
Abstract
Type 2 diabetes (T2D) is a group of metabolic disorders characterized by chronic hyperglycemia and long-term carbohydrate, fat, and protein metabolism disruptions. This study aimed to identify biomarker of T2D and analyze immune cell infiltration in the islets of T2D patients. Using the GSE76895 dataset, 112 differentially expressed genes (DEGs) were identified between islet samples from T2D and non-diabetic (ND) individuals. Then, 112 DEGs were used for functional enrichment and Gene Set Enrichment Analyses (GSEA). Through the least absolute shrinkage and selection operator (LASSO) and support vector machine-recursive feature elimination (SVM-RFE), SLC2A2 emerged as the most likely candidate biomarker of T2D. Moreover, the distribution of tissue-infiltrating immune cells between T2D and ND islet samples was assessed using the CIBERSORT algorithm. The result revealed that resting CD4+ memory T cells might play an important role in T2D and exhibited a positive correlation with SLC2A2. Single-cell RNA sequencing (scRNA-seq) data indicated that SLC2A2 was highly expressed in beta cells of T2D islets and down-regulated in T2D group. Finally, in vivo studies confirmed decreased level of SLC2A2 expression in T2D models. To sum up, these findings highlight SLC2A2 as potential biomarkers, aiding early diagnosis and pharmaceutical advancements in T2D.
Similar content being viewed by others

Introduction
Diabetes mellitus (DM) encompasses a group of metabolic disorders characterized by chronic hyperglycemia and long-term disruptions in carbohydrate, fat, and protein metabolism caused by various factors1,2. The two most common types of DM are type 1 diabetes (T1D), resulting from absolute insulin deficiency due to pancreatic β-cell destruction, and type 2 diabetes, characterized by insulin resistance and inadequate insulin secretion3,4. Type 2 diabetes (T2D) is the predominant form, accounting for 90–95% of all diabetes cases5. This condition is associated with a range of complications, including cardiovascular disease, retinopathy, nephropathy, neuropathy, and foot ulcers that may lead to amputation, significantly impairing patients’ quality of life and life expectancy2,6. As a growing global public health concern, the prevalence of T2D continues to rise, placing substantial burdens on human health and socioeconomic systems4. Therefore, new insights into the etiology, diagnosis, and prognosis of diabetes are urgently needed.
The current gold standard for diagnosing T2D is the measurement of venous plasma glucose levels1. However, research indicates that diabetes-related metabolic changes may occur decades before the onset of hyperglycemia. During this pre-diabetic phase, although blood glucose levels remain below the diagnostic threshold, insulin resistance, β-cell dysfunction, and other metabolic abnormalities are already present. This stage is linked to an increased risk of early kidney disease, chronic kidney disease, small-fiber neuropathy, diabetic retinopathy, and macrovascular disease5,7. Conventional treatments for T2D include dietary management, exercise therapy, oral antidiabetic medications, and insulin injections. However, these strategies neither cure diabetes nor fully prevent its complications4. Thus, early diagnosis and timely intervention are essential for delaying diabetes progression and minimizing complication risks, facilitating clinical decision-making, and benefiting high-risk individuals. Nonetheless, critical diagnostic genes and effective therapeutic targets remain to be identified.
Over the past few decades, diabetes has attracted significant research interest. For instance, Sun et al. demonstrated that exosomes can function not only as biomarkers for the early diagnosis of diabetes but also as potential therapeutic agents for managing diabetes and its complications8. Additionally, several studies have identified associations between specific metabolites and T2D. For example, mannose has been linked to T2D, while fatty acids, proinsulin, inflammatory markers (such as glycoprotein acetylation, interleukin-1 receptor antagonist, and hs-CRP), and amino acids have been associated with an increased risk of developing the disease9.
Machine learning algorithms have been used more than once in diabetes and related diseases. In the search for diagnostic markers for diabetic nephropathy, Ming Zhong et al. used the least absolute shrinkage and selection operator (LASSO) regression algorithm, support vector machine-recursive feature elimination (SVM–RFE) algorithm and random forest (RF) algorithm that eventually obtained four genes as diagnostic markers10. Moreover, the LASSO logistic regression and SVM-RFE algorithms were also applied to identify potential diabetic peripheral neuropathy diagnostic markers11. In addition to these, machine learning algorithms have been used in the study of other diseases such as COPD12 , AD13 , SLE14 and glioma15. All the above studies have shown that machine learning algorithms have high utility and accuracy.
