
Abstract
Thalasseus bernsteini (Chinese crested tern), known as the rarest tern in the world, has become critically endangered with a severely diminished population. Understanding its genetic diversity is crucial for conservation efforts. However, no genomic resource has been publicly reported for this species. In this study, we discuss the first complete mitochondrial genome (mitogenome) of T. bernsteini assembled from an egg sample collected from a newly established breeding site on Yuksan Island, South Korea. The 16,737 bp mitogenome contains 37 genes, including 13 protein-coding genes, 22 tRNAs, and two rRNAs, exhibiting a typical gene order of avian mitogenomes. Phylogenetic analyses based on mitogenomic data confirmed the phylogenetic relationships between T. bernsteini within the Laridae family and other Charadriiformes species. Additionally, the divergence timeline of the terns was estimated, highlighting the evolutionary events that shaped their lineage. Our findings provide valuable genomic resources for future conservation genetic and phylogenetic studies of this critically endangered species, aiding in efforts to protect and manage dwindling populations.
Similar content being viewed by others

Introduction
Global climate change, human-driven habitat destruction, and illegal poaching of countless wildlife species are driving numerous species toward extinction worldwide1,2. To conserve the biodiversity of our planet in the face of these threats, the genetic resources of species serve as the cornerstone of conservation biology3,4,5. The genomes of many endangered species worldwide have been continuously decoded to document their genetic material in preparation for their potential extinction6,7,8. Moreover, recent advances in sequencing technologies have even made it possible to investigate the genomes of extinct species such as the woolly mammoth and the Tasmanian tiger9,10.
Small organellar genomes (i.e., plastid and mitochondrial genomes) are invaluable resources that offer better phylogenetic resolution than single-locus DNA barcodes and provide robust insights into genetic diversity. Preserving global biodiversity is an urgent priority, and mitochondrial genomes (mitogenomes) play a pivotal role in this challenge due to their unique characteristics. The mitogenome of vertebrates, typically 16–20 kb in length, is a compact, maternally inherited, non-recombining genome that evolves more rapidly than nuclear DNA11. This makes it a powerful tool for studying genetic diversity, population structure, and evolutionary relationships among species12,13. The high conservation of mitochondrial genes, combined with their faster mutation rates, allows for detailed phylogenetic analysis that can elucidate both recent and ancient divergence14,15. In avian research, the mitogenome has been extensively explored to reveal intra- and interspecies evolutionary relationships. For example, studies on various bird species have demonstrated that mitogenomes provide critical insights into the genetic diversity of species, aiding conservation efforts, and enhancing our understanding of phylogenetic lineages16,17,18.
Thalasseus bernsteini (Chinese crested tern), belonging to the Laridae family of the order Charadriiformes, is a globally endangered species classified as Critically Endangered on the IUCN Red List BirdLife 19. It was presumed extinct because of the lack of sighting records for 63 years, from 1937 until its rediscovery in July 2000 in Matsu, East China Sea. Since then, its known breeding grounds have been limited to four uninhabited islands in the East China Sea: Matsu, Jiushan, Wuzhishan, and Penghu20,21. However, in April 2016, two breeding pairs were discovered for the first time on Yuksan Island in the Yellow Sea, off Korea22. Since 2016, six to eight Chinese crested terns have arrived at breeding grounds in Korea annually23. The discovery of Korean breeding grounds is significant for species conservation, as it marks an expansion of the Chinese crested tern breeding range by approximately 700–1,100 km North.
Despite the demand for the conservation of this species, the mitochondrial genome of T. bernsteini has not been reported. Furthermore, the phylogenetic relationships within the Laridae family remain unclear, with only a handful of studies in terms of molecular phylogeny involving several mitochondrial and nuclear genes24,25. Given its critically endangered status and severe population decline, mitogenomic studies are crucial for conservation management and provide essential data for understanding its broader phylogenetic and evolutionary context within the Charadriiformes16,26. In this study, we report the first complete mitochondrial genome of T. bernsteini and investigate its phylogenetic relationships with 18 complete mitogenomes within the order Charadriiformes. The divergence timelines of Laridae have been inferred using relaxed molecular clock analysis based on mitogenomes to demonstrate the divergence time of the terns.
