintain energy supply, and the integrity of the respiratory chain is crucial for maintaining stable cardiac function. Therefore, to further determine the mechanism by which dysfunction occurs in myocardial mitochondria of 8-week-old Sirt3−/− mice, we examined the protein levels of the MTND1, SDHB, UQCRC2, MTCO2, and ATP5A genes encoding the subunits of the respiratory chain complex of the mitochondria, and found that the protein expression of MTND1, SDHB, MTCO2, and ATP5A was decreased in combination with the decreased myocardial contractile function and altered mitochondrial morphology, as previously described, this demonstrated decreased mitochondrial OXPHOS levels.
Decreased mitochondrial biosynthesis is one of the causes of mitochondrial dysfunction in cardiac insufficiency19.SIRT3 regulates mitochondrial biosynthesis and maintains mitochondrial mass and number by stimulating the expression of PGC-1α20.PGC-1α is a key molecule in mitochondrial biosynthesis, and PGC-1α can be found in the nucleus where it binds to downstream molecules NRF1 and NRF2, stimulating the expression of TFAM and mediating mitochondrial replication19.TFAM also stabilizes mtDNA and promotes the synthesis of the respiratory chain complex subunit encoded by mtDNA after entering mitochondria21. Thus, PGC-1α can indirectly regulate OXPHOS levels22.The stability of PGC-1α at the protein level is dependent on the deacetylation of SIRT3, so knockdown of the SIRT3 gene results in a significant reduction in the protein level of PGC-1α, which affects mitochondrial biosynthesis and energy metabolism23. Therefore, we believe that the decreased level of OXPHOS in myocardial mitochondria of Sirt3−/− mice is related to PGC-1α. The protein expression of PGC-1α and its downstream molecules NRF1, NRF2 and TFAM were found to be decreased in myocardial tissues of 8-week-old Sirt3−/− mice by experimental assays, so we hypothesized that the PGC − 1α protein in Sirt3 − /− mice was degraded due to the deletion of the SIRT3 gene, which resulted in an increase in the acetylation of the PGC − 1α protein, and that by affecting the expression of TFAM, this reduced the OXPHOS levels, which finally led to insufficient energy supply and decreased cardiac function.
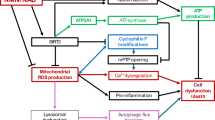
It has been shown that SIRT1 expression is compensatorily elevated in the presence of SIRT3 knockout to maintain the stability of the protein within the mitochondria, thus playing a neuroprotective role in the brain of stroke mice12.SIRT1 is another important deacetylating enzyme in the family of sirtuins, which is abundantly expressed in the mammalian heart, and which shuttles between various intracellular fractions to exert deacetylation24. acetylation24. Studies have shown that SIRT1 can deacetylate and activate PGC-1α to promote mitochondrial biosynthesis in cardiac insufficiency, thereby completing the metabolic pathway and improving cardiac function25(Fig. 7).NAD + is an important substrate for sirtuins to exert deacetylation in vivo26 and the expression of SIRT1 is regulated by the NAD+/NADH ratio. In cardiac insufficiency, the NAD+/NADH ratio is reduced due to metabolic disorders27,28 and increasing NAD + levels can elevate the NAD+/NADH ratio and stimulate the expression of SIRT113.NMN is a precursor for the synthesis of NAD+, and it has been demonstrated that the administration of NMN improves the mitochondrial function through the upregulation of NAD + levels15. We hypothesized that NMN could activate SIRT1 by increasing the NAD+/NADH ratio, thereby compensating for SIRT3 deficiency and improving mitochondrial function in Sirt3−/− mice.

The process of SIRT1 and SIRT3 regulating PGC-1α.
Mitochondrial function and cardiac function were improved in Sirt3−/− mice after administration of NMN. The protein expression of MTND1, SDHB, MTCO2, and ATP5A was restored in myocardial tissues of Sirt3−/− mice after administration of NMN, while ATP content in myocardial tissues was significantly increased. By examining the biosynthetic capacity of mouse myocardial mitochondria we found that the protein expression of PGC-1α, NRF1, NRF2, and TFAM were all increased by the administration of NMN. At the same time, administration of NMN upregulated the expression of SIRT1 in mouse myocardial tissues, suggesting that SIRT1 can compensate for SIRT3 deficiency. Therefore, the possible mechanisms are as follows: NMN upregulated the NAD+/NADH ratio in cardiomyocytes through NAD + supplementation, activated SIRT1, which deacetylated PGC-1α and promoted its expression, thereby promoting mitochondrial biosynthesis, while PGC-1α promoted respiratory chain protein synthesis through the modulation of the downstream molecules NRF1, NRF2, and TFAM, which raised the OXPHOS levels, maintained myocardial metabolic function, and ultimately improved cardiac insufficiency.
