
Abstract
Lung adenocarcinoma (LUAD) is a common and deadly subtype of lung cancer with high mortality and limited treatment options. Enhancer RNAs (eRNAs) have emerged as important regulators in cancer biology, but their specific roles in LUAD have not been fully explored. This study aimed to investigate the role of eRNAs in LUAD pathogenesis and evaluate their potential as diagnostic biomarkers and therapeutic targets. Through integrated bioinformatics and experimental approaches, we identified differentially expressed eRNAs associated with LUAD progression. Functional analysis demonstrated that these eRNAs regulate critical pathways related to cell growth, division, repair, immune response, and tumor development. We constructed eRNA-centric regulatory networks, elucidating their interactions with transcription factors, enhancer-promoter loops, and RNA-binding proteins. Notably, eRNA ENSR00000188682 was highlighted for its central role, along with its associated transcription factor and downstream target gene ADRB2. Additionally, we developed a diagnostic prediction model based on eRNA expression profiles, which showed promising diagnostic accuracy. Our findings provide a comprehensive understanding of eRNA-mediated regulation in LUAD and suggest that eRNAs hold significant potential as prognostic biomarkers and therapeutic targets for improved clinical outcomes.
Similar content being viewed by others


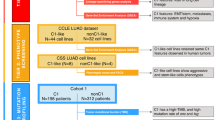
Introduction
Lung cancer, a prevailing malignancy globally, stands as the foremost cause of cancer-related mortality1,2. Lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), together constituting non-small cell lung cancer (NSCLC), dominate its landscape3,4. LUAD, in particular, commands nearly 40% of lung cancer cases worldwide, experiencing a steady rise in incidence alongside diminished overall survival rates vis-à-vis its counterparts5,6. Thus, unraveling its mechanistic intricacies and forging precise diagnostic maodalities endure as formidable imperatives.
Once relegated to the realm of "junk DNA," non-coding regions of the human genome, encompassing roughly 98% of its expanse, have undergone a paradigmatic shift in perception7,8,9. High-throughput sequencing has revealed the vital functions of non-coding RNAs in various biological processes. Enhancer RNAs (eRNAs), transcribed from enhancers, play significant roles in gene regulation by forming chromatin loops with target promoters10 and are linked with active enhancer modifications like H3K27ac and H3K27me11,12. Research increasingly highlights eRNAs as potential biomarkers and therapeutic targets in diseases such as cancer, neurodegenerative, and cardiovascular diseases13,14,15,16. Their disease-specific expression patterns and regulatory plasticity make them particularly attractive as therapeutic targets, enabling selective modulation of pathogenic pathways while sparing normal physiology.
Importantly, eRNAs have been shown to influence various signaling pathways, including G protein-coupled receptor (GPCR) signaling, which plays a pivotal role in tumor biology. One such key GPCR family member is the β2-adrenergic receptor (β2-AR), encoded by ADRB2, which regulates cell proliferation, immune responses, and metastasis in cancers including LUAD.
ADRB2 encodes the β2-adrenergic receptor (β2-AR), a member of the G protein-coupled receptor superfamily (GPCR)17. Its dysregulation has been implicated in tumor biology, including processes such as cell proliferation, immune modulation, and metastasis. Specifically in lung adenocarcinoma (LUAD), Ji et al. reported that low ADRB2 expression in tumor tissues is associated with gender, smoking history, overall survival (OS), and disease-specific survival (DSS), highlighting its relevance as a potential prognostic biomarker17. Our study further provides strong evidence of a regulatory link between ADRB2 and eRNAs in LUAD, suggesting eRNA-mediated modulation of ADRB2 may contribute to LUAD progression and therapeutic response.
Methods
Data sources
Enhancer RNA (eRNA) chromatin coordinates and clinical relevance (survival time and smoking history) in lung adenocarcinoma (LUAD) were collected from the eRic database (https://hanlaboratory.com/eRic/, accessed May 2023)14. The eRNA expression levels and regulatory interactions were obtained from the eRNA-IDO server18. H3K27ac ChIP-seq data in the LUAD A549 cells were obtained from the Gene Expression Omnibus (GEO) database (GSM2359488, GSE89128)19. For consistency, the reference genome used in our study was unified to human hg38 (GENCODE v33)20.