Materials and methods
Field research and ethical approval
The fieldwork was conducted on Yuksan Island (35° 19′ 22.3″ N, 126° 16′ 38.4″ E), one of the islands belonging to the Chilsan Islands, located in Yeonggwang-gun, Jeollanam-do, Korea. Yuksan Island, which hosts a breeding colony of crested Chinese terns, is an important breeding ground for black-tailed gulls (Larus crassirostris) (Fig. 1A,B), with an estimated population of approximately 26,000 birds in Korea. We visited the island two to four times a month from March to July 2023 to monitor the arrival and breeding status of Chinese crested terns. To document bird behavior, egg-laying dates, and hatching dates during the breeding season, 24-h still images and video data were collected using unmanned sensor cameras (HC600; Reconyx, Holmen, WI, USA) and CCTV cameras (SCO-6020R and XNO-6090RH; Hanwha Techwin, Seongnam, South Korea). Unmanned sensor cameras were programmed to capture one image per minute throughout the breeding period (March to July) using a time-lapse function.

Photographs of the breeding habitat of T. bernsteini on Yuksan Island and the collected egg sample. (A) Overview of the breeding habitat on Yuksan Island. (B) Chinese crested terns (T. bernsteini) observed within the breeding habitat. (C) Egg specimen (~ 5.5 cm in height) collected in this study. (D) Embryo specimen preserved in alcohol.
For fieldwork involving the capture and banding of birds, as well as the collection of biological samples (including unhatched eggs that were confirmed to be non-viable), approval for animal research was obtained from the Institutional Animal Care and Use Committee (IACUC) of the National Institute of Ecology (NIE), Republic of Korea (NIEIACUC-2021–007-change-001). All field researchers completed the required animal ethics training program (C02827-278969) at NIE. No experiments on live animals were performed as part of this study, and efforts were made to minimize stress and reduce visiting time during fieldwork to minimize potential harm to the birds.
Sample collection, DNA extraction, and sequencing
On May 23, 2023, an abandoned egg of T. bernsteini that had been left unincubated by the parent birds was found within 2 m of the original nest on Yuksan Island. Based on our camera recordings, the egg was laid on April 25, 2023, and therefore was collected 28 days after oviposition. The egg was carefully collected using sterile forceps, and the embryo in the egg was preserved in 99% ethanol at −20 °C (Fig. 1C,D). To obtain the genomic DNA of T. bernsteini, we fragmented the amniotic tissue of the alcohol-preserved embryo into tiny pieces approximately 2–3 mm wide. Approximately 80 mg of the amnion pieces were lysed in 800 μL of 2% CTAB lysis buffer (2% CTAB, 0.1 M Tris–HCl pH 8.0, 20 mM EDTA pH 8.0, 1.4 M sodium chloride, and 1% beta-mercaptoethanol) and 50 μL of proteinase K (20 mg/mL) solution on a Thermomixer at 65 ℃, 750 rpm for 16 h. Genomic DNA was extracted from the cell lysate using phenol/chloroform/isoamyl alcohol (25:24:1) and ethanol precipitation. The resulting gDNA pellet was dissolved in a 10 mM Tris solution. The quantity of gDNA was measured using a Qubit 4.0 fluorometer (Thermo Fisher, Waltham, MA, USA), and the DNA size distribution was determined using a FEMTO Pulse system (Agilent, Santa Clara, CA). The Illumina sequencing library was constructed using the TruSeq DNA PCR-Free Library Preparation Kit (Illumina, San Diego, CA, USA) following the manufacturer’s protocol, with an insertion size of 350 bp. Finally, the sequence data were generated on the Illumina NovaSeq6000 platform with 151 bp paired ends (PE150).
Mitogenome assembly and annotation
Raw reads of the Illumina sequencing were trimmed using Trimmomatic v0.3627 with a ‘MINLEN:36’ option. The high-quality reads were used to assemble the T. bernsteini mitogenome (Table 1). To assemble the mitogenome, we ran the MitoZ v3.6 assembler28 with default parameters, and the resultant assembly was then polished twice using Pilon v1.2429. Mitogenome annotation was performed using the GeSeq web program30 with the reference genome of the closest species, Sterna hirundo (GenBank accession: NC_036345.1)31. In detail, protein-coding genes and rRNAs were searched using BLAT with 35% and 60% of minimum identity options, respectively. All tRNAs were annotated using tRNAscan-SE v2.0.732 with a “vertebrate mitochondrial tRNAs” genetic code option. Finally, non-typical stop codons (T-- or TA-) and other coding frames were manually checked by comparison with Laridae reference mitogenomes33.