In summary, the present experiments suggest that NMN can improve cardiac function in Sirt3−/− mice by compensating for SIRT3 deficiency through the SIRT1/PGC-1α pathway and improving mitochondrial biosynthesis and oxidative respiration. Sirt3−/− mice were found to develop cardiac insufficiency at 8 weeks of age, demonstrating the importance of SIRT3 in maintaining cardiac function and suggesting that SIRT3 may be able to serve as a target for the treatment of cardiac insufficiency. In addition, this experiment confirmed the role of NMN in improving cardiac function, which could provide a theoretical basis for the role of NMN in the treatment of cardiac diseases. NMN can improve cardiac insufficiency as well as alleviate primary diseases such as diabetes mellitus and hypertension, and may be used as a therapeutic agent for a variety of metabolic syndromes in the future, and as a molecule that exists in the body, it can be used to minimize the adverse effects of drugs, but these still need further clinical studies to be developed.
Data availability
All data generated or analysed during this study are included in this published article.
References
Baman, J. R. & Ahmad, F. S. Heart Fail. JAMA 324, 1015 https://doi.org/10.1001/jama.2020.13310 (2020).
Daubert, C. Heart failure: A major public health problem. Presse Med. 53, 104224. https://doi.org/10.1016/j.lpm.2024.104224 (2024).
Karwi, Q. G., Uddin, G. M., Ho, K. L. & Lopaschuk, G. D. Loss of metabolic flexibility in the failing heart. Front. Cardiovasc. Med. 5, 68. https://doi.org/10.3389/fcvm.2018.00068 (2018).
Vite, A. et al. Functional impact of alternative metabolic substrates in failing human cardiomyocytes. JACC Basic. Transl Sci. 9 https://doi.org/10.1016/j.jacbts.2023.07.009 (2024).
Saddik, M. & Lopaschuk, G. D. Myocardial triglyceride turnover and contribution to energy substrate utilization in isolated working rat hearts. J. Biol. Chem. 266, 8162–8170 (1991).
Sharov, V. G., Goussev, A., Lesch, M., Goldstein, S. & Sabbah, H. N. Abnormal mitochondrial function in myocardium of dogs with chronic heart failure. J. Mol. Cell. Cardiol. 30, 1757–1762 (1998).
Casademont, J. & Miró, O. Electron transport chain defects in heart failure. Heart Fail. Rev. 7, 131–139 (2002).
Karamanlidis, G. et al. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ. Res. 106, 1541–1548. https://doi.org/10.1161/CIRCRESAHA.109.212753 (2010).
Green, D. R. & Reed, J. C. Mitochondria and apoptosis. Science 281, 1309–1312 (1998).
Jeong, S. M. & Haigis, M. C. Sirtuins in cancer: a balancing act between genome stability and metabolism. Mol. Cells. 38, 750–758. https://doi.org/10.14348/molcells.2015.0167 (2015).
Fu, X. et al. The mTOR/PGC-1α/SIRT3 pathway drives reductive glutamine metabolism to reduce oxidative stress caused by ISKNV in CPB cells. Microbiol. Spectr. 10, e0231021. https://doi.org/10.1128/spectrum.02310-21 (2022).
Koentges, C. et al. SIRT3 deficiency impairs mitochondrial and contractile function in the heart. Basic. Res. Cardiol. 110, 36. https://doi.org/10.1007/s00395-015-0493-6 (2015).
Zhang, X. et al. MicroRNA-195 regulates metabolism in failing myocardium via alterations in Sirtuin 3 expression and mitochondrial protein acetylation. Circulation 137, 2052–2067. https://doi.org/10.1161/CIRCULATIONAHA.117.030486 (2018).