In this study, to uncover the potential mechanism and biomarkers of T2D, bulk-seq and single-cell seq data from the Gene Expression Omnibus (GEO) database were analyzed. Key genes (SLC2A2) was identified as the novel biomarker of T2D. Functional enrichment analyses and immune cell infiltration analysis were performed to reveal the molecular mechanism of T2D. Finally, in vivo studies initially validated the results of bioinformatic analysis. Our study provides novel insights by combining differential analysis with machine learning (LASSO and SVM-RFE) to robustly identify SLC2A2 as a key T2D gene. This integrated approach revealed SLC2A2’s dual role in beta cell dysfunction (via cAMP/insulin resistance pathways) and immune modulation (correlating with CD4+ memory T cells), offering new mechanistic understanding of T2D.
Materials and methods
Data collection and download
Type 2 diabetic and non-diabetic gene expression datasets containing GSE76894, GSE76895 and GSE86468 were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). The GSE76894 dataset containing 19 T2D and 84 non-diabetic (ND) islet samples was derived from the GPL570 platform of Affymetrix Human Genome U133 Plus 2.0 Array. And the GSE76895 dataset contains 36 T2D, 32 ND, 15 impaired glucose tolerance (IGT), and 20 type 3c diabetes (T3cD) cases. GSE86468 dataset includes 9 T2D and 15 ND islet samples. We used GSE76895 as a training cohort, while GSE76894 and GSE86468 were used as validation cohorts to confirm the analysis results. Applying the “limma” package, differentially expressed genes (DEGs) from the GSE76895 data were with the cut-off criteria of |fold change| ≥ 1.5, adj.P. Val. Filter < 0.05. A linear model was then fitted using the ‘lmFit’ function, and differential expression was assessed with the “eBayes” function. The LASSO algorithm was implemented using the “glmnet” R package, along with the SVM-RFE algorithm based on the “e1071”, “kernlab”, and “caret” packages to analyze key genes among the DEGs15.
Functional enrichment analysis
With the help of the “ClusterProfiler” package in RStudio, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG)16,17 enrichment analysis was performed to further understand the biological significance of DEGs. Statistical significance was defined as a P value less < 0.05. Then, the “GOplot” package was used to visualize the results of the enrichment analysis. Besides, GSEA analysis was performed with the criteria of adj.P-value < 0.05. Top3 GSEA terms of the two groups were visualized by “enrichplot” package.
Feature selection by LASSO and SVM-RFE algorithms
Two machine-learning algorithms (LASSO and SVM-RFE) were used to screen characteristic genes. LASSO, which was performed to find genes significantly associated with different samples, is an algorithm that can be used for regression analysis and regularization for variable selection with the help of the R software package “glmnet”18. For LASSO analysis, the parameters α = 1, 10-fold cross-validation, and λ selection via minimum criterion were selected. We performed SVM-Recursive Feature Elimination (SVM-RFE) using the “caret” package in R with a radial basis function kernel, evaluating feature subsets at sizes of 2, 4, 6, 8 and sequentially from 10 to 40 features (in increments of 3). The analysis employed 10-fold cross-validation (method = “cv”) with AUC as the evaluation metric, using default parameters for gamma (1/(data dimension)) and cost (C = 1). Feature importance was calculated by averaging variable importance across all cross-validation folds, with the optimal feature subset selected based on maximum mean AUC performance. The random seed was set to 123 to ensure reproducibility of the feature selection process16.
For further analysis, we combined the overlapping genes between LASSO and SVM-RFE. In order to evaluate their power as candidate diagnostic biomarkers, we verified their expression levels.
Evaluation of immune cell infiltration
As a highly applicable analytical tool and a deconvolution algorithm, CIBERSORT can analyze 547 genes. A linear support vector regression, as a machine learning method, was used to deconvolve the bulk gene expression matrix to denoise. Immune cell infiltration (22 various cell types) was precisely quantified in each T2D and ND gene expression profile17 through the “CIBERSORT” package in RStudio. Further analysis was limited to samples that had CIBERSORT outputs of P < 0.05. Using the R packages “reshape 2” and “ggExtra”, spearman correlation coefficients were performed between candidate diagnostic biomarkers and significantly changed immune cells.
Single-cell transcriptome data processing and analyzing
Single-cell RNA sequence (scRNA-seq) data of T2D and ND was downloaded from ArrayExpress (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-5061/). Single-cell cDNA libraries were generated using the Smar-sea2 protocol. The scRNA-seq data contains 4 T2D and 6 ND samples19. During the processing of single-cell transcriptome data, we performed normalization, scaling, and cell clustering. Utilizing the “Seurat” package (http://satijalab.org/seurat/), we successfully identified 12 primary cell categories. Single cells were extracted with the criteria of nFeature_RNA > 500 and < 7000, percent.mt < 10%, nCount_RNA > 1000 to remove doublet and dead cells. Using the “LogNormalize” method with the “NormalizeData” function, we normalized the filtered gene-barcode matrices. The top 2000 highly variable genes were discovered by the “FindVariableFeatures” function using the “vst” method, which was previously centered and scaled using “ScaleData”. Based on these 2000 highly variable genes, we performed principal component analysis (PCA) and used the Harmony package to remove batch effects and perform dimensionality reduction. Functions “FindNeighbors”, “FindClusters” and “runTSNE” from Seurat were then used to display dimensionality-reduced clusters on the 2D maps generated by the t-distributed t-SNE. Moreover, expression levels of diagnostic biomarkers in different cell types are represented by bubble diagrams. Finally, “Vlnplot” function of “Seurat” package was used to show the expression level of key gene in scRNA-seq data.