Phylogenetic and molecular clock analyses
To construct the mitogenome phylogeny of the family Laridae, we collected 13 mitogenomes of Laridae and five mitogenomes of Charadriiformes outgroups from the NCBI Organelle Genome and GenBank databases (Supplementary Table S1). Nucleotide sequences of 13 protein-coding genes were extracted from the GenBank files of each genome, and only these sequences were used in further analyses. The coding sequences were aligned using the codon-aware alignment program MACSE v2.0734 with the vertebrate mitochondrial genetic code option (-gc_def 2). To explore the mitogenomic species tree of Laridae, we inferred maximum likelihood (ML) phylogenetic trees using IQ-TREE2 v2.0.735 from both a single concatenated alignment and partitioned alignments based on individual genes. An adequate DNA substitution model for each alignment was determined using the ModelFinder algorithm (-m MFP), and ultrafast bootstrapping was performed with 1,000 replicates (-bb 1000). Gene concordance factors (gCFs) were analyzed using IQ-TREE2 to assess gene tree conflicts. In addition to the ML tree, we inferred a species tree based on the Bayesian inference (BI) method and the multispecies coalescence (MSC) method using PhyloBayes v4.136 and ASTRAL-III37, respectively. For the BI tree reconstruction using ‘pb’ script of PhyloBayes, we ran two independent MCMC (Markov Chain Monte Carlo) chains for at least 20,000 cycles with the CAT-GTR substitution model (-cat -gtr) which is a site-heterogeneous model accounting for variation in substitution patterns across sites. Convergence between both chains was assessed using ‘bpcomp’ script sampling every 10 cycles after discarding the first 1,000 cycles as burn-ins. The MSC tree was inferred using ASTRAL-III with 13 gene ML-tree inputs. All of best substitution model information from ModelFinder analyses was listed in Supplementary Table S2. The phylogenetic distance (i.e., Kendall–Colijn distance) between the species tree and each gene tree of Laridae was calculated using the TREESPACE R package38.
We further demonstrated the mitogenome-based diversification timeline of the Laridae family using fossil-calibrated relaxed molecular clock analysis. To this end, we employed PhyloBayes utilizing the CAT model (the default option for nucleotides) for relative rates of substitution, and the log-normal (-ln) relaxed molecular-clock model. Two independent MCMC chains were run for at least 10,000 cycles. The assessment of chain convergence and tree sampling followed the same method as described above. Two fossil calibration points were adopted from previous studies39,40, which were P. impennis-S. maccormicki (34.2–36.0 mya) and L. dominicanus-S. albifrons (23.6–24.1 mya). The ML tree in Fig. 2 was imported as a backbone species tree.

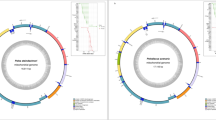
Mitogenome map of T. bernsteini and mitogenomic species-tree of the Laridae family. (A) Circular map of the mitogenome. (B) Maximum likelihood phylogenetic tree of the Laridae family. This tree was inferred from the partitioned nucleotide alignments of 13 protein-coding genes. Nodes without numerical labels represent fully supported ultrafast-bootstrapping values, and scale bar indicates the substitution rate.
Results and discussion
Field research and sample collection on an inhabited island
To determine the causes of nesting failure and egg-laying dates in Chinese crested terns, we collected data using unmanned sensor cameras during the breeding season and analyzed the photographic data in July after the breeding season concluded (see Methods). We observed two pairs of eggs laid among seven Chinese crested terns that arrived on Yuksan Island in 2023. The first nesting pair (male banding label “PA” and female no-banding, hereafter referred to as the “PA pair”) laid egg on April 25, whereas the second nesting pair (hereafter the “no-banding pair”) laid egg on May 2, resulting in a seven-day difference in laying dates. Their nests were within 0.5 m of each other.
On May 9, a confrontation occurred between incubating and non-breeding individuals in the nest of the no-banding pair, causing the egg of the no-banding pair to move closer to the nest of the PA pair (11:18 AM, May 9). The egg of the PA pair was later displaced and abandoned approximately 2 m away at 9:36 PM. The egg was left unattended for approximately 3 h before it disappeared. On May 10, no Chinese crested tern egg was observed. It appears that the egg was pulled into the nest of a nearby black-tailed gull that was incubating in an area where the egg had rolled. The abandoned egg of the PA pair was later reported on May 23, approximately 2 m from the original nest on the ground. We collected the embryo sample from the abandoned egg and extracted genomic DNA from the amniotic tissue (Fig. 1C,D).