ChIP-seq data processing
ChIP-seq data was analyzed using the published strategy, including read trimming, alignment, and signal visualization19. Briefly, raw data was first trimmed using Cutadapt V4.021. Next, the clean reads were mapped to human hg38 reference by using bowtie V1.3.1 (parameter: -p 4 –best -k 2 -m 2 –sam -l 40). MACS2 v2.2.522 with default parameters were used to identify the peaks. Finally, the read coverages were converted to BigWig format and the signals of eRNA regions and the adjacent regions were visualized by the bamCoverage function of deepTools23.
Differentially expressed eRNAs identification
The eRNA chromatin coordinates were uploaded to eRNA-IDO (https://bioinfo.szbl.ac.cn/eRNA_IDO/) to obtain the eRNA expression levels in normal lung samples obtained from the Genotype-Tissue Expression (GTEx) database (N = 579) and in LUAD samples obtained from The Cancer Genome Atlas (TCGA) database (N = 510). Student’s t-test was used to identify significant differences in eRNA expression between LUAD and normal lung samples. We identified differentially expressed eRNAs using the following standards: adjusted p-values < 0.05 and |Log2FC|> 1.5. Volcano plots showing DEGs were drawn using the ‘ggplot2’ v3.5.0 R package.
Co-expression network construction
The co-expression network was obtained from the eRNA-IDO, which was calculated based on the TCGA expression profiles (N = 510). GCEN, a command-line toolkit, was used to calculate gene expression correlations24. The significantly correlated pairs between the differentially expressed eRNAs and the annotated genes were determined with the following standards: absolute Pearson correlation coefficients > 0.5 and false discovery rates (FDRs) < 0.05. Considering the role of eRNAs in activating target gene transcription, only positively correlated pairs were retained. For correlations between the annotated genes, an absolute Pearson correlation coefficient threshold of 0.8 was applied. Subsequently, all significantly correlated pairs were amalgamated to construct the gene co-expression network.
eRNA-centric regulatory network construction
eRNA-IDO server was used to construct the eRNA-centric regulatory network. eRNA-IDO identified transcription factor (TF) binding events on eRNA by analyzing 11,356 TF ChIP-seq datasets from Cistrome database25, and captured eRNA-associated Enhancer-Promoter (E-P) loops by analyzing 200 HiChIP datasets from HiChIPdb26. TF-eRNA and eRNA-loop relationships were connected to form TF-eRNA-loop regulatory axes. These regulatory interactions were integrated with protein–protein interactions from the STRING database27 to construct the overall eRNA-centric regulatory network.
Module extraction and network visualization
To extract the tightly connected gene modules from the overall regulatory network, SPICi28 in the default mode was used. Modules were selected based on the following criteria: cluster density greater than 0.5, support threshold greater than 0.5, and cluster size larger than 2. Each module represents a sub-network. The sub-networks containing at least one eRNA were retained for further analyses. The networks and subnetworks were visualized using Cytoscape v3.9.129.
GO and KEGG enrichment analyses
Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG)30 analyses on the eRNA-connected protein-coding genes within the same network or subnetworks were performed using the script from ncFANs v2.0 (https://github.com/zhangyw0713/FunctionEnrichment)31.
A549 cell culture and treatments
The human non-small cell lung cancer (NSCLC) cell line A549 was obtained from the Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences. The cells were cultured in Complete RPMI 1640 medium supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin solution (Solarbio, China). All cell lines were maintained in a controlled environment at 37 °C with 5% CO2 concentration. Regular assessments for mycoplasma contamination were conducted using PCR techniques.
The overexpression of eRNA in the A549 cell line
We reanalyzed the published PRO-seq from GEO database (accession ID: GSM1055806) and found the exact eRNA trancripts from the ENSR00000188682 sites, which was used for the following experimental validation. Specifically, use this predicted eRNA position to intersect with the chromosomal position measured by gro-seq and the enhancer peak of H3K27ac to obtain high-confidence transcript fragments.
To establish cells with stable expression of this eRNA, we infected the A549 cell line with lentiviral particles obtained from GenePharma (Shanghai, China). Following a 48-h incubation period, cells were subjected to selection for 7 days in culture medium containing 5 μg/ml puromycin to enhance expression levels. The specific primers utilized in this experiment are provided in the Supplementary table1.