T2D mouse model
C57BL/6J wild-type mice, aged 6–7 weeks, were randomly assigned to two groups: (1) a control group receiving a standard diet (ND group) and (2) a T2D group subjected to a high-fat diet and treated with streptozotocin. The T2D group was fed a high-fat diet (#D12492) for a period of 4 weeks before receiving streptozotocin injections (30 mg/kg, Sigma-Aldrich) for three consecutive days to induce T2D. Mice with blood glucose levels above 16.7 mmol/L, as determined by a blood glucose meter (LifeScan), were identified as having T2D. All animal experiments were approved by the Institutional Animal Care and Use Committee of Fudan University and were consistent with the governmental regulations of China for the care and use of animals. All methods are reported in accordance with ARRIVE guidelines.
Intraperitoneal insulin tolerance tests and intraperitoneal glucose tolerance tests
Tail vein blood glucose levels in mice were measured using a blood glucose meter (LifeScan) following a fasting period of 12 h. For the intraperitoneal glucose tolerance test (IPGTT), mice received an intravenous injection of glucose (2 g/kg body weight) after the same fasting interval, and tail vein blood samples were taken at 0, 30, 60, 90, and 120 min post-injection. In a similar fashion, the intraperitoneal insulin tolerance test (IPITT) was conducted by administering an insulin injection (0.75 U/kg body weight) to the fasted mice, with subsequent blood samples collected from the tail vein at 0, 15, 30, 60, and 90 min following the injection.
Real-time quantitative PCR
Total RNA was purified from cells or tissues with the RNAiso Plus Reagent (Catalog#9109, TaKaRa), after which complementary DNA (cDNA) was synthesized using the PrimeScript™ RT Reagent Kit with gDNA Eraser (Catalog#RR047A, TaKaRa). Real-time quantitative PCR was performed on a ViiA 7 Real-Time PCR System (Applied Biosystems) using QuantiNova SYBR® Green (Catalog#208056, QIAGEN) and primers specific to the target genes. The sequences of the primers: Actin: forward - CGCCACCAGTTCGCCATGGA-, reverse - TACAGCCCGGGGAGCATCGT-. SLC2A2: forward - TCAGAGCGAGTCACAAACCT-, reverse - TCTGCCTGGCCAATCCTTTA-.
Statistics
The data were analyzed using the R programming language (version 4.1.1) and are expressed as mean ± standard deviation (SD). To assess the statistical significance between the two groups, an unpaired Student’s t-test or the nonparametric Mann-Whitney U test was employed. A p-value of less than 0.05 was considered indicative of statistical significance.
Results
Screening of DEGs in the T2D and ND islet samples
The flow of this study is shown in Fig. 1. After data normalization (Supplementary Figs. 1 and 2), differential expression analysis between T2D and ND islet samples in the GSE76895 dataset was performed utilizing the “limma” package. Of the 112 identified DEGs, 67 genes upregulated significantly and 45 genes downregulated significantly, and all DEGs were displayed in the heatmap (Fig. 2A, B & Supplementary Table 1).

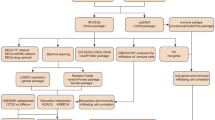
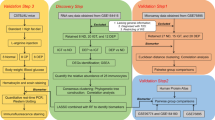
Flow chart of the study.

Differential analysis and functional enrichment analysis between T2D and ND islet samples in GSE76895 datasets. (A) Heatmap plot of all DEGs. Gene expression trends in different tissues were represented by different colors. (B) Volcano plot of DEGs constructed using the fold change values and P-adjust; red dots represented up-regulated differential genes, gray dots represented nonsignificant genes, and blue dots represented down-regulated differential genes. (C) Bubble diagram of DEGs GO analysis. (D) Bubble diagram of DEGs KEGG16,17 analysis. Bubble color was determined by p-value and bubble sized by gene count.
Biological functions of DEGs
GO and KEGG analyses were carried out to find the biological functions of DEGs using the ‘ClusterProfiler’ package. GO analysis includes Biological Process (BP) analysis, Cell Component (CC) analysis, and Molecular Function (MF) analysis. In BP analysis, DEGs were primarily involved with regulation of membrane potential and reactive oxygen species metabolic process. In CC analysis, DEGs were primarily involved with membrane and clathrin-sculpted vesicle. In MF analysis, DEGs were primarily involved with monoatomic ion channel activity, gated channel activity and aldo-keto reductase (NADP) acitivity (Fig. 2C). Moreover, the result of KEGG analysis showed these DEGs were also mainly enriched in the hormone signaling, cAMP signaling pathway, calclum signaling pathway, and insulin resistance (Fig. 2D).