First complete mitochondrial genome of T. bernsteini
We sequenced approximately 109 gigabases of T. bernsteini DNA using the Illumina NovaSeq 6000 platform and retained 97.1% of the reads after low-quality trimming (Table 1). From these high-throughput DNA reads, we successfully assembled the complete mitochondrial genome, which was 16,737 bp in length and included 13 protein-coding, two rRNA, and 22 tRNA genes (Fig. 2A). Most genes were located on the heavy strand, whereas only one protein-coding gene and eight tRNA genes were found on the light strand. The gene arrangement was consistent with that of a typical avian mitogenome, without any structural or compositional deviations (Supplementary Table S1). The A/T content was 58.8% for PCGs, 50.7% for tRNA genes, 58.8% for the srRNA gene, 57.8% for the lrRNA gene, 57.9% for the control region, and 60.3% for the T. bernsteini mitogenome (Supplementary Table S1). The start codons for the 13 protein-coding genes in T. bernsteini included typical ATN, with a few exceptions, such as GTG for COX1. The stop codons were primarily TAA and TAG, although some genes exhibited an incomplete stop codon (T--). This genomic structure aligned with previously documented patterns in Charadriiformes species (Table 2 and Supplementary Table S1).
Phylogenetic analysis of the family Laridae including T. bernsteini
To reconstruct a mitogenome phylogenetic tree for the family Laridae, we collected 18 complete mitogenomes from the NCBI GenBank database (see Methods and Supplementary Table S3). The genome set was comprised by five outgroup species (two Alcidae, two Scolopacidae, and one Stercorariidae) and 13 Laridae species (eight Larinae and five Sterninae). We aligned the nucleotide sequences of 13 protein-coding genes from these mitogenomes, including the newly sequenced T. bernsteini mitogenome, resulting in a concatenated alignment of 11,400 bp. The resultant ML species tree with gene-wise partitioning scheme showed that T. bernsteini grouped with the Sterna genus within the Sterninae subfamily with full support (Fig. 2B). There was no phylogenetic conflict in the gene concordance analysis, and the result was consistent to partitioning schemes (Supplementary Figure S1). The phylogenetic relationships across the Sterninae and Larinae were consistent with previously reported phylogenetic trees based on mitochondrial markers25. Therefore, our phylogenetic inference provides the Laridae’s most robustly supported species tree with mitogenome-level phylogenetic resolution. This tree topology was strongly supported by other species tree inference methods the multispecies coalescent method, and Bayesian inference method (Supplementary Figure S2). The ML tree with amino-acid alignments showed a few topological changes, but with weak phylogenetic supports (Supplementary Figure S2). Therefore, we used nucleotide alignments for further analyses.
We investigated the phylogenetic distances between the species tree and each mitochondrial gene tree to identify the gene that provided the highest phylogenetic resolution in the family Laridae38. The MDS plot of phylogenetic distance showed that the ATP6 and ND4L genes exhibited no phylogenetic distance from the species tree, and the COX1, ND1, and ND2 genes showed subsequently short distances (Fig. 3A), suggesting that these five mitochondrial genes are the most suitable phylogenetic marker genes within the Laridae family. In particular, COX1 and ND2 were more suitable as markers because of their sufficient coding sequence lengths of 1,551 and 1,039 bp, respectively. These results indicate that these genes are the most informative markers when the complete mitogenome is not available. In terms of phylogenetic support, the complete mitogenome consistently outperforms individual genes despite showing the same tree topology.

Phylogenetic distances and relaxed molecular-clock analysis of Laridae. (A) MDS (multidimensional scaling) plot of TREESPACE landscape analysis result among the species and each gene tree. “SPECIES” with red text indicates the mitogenome species tree. (B) Estimated divergence timelines of the Laridae family. Blue bar on each node indicates the 95% confidence interval of the estimated divergence time with “million years ago” unit, and the nodes with an asterisk indicate the fossil calibration points.