Western blot
The cells were lysed in ice-cold RIPA lysis buffer containing 1 mM phenylmethylsulfonyl fluoride (PMSF) to extract proteins. The protein concentration in the lysates was determined using a BCA protein assay kit. Equal amounts of total protein were separated on 10% SDS-PAGE and transferred onto PVDF membranes. Subsequently, the membranes were incubated with specific antibodies against ADRB2 and β-actin. After washing with TBST, the membranes were exposed to an HRP-conjugated secondary antibody, and visualization was performed using the ChemiScopeTouch imaging system. The intensity of the immunoblot bands was quantified using ImageJ 1.8.0 software.
Colony formation assay
To assess the colony-forming ability of A549 cells under different treatments, cells from different experimental groups were seeded in 6-well plates at a density of 5000 cells per well and cultured for 10 to 14 days. The culture medium was changed every two days to support colony growth. After the incubation period, colonies were washed with PBS, fixed with 4% formaldehyde for 30 min, and then stained with a 0.5% crystal violet solution in 10% methanol for an additional 30 min. Subsequently, the plates were washed, air-dried, and colonies were visualized.
Cell growth assay
Following transfection with siRNA and negative control RNA, A549 cells were seeded into 6-well plates at a density of 10,000 cells per well. Subsequently, the cell number was quantified on days 2, 4, and 6 post-seeding to compare the growth rate.
Diagnostic prediction model development and evaluation
The eRNA expression levels of the GTEx normal lung cortex samples32 and the TCGA LUAD samples (https://portal.gdc.cancer.gov/) were used to develop the diagnostic prediction model. Half of the samples were randomly selected to create the training set, while the remaining samples formed the test set. First, the univariate logistic regression with the eRNA expression levels as input variables was used to identify the differentially expressed eRNAs significantly associated with oncogenesis. These cancer-associated eRNAs were further used to construct the diagnostic prediction model using multivariate logistic regression. Next, the test set was used to evaluate the performance of the diagnostic prediction model. Receiver operating characteristic (ROC) curves were generated using the R package ‘pROC’.
Declaration of generative AI and AI-assisted technologies in the writing process
During the preparation of this work the authors used chatGPT in order to polish. After using this tool, the authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.
Results
Revealing the potential role of eRNA in LUAD
Our study is based on 1202 eRNAs associated with human LUAD, as identified from the eRic database (Fig. 1A), noting that these eRNAs show H3K27ac signals, indicating active transcription (Fig. 1B). Comparative analysis utilizing TCGA and GTEx repositories unveiled striking expression disparities among eRNAs between LUAD samples and normal lung tissues, with 54% exhibiting significant differences in expression levels (Fig. 1C). This observation alludes to the putative functional involvement of eRNAs in LUAD pathogenesis.

Differentially expressed eRNAs in LUAD. (A) Research technology roadmap; (B) profile of the H3K27ac around TSS and TES; (C) Pie chart showing the proportion of differentially expressed eRNAs; (D) The volcano plots of the differentially expressed eRNAs between LUAD samples and normal samples; (E) Barplot of the proportion of eRNA regions with COSMIC mutations; Pie chart illustrating the proportion of eRNAs significantly associated with smoking (F) and survival (H) in LUAD; (G) Bar chart depicting the distribution of eRNAs significantly associated with smoking; (I) The Kaplan–Meier survival curve of the eRNA(ENSR0000009256)1 in LUAD.
The volcano plot depicted in Fig. 1D vividly illustrates the landscape of differentially expressed eRNAs, comprising 228 upregulated and 421 downregulated transcripts. Intriguingly, scrutiny of eRNA regions for cancer-related mutations in the COSMIC database revealed a markedly higher proportion of mutations among upregulated eRNAs compared to their downregulated or non-significantly different counterparts (Fig. 1E), hinting at a potential role of COSMIC mutations in driving eRNA upregulation. Given the well-established association between smoking and LUAD risk, we undertook a meticulous examination of the correlation between smoking history and eRNA expression. Notably, 22.8% of the eRNAs exhibited significant associations with smoking (Fig. 1F), with a conspicuous prevalence of upregulated eRNAs among the smoking-associated subset (Fig. 1G), thereby implicating smoking in the modulation of eRNA expression levels.