Moreover, GSEA results showed cytokine-cytokine receptor interaction, complement and coagulation cascades, and ECM-receptor interaction were up-regulated in T2D samples (Fig. 3A). RNA polymerase, ribosome biogenesis in eukaryotes, and oxidative phosphorylation were mainly down-regulated in T2D samples (Fig. 3B). The functional enrichment analyses in GSE76894 displays the similar results (Supplementary Figs. 3–4).

The results of GSEA analysis in GSE76895 datasets. (A) GSEA enrichment analysis results in T2D islet samples. (B) GSEA enrichment analysis results in ND islet samples.
Nine genes were identified as the biomarkers by LASSO and SVM-RFE
Two algorithms were used to screen biomarkers in the GSE76895 dataset. For Fig. 4A (LASSO analysis), the x-axis represents log(λ) values, where λ is the regularization parameter controlling feature sparsity. The y-axis shows standardized coefficient magnitudes. The left vertical dotted line, which indicating the optimal λ (minimum deviance criterion) was selected. Therefore, we got 21 candidate genes (Supplementary Table 2). For Fig. 4B (SVM-RFE analysis), the x-axis now displays the number of retained features and the y-axis presents the AUC performance metric. The curve shows maximal AUC (0.957) achieved with 19 features (Supplementary Tables 3, 4). Then, Veen plot indicated 9 overlapping meaningful features (Fig. 4C). Moreover, we analyzed the expression level of these 9 genes, which showed their significantly different expression levels between T2D and ND islet samples (Fig. 4D).
As a validation cohort, the GSE76894 dataset was used to assess the accuracy of the above analytic results and the expression levels for the 9 candidate biomarkers. The expression levels of ASCL2, ARG2, SLC2A2, OPRD1, and HS6ST2 were significantly different between T2D and ND islet samples (Fig. 5). Compared to ND islet samples, T2D islet samples exhibited significantly lower expression levels of ASCL2 (p value = 2.9e-06), ARG2 (p value = 1.3e-07), SLC2A2 (p value = 1.6e-06), OPRD1 (p value = 3.2e-06), and HS6ST2 (p value = 0.0094) (Fig. 5). The gene PCOLCE2 and PRIMA1 were not found in the validation cohort, so it was not shown.
Moreover, the GSE86468 dataset was also used to verify the above results. As shown in the Fig. 6, PCOLCE2 (p value = 0.034) and SLC2A2 (p value = 0.013) were significantly different between T2D and ND islet samples. Combining three datasets, only one gene SLC2A2 directly differs between T2D and ND. Therefore, SLC2A2 is considered a biomarker for T2D.

Screening candidate biomarkers from DEGs between the T2D and ND islet samples in GSE76895. (A) 21 genes were screened by the LASSO logistic regression algorithm. (B) 19 genes were screened by the SVM-RFE algorithm. (C) Venn diagram of 9 features including PCOLCE2, ASCL2, ARG2, PRIMA1, SLC2A2, OPRD1, SLC26A4, HS6ST2, and CYS1 intersected by LASSO and SVM-RFE algorithms. (D) The expression level of candidate markers PCOLCE2, ASCL2, ARG2, PRIMA1, SLC2A2, OPRD1, SLC26A4, HS6ST2, and CYS1 in GSE76895 datasets between the T2D and ND islet samples.

Validating candidate biomarkers in GSE76894. (A–G) The expression level of candidate markers ASCL2, ARG2, SLC2A2, OPRD1, SLC26A4, HS6ST2, and CYS1 in GSE76894 datasets between the T2D and ND islet samples. ASCL2, ARG2, SLC2A2, OPRD1, and HS6ST2 were the significant genes among the seven genes.

Validating candidate biomarkers in GSE86468. (A–I) The expression level of candidate markers PCOLCE2, ASCL2, ARG2, PRIMA1, SLC2A2, OPRD1, SLC26A4, HS6ST2, and CYS1 in GSE86468 datasets between the T2D and ND islet samples. PCOLCE2 (A) and SLC2A2 (E) were the significant genes among the nine genes.
Analysis of immune cell infiltration showed significant changes in immune cells between T2D and ND islet samples
Using the CIBERSORT algorithm, we first calculated the proportion of immune cell infiltration in the T2D and ND islet samples (Fig. 7A). Following that, we calculated the correlation between the 22 types of infiltrating immune cells and SLC2A2 (Fig. 7B). Mast cells activated was correlated positively with NK cells resting but negatively with mast cells resting and NK cells activated. However, mast cells resting was correlated positively with NK cells activated. Neutrophils were correlated negatively with monocytes. Moreover, we can notice that SLC2A2 had a positive correlation with resting CD4+ memory T cells.