To infer the divergence time of the Laridae, we performed a relaxed molecular clock analysis with fossil calibration using our mitogenome datasets (see Methods section). The resultant chronogram shows that Laridae diverged from its sister groups 34.4–37.0 million years ago during the late Eocene epoch. Moreover, T. bernsteini species diverged within the Sterninae subfamily 4.2 to 7.2 million years ago (Fig. 3B). The inference results were comparable to the estimates from TimeTree5 (Supplementary Figure S3)41 and previous study25, but we consider our findings more robust because of the use of mitogenome-scale genetic data. This inference suggests that the black-crested head, a unique morphological trait of the Thalasseus genus, emerged 4.2–7.2 million years ago.
Genetic studies of the world’s most endangered tern
The Chinese crested tern, recognized as the world’s most endangered tern, has been observed breeding on several islands in China, Taiwan, South Korea, and Japan20,21,42. Despite its critically endangered status and increasing conservation importance, its genomic background remains largely unexplored owing to constraints on the collection of genetic resources. Previous studies have suggested genetic introgression between T. bernsteini and the great crested tern (Thalasseus bergii), based on the haplotype networks of three genes (ND2, NHS, and RP2)43. Gu et al. developed microsatellite markers for T. bernsteini genotyping using a draft genome of T. bergii and reported low genetic diversity alongside evidence of introgression44. A whole-genome study analyzing the population genomics of both species has been reported in a preprint archive45. However, a major limitation of these previous studies is the lack of publicly available genomic data, which restricts further genomic research on conservation.
The complete mitochondrial genome of T. bernsteini or any other species within the Thalasseus genus had not yet been reported. By successfully sequencing and assembling the complete mitochondrial genome of T. bernsteini, this study fills a critical gap in genomic research on this species. Our findings provide new insights into the phylogenetic placement within Laridae and offer a valuable genetic resource for future conservation efforts. Furthermore, the availability of this mitochondrial genome allows comparative analyses with other seabirds, facilitating studies on genetic diversity, adaptation, and population structure. Future studies should integrate nuclear genomic data to elucidate the evolutionary dynamics and conservation requirements of T. bernsteini.
Data availability
The complete mitochondrial genome of T. bernsteini was deposited in GenBank database with an accession number PV440435. Due to the critically endangered status of this species, the raw Illumina sequencing data are not publicly available. However, the data may be made available upon reasonable request to the corresponding author, particularly for bona fide academic or conservation-related research purposes.
References
Seney, E. E., Rowland, M. J., Lowery, R. A., Griffis, R. B. & McClure, M. M. Climate change, marine environments, and the US Endangered species act. Conserv. Biol. 27, 1138–1146. https://doi.org/10.1111/cobi.12167 (2013).
Pauls, S. U., Nowak, C., Balint, M. & Pfenninger, M. The impact of global climate change on genetic diversity within populations and species. Mol. Ecol. 22, 925–946. https://doi.org/10.1111/mec.12152 (2013).
Ryder, O. A., McLaren, A., Brenner, S., Zhang, Y. P. & Benirschke, K. DNA banks for endangered animal species. Science 288, 275–277. https://doi.org/10.1126/science.288.5464.275 (2000).
Brown, W. Y. Conservation: Invest in a DNA bank for all species. Nature 476, 399. https://doi.org/10.1038/476399a (2011).
Morin, P. A. Preservation of DNA from endangered species. Science 289, 725–726; author reply 726–727, https://doi.org/10.1126/science.289.5480.725d (2000).
Khan, S. et al. Overview on the role of advance genomics in conservation biology of endangered species. Int. J. Genom. 2016, 3460416. https://doi.org/10.1155/2016/3460416 (2016).
Hogg, C. J. et al. Threatened species initiative: Empowering conservation action using genomic resources. Proc. Natl. Acad. Sci. USA https://doi.org/10.1073/pnas.2115643118 (2022).
Kryukov, K., Nakahama, N. & Kuraku, S. Genome assembly catalog for species in the Japanese Red List: Unlocking endangered biodiversity through genomic inventory. F1000Res 13, 583. https://doi.org/10.12688/f1000research.149793.2 (2024).
Feigin, C. Y. et al. Genome of the Tasmanian tiger provides insights into the evolution and demography of an extinct marsupial carnivore. Nat. Ecol. Evol. 2, 182–192. https://doi.org/10.1038/s41559-017-0417-y (2018).
Miller, W. et al. Sequencing the nuclear genome of the extinct woolly mammoth. Nature 456, 387–390. https://doi.org/10.1038/nature07446 (2008).