Moreover, a compelling revelation emerged from our survival analysis, wherein 10.2% of the 1202 eRNAs were found to bear prognostic significance (Fig. 1H). Illustrated by the example of ENSR00000092461, a putative prognostic eRNA, the Kaplan–Meier survival curve (Fig. 1I) delineates a stark divergence in overall survival (OS) between LUAD patients stratified based on eRNA expression levels, with the high-expression cohort exhibiting significantly worse outcomes than their low-expression counterparts (p = 0.0036), thereby underscoring the prognostic potential of select eRNAs as biomarkers in LUAD management.
Up-regulated eRNAs in LUAD potentially regulate cell growth, division, and repair, as well as immune processes
Among the 649 differentially expressed eRNAs, 75.7% showed a positive correlation with the expression of their target genes (Fig. 2A). Previous research findings have demonstrated that eRNAs regulate E-P loops, thereby stimulating the expression of target genes. Therefore, these eRNAs positively correlated with target gene expression are likely to play a regulatory role. In subsequent studies, we focused solely on these eRNAs.

Regulatory network of the up-regulated eRNAs in LUAD. (A) Pie chart illustrating the proportion of eRNAs positively or negatively correlated with E-P loop target genes; (B) The co-expression network between up-regulated eRNAs and positively correlated targets; (C) The barplot of GO enrichment analysis results of the positively correlated targets; The most densely connected module from the subnetwork1 (D) and subnetwork2 (G); (E, H) The barplot of GO enrichment analysis results of these two modules; (F, I) The barplot of KEGG enrichment analysis results of these two modules.
To explore the functionality of these eRNAs, we constructed a co-expression network of up-regulated eRNAs and their target protein-coding genes. (Fig. 2B). This network involved 631 protein-coding genes, through which we could infer the functions of these up-regulated eRNAs. The GO enrichment analysis results showed that these protein-coding genes significantly enriched in pathways related to "cell division," "cell cycle," and "sister chromatid segregation" (Fig. 2C). This suggests that these genes may play important roles in cell growth, division, repair, and regulation.
In Fig. 2B, it is evident that two modules exhibit significantly higher degrees of connectivity. Module 1, the most densely regulated module, consists of 2 eRNAs and 141 protein-coding genes (Fig. 2D). The GO enrichment analysis of this module showed that this module likely contributes significantly to the enrichment results of the entire network (Fig. 2E). Notably, the KEGG enrichment analysis of this module suggests involvement in the "P53 signaling pathway," which may indicate a certain correlation with tumorigenesis or its involvement in processes such as apoptosis and DNA repair (Fig. 2F). Module 2, composed of 4 eRNAs and 21 protein-coding genes (Fig. 2G), exhibits different characteristics. The results of GO and KEGG analyses suggest that the target genes in this module may be involved in immune regulation (Fig. 2H, I). This suggests that upregulated eRNAs may also have potential functions in immune regulation.
Down-regulated eRNAs have potential effects on tumor development and metastasis
Applying a similar methodology, we established a co-expression network for downregulated eRNAs, encompassing 1380 target protein-coding genes and manifesting 35,848 connections (Fig. 3A). Analysis of gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment unveiled the potential involvement of these protein-coding genes in diverse pivotal regulatory mechanisms. These mechanisms spanned cell migration, multicellular tissue regulation, cell–cell communication, cellular response to stimuli, vascular development, and the formation of the tumor microenvironment. Notably, a majority of the enriched pathways were closely associated with vascular formation and cell migration, hinting at their plausible roles in tumor development and metastasis (Fig. 3B, C).

Regulatory network of the down-regulated eRNAs in LUAD. (A) The co-expression network between down-regulated eRNAs and positively correlated targets; The barplot of KEGG(B) and GO(C) enrichment analysis results of the positively correlated targets; (D, G) The two most densely connected modules from the co-expression network; (E, H) The barplot of GO enrichment analysis results of these two modules; (F, I) The barplot of KEGG enrichment analysis results of these two modules.