Evaluation and visualization of immune cell infiltration and correlation with 22 types of immune cell infiltration between T2D and ND islet samples. (A) The proportion of infiltrating immune cells in the T2D and ND islet samples. (B) Correlation matrix of 22 immune cell infiltration between the T2D and ND islet samples. Blue and red represented positive and negative correlations, respectively. Darker color indicated stronger correlation.
Expression level of SLC2A2 in single-cell transcriptome data
The scRNA-seq data was performed normalization, scaling, clustering, highly variable genes screening, and so on (Fig. 8A-B). Based on these 2000 highly variable genes, we then demonstrated the 12 clusters. Based on the expression of featural markers (Supplementary Fig. 2C), we identified them into 10 cell types, including gamma cells, delta cells, stellate cells, beta cells, alpha cells, acinar cells, immune cells, endothelial cells, ductal cells, and other cells (Fig. 8C). Interestingly, of the 10 cell types, the SLC2A2 presented the highest expression in the beta cells, which is the most important cell type in the islet (Fig. 8D). In beta cells, the SLC2A2 was significantly down-regulated in T2D group (Fig. 8E).

The SLC2A2 expression analysis in the single-cell transcriptome data. (A, B) The vlnplot of single-cell transcriptome data before (A) and after (B) quality control. (C) The t-SNE plot of the 10 identified main cell types. (D) The bubble plot showed the expression of the HHATL and SLC2A2 related to different cell types. Bubble color was determined by average expression and bubble sized by percent expressed. (E) The expression level of SLC2A2 in beta cells between control group and T2D group.
Validation of key genes
SLC2A2 were identified as the key genes in this study, so an in vivo study was performed to validate their expression levels. After confirming the successful establishment of T2D (Fig. 9A–D), qPCR analysis was performed to test the expression level of SLC2A2. The result showed that SLC2A2 were down-regulated in the T2D group (Fig. 9E).

Validation of SLC2A2 in mouse T2D model. (A) Body weight of the ND and T2D group. (B) Fasting blood glucose levels of the ND and T2D group. (C) IPGTT of the ND and T2D group. (D) IPITT of the ND and T2D group. (E) The relative expression level of SLC2A2.
Discussion
Type 2 diabetes (T2D) is a chronic metabolic disorder characterized by insulin resistance and relative insulin deficiency, leading to hyperglycemia20. It represents the most prevalent form of diabetes, accounting for approximately 90–95% of all diagnosed cases globally21. The pathophysiology of T2D is complex and multifactorial, involving genetic predisposition, environmental factors, and lifestyle choices22. Key risk factors include obesity, physical inactivity, and advancing age, which contribute to the disruption of glucose homeostasis23. Over the past few decades, the rising prevalence of T2D has been linked to lifestyle changes, particularly the increase in sedentary behavior and unhealthy dietary patterns24. If left unmanaged, T2D can lead to serious complications, including cardiovascular diseases, neuropathy, nephropathy, and retinopathy, significantly impairing the quality of life, and increasing healthcare costs25.
In this study, one biomarker of T2D were identified. Firstly, we identified a gene named SLC2A2, which encodes glucose transporter 2 (GLUT2). GLUT2 is widely expressed in the liver, intestinal epithelial cells, and pancreatic β-cells, functioning as a key transporter mediating transmembrane glucose transport26,27. Dysregulated expression or impaired function of GLUT2 has been strongly linked to impaired fasting glucose and type 2 diabetes, contributing to an increased risk of developing diabetes28,29,30. Whole-genome bisulfite sequencing of human pancreatic islets identified 457 genes exhibiting differentially methylated regions (DMRs) in T2D islets, including NR4A3, PARK2, PID1, SLC2A2, and SOCS231. Notably, SLC2A2 has been further implicated as a potential therapeutic target for T2D through genome-wide association studies, suggesting its utility in tailored therapy design32. Mechanistically, SLC2A2 dysfunction contributes to impaired insulin secretion in pancreatic islets—a key pathological feature of T2D33. The aforementioned study provides multi-faceted evidence supporting SLC2A2 as a potential biomarker for T2D, while simultaneously validating the reliability of our research findings. Among the nine candidate genes identified in our study, several, including SLC2A2, have been previously linked to type 2 diabetes and glycemic traits in genome-wide association studies (GWAS)34. In contrast, others such as PRIMA1 and CYS1 have not shown prior associations with T2D, highlighting their potential as novel biomarkers. These findings demonstrate the strength of our integrated approach in uncovering both established and underexplored genetic contributors to T2D.