Verma, R., Ritzel, R. M., Crapser, J., Friedler, B. D. & McCullough, L. D. Evaluation of the neuroprotective effect of Sirt3 in experimental stroke. Transl Stroke Res. 10, 57–66. https://doi.org/10.1007/s12975-017-0603-x (2019).
Liu, X. et al. Nicotinamide mononucleotide promotes pancreatic islet function through the SIRT1 pathway in mice after severe burns. Burns 48, 1922–1932. https://doi.org/10.1016/j.burns.2022.01.013 (2022).
van de Ven, R. A. H., Santos, D. & Haigis, M. C. Mitochondrial sirtuins and molecular mechanisms of aging. Trends Mol. Med. 23, 320–331. https://doi.org/10.1016/j.molmed.2017.02.005 (2017).
Wei, T. et al. Sirtuin 3 deficiency accelerates hypertensive cardiac remodeling by impairing angiogenesis. J. Am. Heart Assoc. 6 https://doi.org/10.1161/JAHA.117.006114 (2017).
Su, H., Cantrell, A. C., Chen, J. X., Gu, W. & Zeng, H. SIRT3 deficiency enhances ferroptosis and promotes cardiac fibrosis via p53 acetylation. Cells 12 https://doi.org/10.3390/cells12101428 (2023).
Finck, B. N. & Kelly, D. P. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J. Clin. Invest. 116, 615–622 (2006).
Xin, T. & Lu, C. SirT3 activates AMPK-related mitochondrial biogenesis and ameliorates sepsis-induced myocardial injury. Aging (Albany NY). 12, 16224–16237. https://doi.org/10.18632/aging.103644 (2020).
Quan, Y., Xin, Y., Tian, G., Zhou, J. & Liu, X. Mitochondrial ROS-Modulated mtdna: A potential target for cardiac aging. Oxid. Med. Cell. Longev. 2020 (9423593). https://doi.org/10.1155/2020/9423593 (2020).
Wang, C., Wang, Y. & Shen, L. Mitochondrial proteins in heart failure: the role of deacetylation by SIRT3. Pharmacol. Res. 172, 105802. https://doi.org/10.1016/j.phrs.2021.105802 (2021).
Ding, Y. et al. Sirtuin 3 is required for osteogenic differentiation through maintenance of PGC-1ɑ-SOD2-mediated regulation of mitochondrial function. Int. J. Biol. Sci. 13, 254–264. https://doi.org/10.7150/ijbs.17053 (2017).
Aquilano, K. et al. Peroxisome proliferator-activated receptor gamma co-activator 1alpha (PGC-1alpha) and Sirtuin 1 (SIRT1) reside in mitochondria: possible direct function in mitochondrial biogenesis. J. Biol. Chem. 285, 21590–21599. https://doi.org/10.1074/jbc.M109.070169 (2010).
Park, S. J. et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 148, 421–433. https://doi.org/10.1016/j.cell.2012.01.017 (2012).
Wu, Y., Pei, Z. & Qu, P. NAD+-A hub of energy metabolism in heart failure. Int. J. Med. Sci. 21, 369–375. https://doi.org/10.7150/ijms.89370 (2024).
Lee, C. F. et al. Normalization of NAD + Redox balance as a therapy for heart failure. Circulation 134, 883–894. https://doi.org/10.1161/CIRCULATIONAHA.116.022495 (2016).
Karamanlidis, G. et al. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell. Metab. 18, 239–250. https://doi.org/10.1016/j.cmet.2013.07.002 (2013).
Funding
This work was supported by grants from National Natural Science Foundation of China (82102733, 82303668), Jilin Provincial Research Foundation for Health Technology Innovation (2022JC080,2023JC018,2023JC019). Jilin Provincial Research Foundation for the Development of Science and Technology Projects (20220505002ZP, 20220303003SF).
Author information
Authors and Affiliations
Contributions
Xiyao Zhao wrote the main manuscript text and conducted the experiments, Mengrui He prepared figures. Lina He and Ruijie Han conducted the experiments. Yanan Liu carried out the editorial review and experimental design.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
All authors agree to publish in this journal.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, X., He, M., He, L. et al. NMN improves cardiac function in SIRT3 knockout mice via the SIRT1/PGC-1α pathway. Sci Rep 15, 28975 (2025). https://doi.org/10.1038/s41598-025-14349-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-14349-6