Within this network, we identified two noteworthy modules. Module 1 emerged as the most interconnected module, with its functional enrichment aligning with the overarching network characteristics (Fig. 3D–F). Conversely, Module 2 comprised one eRNA and 96 protein-coding genes. The results from GO and KEGG enrichment analyses underscored pathways such as Axoneme assembly, cilium movement, microtubule-based processes, and microtubule bundle formation (Fig. 3G–I). These findings suggest the potential roles of these genes in governing cellular structure and function.
eRNA-centered regulatory network
To delve deeper into the regulatory mechanisms governed by eRNAs, we meticulously constructed a TF-eRNA-EP loop regulatory axis network. Leveraging the analysis of 11,356 TF ChIP-seq datasets sourced from the Cistrome database, we discerned TF binding events on eRNAs. Additionally, by scrutinizing 200 HiChIP datasets from the HiChIPdb, we identified eRNA-associated enhancer-promoter (E-P) loops26. The amalgamation of TF-eRNA interactions and eRNA loops facilitated the delineation of the TF-eRNA loop regulatory axis. These regulatory dynamics were seamlessly integrated with protein–protein interactions from the STRING database27, thus culminating in the creation of a comprehensive regulatory network anchored around eRNAs. Figure 4A encapsulates the regulatory network focalized on upregulated eRNAs, while Fig. 4B delineates the counterpart network spotlighting downregulated eRNAs.

The analysis of the TF-eRNA-EP loop regulatory axis network. The TF-eRNA-EP loop regulatory network centered on up-regulated RNA(A) and down-regulated RNA(B); Bar chart depicting the proportions of TFs(C), E-P loops(D), and RBPs(E) in two networks (***P < 0.001); The barplot of KEGG enrichment analysis results of up-(F) and down-regulated(G) eRNA-centric regulatory network; (H) Visualization of eRNA (ENSR00000038731) along with its connected TFs, E-P loops, and RBPs.
A comparative analysis of transcription factors (TFs), E-P loops, and RNA-binding proteins (RBPs) between these two networks unveiled intriguing insights. Remarkably, the integrated regulatory network revolving around upregulated eRNAs showcased a significantly higher proportion of TFs, juxtaposed with a lower proportion of E-P loops and RBPs vis-à-vis the network centered around downregulated eRNAs (Fig. 4C–E). This hints at the potentially greater functional impact wielded by downregulated eRNAs. Delving deeper into the biological underpinnings of these networks, we conducted KEGG pathway enrichment analyses on the implicated genes. Both the upregulated and downregulated networks predominantly exhibited enrichment in pathways associated with cancer, with both networks converging on the “cell cycle” pathway (Fig. F,G). This observation is comprehensible, given that cancer cells often exhibit unbridled proliferation, consequently fostering aberrations in the cell cycle33,34.
Illustrating the regulatory mechanism of eRNAs using a notably upregulated eRNA (ENSR00000038731) in LUAD, Fig. 4H portrays the potential interplay between the transcription factor ATF3 and ENSR00000038731, orchestrating the formation of the E-P loop and thereby modulating the expression of the target gene CD44. Furthermore, discernible H3K27ac signals at this locus attest to its enhancer activity. Of paramount significance, the expression of the target gene CD44 bears significant clinical implications and survival indicators in cancer35,36. Specifically, Chen et al. found that the expression of CD44 is significantly correlated with TMB and MSI in 10 and 6 types of cancers, respectively, based on data from the TCGA database. This suggests that CD44 could serve as a potential biomarker for predicting the outcomes of immunotherapy35. Additionally, a positive correlation between CD44 and PD-L1 has been found in patients with lung adenocarcinoma, and some mechanistic studies have been conducted37,38. In summary, these findings provide new insights into CD44 as a critical therapeutic target for inhibiting the intrinsic function of PD-L1 in tumors. Based on our study, the high expression of CD44 in LUAD may be associated with the upregulation of eRNA (ENSR00000038731). Therefore, the regulation of this eRNA may represent a more upstream mechanism.