Our findings suggest that resting CD4+ memory T cells may contribute significantly to the pathogenesis of T2D, showing a positive correlation with SLC2A2 expression. These cells, which are essential for adaptive immunity, maintain long-term immunological memory through a quiescent state (characterized by low proliferation and metabolic activity) while retaining rapid responsiveness to antigen re-exposure. Notably, dysregulation of CD4+ memory T cell homeostasis has been implicated in various immune-related disorders, including autoimmune diseases, chronic infections, and impaired vaccine responses35. Intriguingly, recent studies have revealed distinct alterations in immune cell populations in T2D patients, with elevated levels of eosinophils, naïve CD4+ T cells, and regulatory T cells (Tregs), but reduced frequencies of resting CD4+ memory T cells and monocytes in pancreatic islets36. Furthermore, accumulating evidence highlights the critical regulatory role of immune-related genes and immune cell subsets (e.g., resting CD4+ memory T cells) in the onset and progression of DM, potentially offering novel insights for immune-targeted therapeutic strategies37,38.
In this study, a significant decrease in SLC2A2 expression was observed in beta cells of the T2D model, resulting in beta cell dysfunction. This is different from previous literature that transient downregulation of SLC2A2 has a potential beta cell protective effect. However, persistent downregulation of SLC2A2 expression may lead to a decrease in glucose sensing ability of beta cells, thereby exacerbating insulin secretion defects and leading to beta cell dedifferentiation39. Therefore, the protective effect of SLC2A2 transient downregulation on β cells may be a compensatory mechanism, and the pathological significance of SLC2A2 and β cells is different. Further studies are needed to explore the effect of SLC2A2 dynamic changes on T2D.
Recent studies exploring the pathogenesis of diabetes have revealed a potential link between the several signaling pathways and type 2 diabetes. Hedgehog signaling pathway, activated by three ligands—Sonic hedgehog, Desert hedgehog, and Indian hedgehog—plays a critical role in various physiological processes, including embryonic development, tissue regeneration, and adult tissue homeostasis40. Historically recognized as a major oncogenic pathway associated with several human malignancies, it has primarily been studied in the context of cancer research41. Emerging evidence suggests that the Hedgehog signaling pathway can be activated under hyperglycemic conditions, functioning as a regulatory axis for lipid metabolism and insulin signaling42. Yao et al. demonstrated its involvement in insulin resistance by inhibiting PPAR-γ, highlighting the therapeutic potential of targeting the Hedgehog pathway for treating metabolic disorders characterized by insulin resistance43. Moreover, the Hedgehog signaling pathway has been shown to induce metabolic reprogramming toward a Warburg-like glycolytic state via the cilia-dependent Smo-Ca2+-Ampk axis. Activation of this axis promotes insulin-independent glucose uptake in muscle and brown adipose tissue, suggesting that rationally designed Hedgehog pathway modulators could offer innovative therapeutic approaches for obesity and diabetes. Additionally, multiple studies have linked this pathway to the development of diabetic complications, including diabetic nephropathy, retinopathy, neuropathy, and cardiovascular diseases40.Overall, this study bridges gaps in current T2D biomarker research by linking β-cell specificity, immune correlation, and multi-cohort validation into a unified framework.
Several limitations should be acknowledged. First, while preliminary in vivo experiments were performed, a deeper exploration of the underlying mechanisms is still needed. Second, extending this research to pre-diabetic populations would strengthen the clinical applicability of the findings.
Conclusion
SLC2A2 was identified as key gene of T2D through differential analysis, LASSO and SVM-RFE. Notably, the enrichment of key biological processes such as cAMP signaling pathway, insulin resistance, cytokine-cytokine receptor interaction, ECM-receptor interaction, and oxidative phosphorylation underscores the multifactorial nature of T2D, where diverse molecular pathways converge to drive disease progression. Moreover, SLC2A2 had a positive correlation with resting CD4+ memory T cells. Further analysis uncovered that SLC2A2 expressed in beta cells. Finally, in vivo studies confirmed the expressed level of SLC2A2. This study provided a novel vision for the diagnosis and pharmaceutical development of T2D.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request. Bulk RNA-seq data (GSE76894, GSE76895, and GSE86468) were downloaded from GEO database (https://www.ncbi.nlm.nih.gov/geo/) and scRNA-seq data were downloaded from ArrayExpress (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-5061/).
References
Petersmann, A. et al. Definition, classification and diagnosis of diabetes mellitus. Exp. Clin. Endocrinol. Diabetes. 127, S1–s7. https://doi.org/10.1055/a-1018-9078 (2019).
Wu, Y., Ding, Y., Tanaka, Y. & Zhang, W. Risk factors contributing to type 2 diabetes and recent advances in the treatment and prevention. Int. J. Med. Sci. 11, 1185–1200. https://doi.org/10.7150/ijms.10001 (2014).