Boore, J. L. Animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780. https://doi.org/10.1093/nar/27.8.1767 (1999).
Saccone, C., De Giorgi, C., Gissi, C., Pesole, G. & Reyes, A. Evolutionary genomics in Metazoa: The mitochondrial DNA as a model system. Gene 238, 195–209. https://doi.org/10.1016/s0378-1119(99)00270-x (1999).
Schmidt, C. & Garroway, C. J. The conservation utility of mitochondrial genetic diversity in macrogenetic research. Conserv. Genet. 22, 323–327. https://doi.org/10.1007/s10592-021-01333-6 (2021).
Mindell, D. P. et al. Interordinal relationships of birds and other reptiles based on whole mitochondrial genomes. Syst. Biol. 48, 138–152. https://doi.org/10.1080/106351599260490 (1999).
Ballard, J. W. O. & Rand, D. M. The population biology of mitochondrial DNA and its phylogenetic implications. Annu. Rev. Ecol. Evol. Syst. 36, 621–642. https://doi.org/10.1146/annurev.ecolsys.36.091704.175513 (2005).
Yoon, K. B., Cho, C. U. & Park, Y. C. The mitochondrial genome of the Saunders’s gull Chroicocephalus saundersi (Charadriiformes: Laridae) and a higher phylogeny of shorebirds (Charadriiformes). Gene 572, 227–236. https://doi.org/10.1016/j.gene.2015.07.022 (2015).
Liu, D., Zhou, Y., Fei, Y., Xie, C. & Hou, S. Mitochondrial genome of the critically endangered Baer’s Pochard, Aythya baeri, and its phylogenetic relationship with other Anatidae species. Sci. Rep. 11, 24302. https://doi.org/10.1038/s41598-021-03868-7 (2021).
Hu, C. et al. The mitochondrial genome of pin-tailed snipe Gallinago stenura, and its implications for the phylogeny of Charadriiformes. PLoS ONE 12, e0175244. https://doi.org/10.1371/journal.pone.0175244 (2017).
International, B. State of the World’s Birds 2024 Annual Update, https://datazone.birdlife.org/articles/state-of-the-worlds-birds-2024-annual-update (2024).
Chen, S. H., Fan, Z. Y., Chen, C. S. & Lu, Y. W. The breeding biology of Chinese Crested Terns in mixed species colonies in eastern China. Bird Conserv. Int. 21, 266–273. https://doi.org/10.1017/S0959270910000547 (2011).
Hung, C.-H., Chang, L.-N., Chiang, K.-K. & Yuan, H.-W. Trends in numbers of the critically Endangered Chinese Crested Tern Thalasseus bernsteini and sympatrically nesting Greater Crested Tern T bergii in the Matsu Archipelago, Taiwan. Bird Conserv. Int. 29, 386–399. https://doi.org/10.1017/S0959270918000369 (2019).
Lee, Y. et al. Arrival and breeding status of the Chinese Crested Tern (Thalasseus bernsteini) in Korea. Korean J. Ornithol. 28, 66–74. https://doi.org/10.30980/kjo.2021.12.28.2.66 (2021).
Lee, Y., No, D. & Kim, J. Individual identification of Chinese Crested Tern (Thalasseus bernsteini) from image-appearance characteristics. Korean J. Ornithol. 31, 126–133. https://doi.org/10.30980/kjo.2024.12.31.2.126 (2024).
Pons, J. M., Hassanin, A. & Crochet, P. A. Phylogenetic relationships within the Laridae (Charadriiformes: Aves) inferred from mitochondrial markers. Mol. Phylogenet. Evol. 37, 686–699. https://doi.org/10.1016/j.ympev.2005.05.011 (2005).
Bridge, E. S., Jones, A. W. & Baker, A. J. A phylogenetic framework for the terns (Sternini) inferred from mtDNA sequences: Implications for taxonomy and plumage evolution. Mol. Phylogenet. Evol. 35, 459–469. https://doi.org/10.1016/j.ympev.2004.12.010 (2005).
Sun, K. et al. Complete mitochondrial genomes of Pluvialis fulva and Charadrius dubius with phylogenetic analysis of charadriiformes. Genes https://doi.org/10.3390/genes15121642 (2024).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. https://doi.org/10.1093/bioinformatics/btu170 (2014).