The eRNA network potentially involved in regulation in LUAD
To refine our focus on eRNAs and protein-coding genes with potentially substantive functionality, we curated a subset comprising LUAD-DEGs and differentially expressed eRNAs exhibiting positive correlations with these target genes (Fig. 5A,B). By scrutinizing the average number of edges linked to eRNAs, we discerned that networks centralized on upregulated eRNAs exhibited a marginally lower average compared to the overall network (Fig. 5C), whereas for networks centralized on downregulated eRNAs, the average was marginally higher (Fig. 5F).

The analysis of the LUAD-specific eRNA-centric regulatory network. The TF-eRNA-EP loop regulatory network centered on up-(A) and down-regulated(B) RNA, which only includes LUAD DEGs; The barplot of the average number of edges of eRNA and overall in the up-(C) and down-regulated(F) eRNA-centric regulatory network; (D, G) The barplot of GO enrichment analysis results of these two networks; (E, H) The barplot of KEGG enrichment analysis results of these two networks.
Subsequently, we embarked on an enrichment analysis of the genes within the network to glean insights into their regulatory roles. The GO analysis unveiled the engagement of genes in networks revolving around upregulated eRNAs primarily in the positive regulation of protein kinase activity, multicellular organismal processes, developmental processes, and nitrogen compound metabolic processes (Fig. 5D). Interestingly, the top enriched KEGG pathway, "Endocytosis," is a pivotal pathway for regulating metastasis (Fig. 5E)39. The "Wnt signaling pathway" and the "MAPK signaling pathway" stand out as quintessential signaling cascades in lung cancer (Fig. 5E). Aberrant activation of the "Wnt signaling pathway" is closely associated with the proliferation, invasion, and metastasis of tumor cells40. Furthermore, aberrant activation of this pathway may lead to epithelial-mesenchymal transition (EMT) and enhancement of tumor stem cell properties, thereby promoting the malignant progression of lung cancer41,42. Abnormal activation of the "MAPK signaling pathway" can promote the proliferation and invasion of lung cancer cells while inhibiting apoptosis, thereby facilitating the malignant progression of lung cancer43,44.
Conversely, the GO enrichment analysis outcomes pertaining to the genes within the network focalized around downregulated eRNAs unveil pronounced associations with pathways encompassing cell fate specification, cellular responses to external stimuli, skeletal muscle tissue development, among others (Fig. 5G). Additionally, the KEGG enrichment outcomes solely spotlighted enrichment in "Pathways in cancer" (Fig. 5H).
The eRNA (ENSR00000188682) regulates ADRB2 in LUAD
Next, we utilized SPICI to extract a tightly connected network related to ADRB2 from Fig. 5B (Fig. 6A). From the GO and KEGG enrichment analysis results of these genes, we identified an interesting pathway—Transcriptional misregulation in cancer (Fig. 6B). This suggests that these genes may be associated with transcriptional dysregulation in the occurrence and development of cancer.

Regulatory axis associated with ADRB2. (A) Regulatory module associated with ADRB2; (B) The barplot of GO (green) and KEGG (red) enrichment analysis results of genes from this module. (C) The expression level of eRNA (ENSR00000188682); The Kaplan–Meier survival curve of ENSR00000188682(D), ADRB2(F), and ERG(G) in LUAD; (E) The expression level of ADRB2 and ERG (***P < 0.001); (H) Immunoblot detecting the expression of ADRB2 in the ENSR00000188682 OE transduced A549 cell line. Colony formation assays(I) and cell growth curves(J) were performed to assess the proliferation of ENSR00000188682 OE transduced A549 cell line (P values were determined by the one-way ANOVA, **P < 0.01, **** P < 0.0001). (K) Pattern diagram for diagnosis model establishment; (L) Dot plot illustrating the coefficients (β) and corresponding p values for each variable in the statistical model. Each point represents a variable; (M) ROC curve on the test set (AUC = 0.82).
Within this small network, we selected an eRNA (ENSR00000188682) regulated by only one transcription factor (TF) to validate the regulatory mechanism. Figure 6C shows that the expression of eRNA (ENSR00000188682) is significantly lower in cancer samples compared to normal samples. Furthermore, patients exhibiting high expression of ENSR00000188682 demonstrate markedly improved survival outcomes compared to those with low expression (Fig. 6D). Both the TF (ERG) of this eRNA and the target gene of the e-p loop (ADRB2) are substantially downregulated in cancer samples (Fig. 6E). Survival analysis outcomes for ERG and ADRB2 echo those of ENSR00000188682, indicating better survival among patients with elevated expression of ERG or ADRB2 (Fig. 6F,G). Indeed, previous studies have underscored the prognostic significance of ADRB2 in LUAD17,45,46.