Schmidt, A. M. Highlighting diabetes mellitus: the epidemic continues. Arterioscler. Thromb. Vasc Biol. 38, e1–e8. https://doi.org/10.1161/atvbaha.117.310221 (2018).
Kaul, K., Tarr, J. M., Ahmad, S. I., Kohner, E. M. & Chibber, R. Introduction to diabetes mellitus. Adv. Exp. Med. Biol. 771, 1–11. https://doi.org/10.1007/978-1-4614-5441-0_1 (2012).
ElSayed, N. A. et al. 2. Classification and diagnosis of diabetes: standards of care in diabetes-2023. Diabetes Care 46, S19-s40, (2023). https://doi.org/10.2337/dc23-S002
Kanter, J. E. & Bornfeldt, K. E. Impact of diabetes mellitus. Arterioscler. Thromb. Vasc Biol. 36, 1049–1053. https://doi.org/10.1161/atvbaha.116.307302 (2016).
Tabák, A. G., Herder, C., Rathmann, W., Brunner, E. J. & Kivimäki, M. Prediabetes: a high-risk state for diabetes development. Lancet 379, 2279–2290. https://doi.org/10.1016/s0140-6736(12)60283-9 (2012).
Sun, Y. et al. The utility of exosomes in diagnosis and therapy of diabetes mellitus and associated complications. Front. Endocrinol. (Lausanne). 12, 756581. https://doi.org/10.3389/fendo.2021.756581 (2021).
Laakso, M. Biomarkers for type 2 diabetes. Mol Metab. 27s, S139–s146 (2019). https://doi.org/10.1016/j.molmet.2019.06.016
Zhong, M. et al. Identification of diagnostic markers related to oxidative stress and inflammatory response in diabetic kidney disease by machine learning algorithms: evidence from human transcriptomic data and mouse experiments. Front. Endocrinol. (Lausanne). 14, 1134325. https://doi.org/10.3389/fendo.2023.1134325 (2023).
Li, W. et al. Identification of immune infiltration and the potential biomarkers in diabetic peripheral neuropathy through bioinformatics and machine learning methods. Biomolecules 13 https://doi.org/10.3390/biom13010039 (2022).
Zhang, Y. et al. Machine-learning Algorithm-based prediction of diagnostic gene biomarkers related to immune infiltration in patients with chronic obstructive pulmonary disease. Front. Immunol. 13, 740513. https://doi.org/10.3389/fimmu.2022.740513 (2022).
Liu, Z., Li, H. & Pan, S. Discovery and validation of key biomarkers based on immune infiltrates in alzheimer’s disease. Front. Genet. 12, 658323. https://doi.org/10.3389/fgene.2021.658323 (2021).
Zhong, Y. et al. Screening biomarkers for systemic lupus erythematosus based on machine learning and exploring their expression correlations with the ratios of various immune cells. Front. Immunol. 13, 873787. https://doi.org/10.3389/fimmu.2022.873787 (2022).
Fan, W. et al. A novel chemokine-based signature for prediction of prognosis and therapeutic response in glioma. CNS Neurosci. Ther. 28, 2090–2103. https://doi.org/10.1111/cns.13944 (2022).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–d677. https://doi.org/10.1093/nar/gkae909 (2025).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Li, J. et al. Tumor characterization in breast cancer identifies immune-relevant gene signatures associated with prognosis. Front. Genet. 10, 1119. https://doi.org/10.3389/fgene.2019.01119 (2019).
Segerstolpe, Å. et al. Single-cell transcriptome profiling of human pancreatic Islets in health and type 2 diabetes. Cell. Metab. 24, 593–607. https://doi.org/10.1016/j.cmet.2016.08.020 (2016).
Valent, A. M. & Caughey, A. B. Comprehensive management of type 2 diabetes during pregnancy. Jama 329, 1022–1023. https://doi.org/10.1001/jama.2023.0929 (2023).
Rigalleau, V. et al. Comment on gange et alincidence of proliferative diabetic retinopathy and other neovascular sequelae at 5 years following diagnosis of type 2 diabetes. Diabetes Care. 44:2518–2526 (2021). Diabetes Care 45, e60. https://doi.org/10.2337/dc21-2254 (2022).
Bai, S., Yu, H., Wang, B. & Li, J. Retrospective and perspective of rice breeding in China. J. Genet. Genomics. 45, 603–612. https://doi.org/10.1016/j.jgg.2018.10.002 (2018).
Liang, Y. Y. et al. Social isolation, loneliness, and risk of microvascular complications among individuals with type 2 diabetes mellitus. Am. J. Kidney Dis. 84, 557–566e551. https://doi.org/10.1053/j.ajkd.2024.05.004 (2024).
ElSayed, N. A. et al. 3. Prevention or delay of type 2 diabetes and associated comorbidities: standards of care in diabetes-2023. Diabetes Care 46, S41–s48 (2023). https://doi.org/10.2337/dc23-S003
Yu, M. G. et al. Protective factors and the pathogenesis of complications in diabetes. Endocr. Rev. 45, 227–252. https://doi.org/10.1210/endrev/bnad030 (2024).