Meng, G., Li, Y., Yang, C. & Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 47, e63. https://doi.org/10.1093/nar/gkz173 (2019).
Walker, B. J. et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963. https://doi.org/10.1371/journal.pone.0112963 (2014).
Tillich, M. et al. GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. https://doi.org/10.1093/nar/gkx391 (2017).
Yang, C. et al. The mitogenomes of Gelochelidon nilotica and Sterna hirundo (Charadriiformes, Sternidae) and their phylogenetic implications. Mitochondr. DNA B Resour. 2, 601–603. https://doi.org/10.1080/23802359.2017.1372709 (2017).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096. https://doi.org/10.1093/nar/gkab688 (2021).
Donath, A. et al. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 47, 10543–10552. https://doi.org/10.1093/nar/gkz833 (2019).
Ranwez, V., Douzery, E. J. P., Cambon, C., Chantret, N. & Delsuc, F. MACSE v2: Toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol. Biol. Evol. 35, 2582–2584. https://doi.org/10.1093/molbev/msy159 (2018).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. https://doi.org/10.1093/molbev/msaa015 (2020).
Lartillot, N., Lepage, T. & Blanquart, S. PhyloBayes 3: A Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 25, 2286–2288. https://doi.org/10.1093/bioinformatics/btp368 (2009).
Zhang, C., Rabiee, M., Sayyari, E. & Mirarab, S. ASTRAL-III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 19, 153. https://doi.org/10.1186/s12859-018-2129-y (2018).
Jombart, T., Kendall, M., Almagro-Garcia, J. & Colijn, C. treespace: Statistical exploration of landscapes of phylogenetic trees. Mol. Ecol. Resour. 17, 1385–1392. https://doi.org/10.1111/1755-0998.12676 (2017).
Smith, N. A. Sixteen vetted fossil calibrations for divergence dating of Charadriiformes (Aves, Neognathae). Palaeontol. Electron. 18, 1–18 (2015).
Paton, T. A., Baker, A. J., Groth, J. G. & Barrowclough, G. F. RAG-1 sequences resolve phylogenetic relationships within Charadriiform birds. Mol. Phylogenet. Evol. 29, 268–278. https://doi.org/10.1016/s1055-7903(03)00098-8 (2003).
Kumar, S. et al. TimeTree 5: An expanded resource for species divergence times. Mol. Biol. Evol. https://doi.org/10.1093/molbev/msac174 (2022).
Dinets, V. First record of the Chinese crested tern Thalasseus bernsteini in Japan. J. Yamashina Inst. Ornithol. 50, 138–140. https://doi.org/10.3312/jyio.50.138 (2019).
Yang, J. et al. Genetic evidence of hybridization of the world’s most endangered tern, the Chinese Crested Tern Thalasseus bernsteini. Ibis 160, 900–906. https://doi.org/10.1111/ibi.12616 (2018).
Gu, N. et al. Novel microsatellite markers reveal low genetic diversity and evidence of heterospecific introgression in the critically endangered Chinese Crested Tern (Thalasseus bernsteini). Glob. Ecol. Conserv. 28, e01629. https://doi.org/10.1016/j.gecco.2021.e01629 (2021).
Chen, G. et al. Long-term and extensive population decline drives elevated expression of genetic load in a critically endangered seabird, 22 May 2023, PREPRINT (Version 1). Res. Square. https://doi.org/10.21203/rs.3.rs-2960319/v1 (2023).
Funding
This research was supported by grants from the Honam National Institute of Biological Resources (Grant No. HNIBR202402118) and the National Institute of Ecology (Grant No. NIE-B-2024-13). All grants were funded by the Ministry of Environment (MOE) of the Republic of Korea.
Author information
Authors and Affiliations
Contributions
JSJ, KJL, YL, and JJ contributed to the study’s conception and funding acquisition. JK and YL conducted the fieldwork and sample collection. JSJ performed genome assembly, annotation, and formal analysis. JJ performed gDNA extraction, library preparation, phylogenetic analysis, and visualization. JSJ, YL, and JJ wrote the original draft, and all authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jeong, J.S., Kim, J., Lee, K.J. et al. Complete mitochondrial genome of the world’s most endangered tern, Thalasseus bernsteini, and a mitogenomic phylogenetic study of the Laridae family. Sci Rep 15, 29326 (2025). https://doi.org/10.1038/s41598-025-14897-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-14897-x