To validate the biological function of ENSR00000188682 in tumor progression and its association with LUAD, we conducted overexpression experiments of ENSR00000188682 in the human non-small cell lung cancer cell line (A549). Protein immunoblotting corroborated that the overexpression of ENSR00000188682 resulted in the upregulation of the target gene of the E-P loop (ADRB2) (Fig. 6H, Supplementary Fig. 1), effectively delineating the regulatory role of the eRNA. In colony formation assays, we noted a significant inhibition in the formation of tumor cell colonies upon overexpression of ENSR00000188682 (Fig. 6I), indicating its pivotal role in regulating the clonal growth ability of lung cancer cells. These findings were further corroborated by cell proliferation assays, which demonstrated a significant reduction in the growth rate of the lung cancer cell line upon overexpression of ENSR00000188682 (Fig. 6J).
Diagnostic model establishment
In our previous investigations, we elucidated the pivotal role of ADRB2 in both the initiation and progression of LUAD. Furthermore, our findings underscored the robust interactive functionality inherent within this compact network. To delve deeper into the diagnostic potential of eRNAs within this network, we harnessed data from the GTEx and TCGA databases (Fig. 6K). Dividing the dataset into halves, we designated one portion as the testing set and conducted single-variable logistic regression on four eRNAs. Analysis of the coefficient β and p-values provided compelling evidence regarding the diagnostic significance of these four eRNAs (Fig. 6L). Notably, the area under the ROC curves was computed to be 0.82 (Fig. 6M), unequivocally demonstrating the efficacy of this diagnostic model.
Mechanism explanation
In the expression profile data of normal samples, a robust correlation emerged between ENSR00000188682 and ADRB2 expression (R = 0.83, P = 6.83e-27) (Fig. 7A). However, within LUAD samples, this correlation markedly diminished (R = 0.04, P = 0.67) (Fig. 7B). Similarly, the expression of ENSR00000188682 and its transcription factor (ERG) exhibited a strong correlation in normal samples (R = 0.54, P = 5.12e-09) (Fig. 7C), yet demonstrated a notably weaker association in LUAD samples (R = 0.07, P = 0.46) (Fig. 7D).

eRNA (ENSR00000188682) regulatory mechanism. Correlation graph between ADRB2 expression and eRNA (ENSR00000188682) expression in LUAD cohort(A) and normal cohort(B); Correlation graph between ERG expression and eRNA (ENSR00000188682) expression in LUAD cohort(C) and normal cohort(D); (E) Regulatory mechanism of eRNA (ENSR00000188682) in LUAD and normal samples.
Figure 7E delineates the regulatory mechanism of the eRNA (ENSR00000188682) within normal and LUAD contexts. In normal samples, the transcription factor of ENSR00000188682 (ERG) maintains adequate expression levels, fostering the upregulation of eRNA (ENSR00000188682). This, in turn, bolsters the mediated E-P loop strength, ultimately leading to the enhanced expression of the target gene (ADRB2) and exerting an anti-tumor effect. Conversely, within LUAD samples, the under-expression of the TF (ERG) results in the downregulation of eRNA (ENSR00000188682) expression. Consequently, this weakens the mediated E-P loop strength, culminating in decreased expression of the target gene (ADRB2) and facilitating a tumor-promoting effect.
Discussion
LUAD originates from glandular epithelium in the bronchial mucosa, and its early diagnosis remains challenging. Over the past decade, significant research efforts have associated enhancer RNA (eRNA) with the initiation and advancement of various diseases47. Despite this, the exact roles and mechanisms of eRNA in cancer progression continue to elude researchers. Within the vast expanse of the human transcriptome, over 65,000 enhancers have been identified as potential eRNA sources, exhibiting specificity across various tumor types and individual patients48,49,50. This abundance underscores the promising potential of eRNA as both diagnostic biomarkers and targets for therapeutic intervention in cancer research. Our research mainly explores the function of eRNA in LUAD. This study reveals the functional role of eRNA as a ubiquitous cellular mechanism in the pathogenesis of LUAD through network analysis. Specifically, eRNA can enhance the E-P cycle and upregulate the expression of specific target genes.