Hannou, S. A., Haslam, D. E., McKeown, N. M. & Herman, M. A. Fructose metabolism and metabolic disease. J. Clin. Invest. 128, 545–555. https://doi.org/10.1172/jci96702 (2018).
Thorens, B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia 58, 221–232. https://doi.org/10.1007/s00125-014-3451-1 (2015).
Dupuis, J. et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 42, 105–116. https://doi.org/10.1038/ng.520 (2010).
Gaulton, K. J. et al. Comprehensive association study of type 2 diabetes and related quantitative traits with 222 candidate genes. Diabetes 57, 3136–3144. https://doi.org/10.2337/db07-1731 (2008).
Kilpeläinen, T. O. et al. Physical activity modifies the effect of SNPs in the SLC2A2 (GLUT2) and ABCC8 (SUR1) genes on the risk of developing type 2 diabetes. Physiol. Genomics. 31, 264–272. https://doi.org/10.1152/physiolgenomics.00036.2007 (2007).
Volkov, P. et al. Whole-Genome bisulfite sequencing of human pancreatic Islets reveals novel differentially methylated regions in type 2 diabetes pathogenesis. Diabetes 66, 1074–1085. https://doi.org/10.2337/db16-0996 (2017).
Segrè, A. V., Wei, N., Altshuler, D. & Florez, J. C. Pathways targeted by antidiabetes drugs are enriched for multiple genes associated with type 2 diabetes risk. Diabetes 64, 1470–1483. https://doi.org/10.2337/db14-0703 (2015).
Bacos, K. et al. Type 2 diabetes candidate genes, including PAX5, cause impaired insulin secretion in human pancreatic Islets. J. Clin. Invest. 133 https://doi.org/10.1172/jci163612 (2023).
Buniello, A. et al. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–d1012. https://doi.org/10.1093/nar/gky1120 (2019).
Onrust-van Schoonhoven, A. et al. 3D chromatin reprogramming primes human memory T(H)2 cells for rapid recall and pathogenic dysfunction. Sci. Immunol. 8, eadg3917. https://doi.org/10.1126/sciimmunol.adg3917 (2023).
Zhang, Z. et al. Integrated bioinformatics analysis revealed immune checkpoint genes relevant to type 2 diabetes. Diabetes Metab. Syndr. Obes. 17, 2385–2401. https://doi.org/10.2147/dmso.S458030 (2024).
Hellings, W. E. et al. Composition of carotid atherosclerotic plaque is associated with cardiovascular outcome: a prognostic study. Circulation 121, 1941–1950. https://doi.org/10.1161/circulationaha.109.887497 (2010).
Zhao, L. et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 359, 1151–1156. https://doi.org/10.1126/science.aao5774 (2018).
Deng, Z., Kuno, A., Ojima, M. & Takahashi, S. MafB maintains β-cell identity under MafA-deficient conditions. Mol. Cell. Biol. 42, e0054121. https://doi.org/10.1128/mcb.00541-21 (2022).
Ingham, P. W., Nakano, Y. & Seger, C. Mechanisms and functions of Hedgehog signalling across the metazoa. Nat. Rev. Genet. 12, 393–406. https://doi.org/10.1038/nrg2984 (2011).
Skoda, A. M. et al. The role of the Hedgehog signaling pathway in cancer: a comprehensive review. Bosn J. Basic. Med. Sci. 18, 8–20. https://doi.org/10.17305/bjbms.2018.2756 (2018).
Benchoula, K., Parhar, I. S. & Wong, E. H. The crosstalk of hedgehog, PI3K and Wnt pathways in diabetes. Arch. Biochem. Biophys. 698, 108743. https://doi.org/10.1016/j.abb.2020.108743 (2021).
Zhao, C. et al. Rational design of layered oxide materials for sodium-ion batteries. Science 370, 708–711. https://doi.org/10.1126/science.aay9972 (2020).
Funding
This work was supported by the initial funding of Wuxi Taihu University.
Author information
Authors and Affiliations
Contributions
Jinwei Li conceived and headed this project. Yan Cao and Liqi Chen participated in processing raw data and technical analysis. Haoru Dong provided technical support. Haoru Dong, Yurui Zhuang, and Yuzhe Shi finished the in vivo studies. Liqi Chen and Yan Cao drafted the manuscript. Ziyi Guo provided comments and improvements to the paper. All authors approved the final version manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cao, Y., Chen, L., Zhuang, Y. et al. Integrated bulk and single cell sequencing with experimental validation identifies type 2 diabetes biomarkers. Sci Rep 15, 28924 (2025). https://doi.org/10.1038/s41598-025-14915-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-14915-y