Our study delves into the functional role of eRNA in LUAD pathogenesis through network analysis, particularly in enhancing the E-P cycle and upregulating the expression of specific target genes. Initially, our analysis identified approximately 10% of over 1200 examined eRNAs as significantly associated with LUAD patient survival outcomes. Further exploration involved screening for differentially expressed eRNAs between normal and LUAD samples, and constructing co-expression networks comprising these eRNAs and their target genes. Pathway enrichment analysis suggested that upregulated eRNAs in LUAD may regulate cell growth, division, repair, and immune responses, while downregulated eRNAs may impact tumor development and metastasis.
To elucidate the regulatory mechanisms of eRNA, the pathways involving TFs, eRNAs, E-P loops, and RBPs were integrated into the eRNA-centric regulatory network depicted in Fig. 4. The establishment of these two networks, along with the enrichment analysis of the protein-coding genes present in the networks, further underscores the potentially significant impact of the entire regulatory network on tumorigenesis. After filtering the genes in these two networks with LUAD differentially expressed genes, we obtained an eRNA-centric regulatory network more closely associated with LUAD. It is noteworthy that the enrichment analysis of these two networks revealed enrichment in pathways such as "Endocytosis," "Wnt signaling pathway," and the "MAPK signaling pathway." Numerous studies have confirmed the close association of these pathways with LUAD progression and metastasis39,40,41,43,44. This potentially suggests that these eRNAs may influence prognosis by regulating upstream pathways such as these.
We employed the SPICI method to extract a tightly connected module in the downregulated eRNA-centric regulatory network, which includes a potential prognostic biomarker (ADRB2) for LUAD. We validated one of its regulatory axes centered around ENSR00000188682. Data mining from the TCGA database revealed a significant association between low expression of the transcription factor (TF) ERG, eRNA (ENSR00000188682), and the target gene (ADRB2) with adverse prognosis. Further validation in human NSCLC cell lines (A549) by overexpressing ENSR00000188682 confirmed its regulatory role on ADRB2 and its role in promoting lung cancer cell growth. The effectiveness of the diagnostic model also suggests the feasibility of eRNA as a biomarker for LUAD.
Despite these strengths, our study has several limitations. First, it focuses primarily on one regulatory mechanism: eRNAs modulate target gene expression by enhancing the E–P loop through TF binding. However, emerging studies reveal additional modes of eRNA action. For example, eRNAs may promote enhancer acetylation by recruiting cofactors such as CBP/p300 or stabilize E–P interactions through binding with RAD21 or hnRNPU51,52. Future research should investigate whether these alternative mechanisms are active in LUAD and how they interact with the regulatory frameworks identified in this study.
In addition, further validation of our findings in animal models, such as LUAD-prone transgenic mice or xenograft models, would help confirm the biological relevance of eRNA regulatory axes in vivo. Studies using early-stage and metastatic LUAD samples, or single-cell transcriptomics, could reveal stage-specific patterns of eRNA expression. Investigating circulating eRNAs in blood or sputum may also offer insights into their potential as non-invasive biomarkers. Clinical correlation studies evaluating eRNA levels in patients undergoing therapy could further clarify their prognostic or predictive utility.
In conclusion, this study provides a systems-level perspective on the regulatory landscape of eRNAs in LUAD and underscores their potential as mechanistic drivers, diagnostic biomarkers, and therapeutic targets. The integrative analysis presented here lays the groundwork for future mechanistic and translational investigations into eRNA function in lung cancer and beyond.
Data availability
The datasets analysed during the current study are available in the GEO database (accession ID: GSM2359488, GSM1055806, GSE89128), Cistrome database, GTEx database, TCGA database, eRic database, HeRA database, STRING database, and oncoDB database. The specific data used has been explained in the text, and references have been added.
References
Qin, S. et al. Immune, metabolic landscapes of prognostic signatures for lung adenocarcinoma based on a novel deep learning framework. Sci. Rep. 14(1), 527 (2024).
Mullard, A. Addressing cancer’s grand challenges. Nat. Rev. Drug Discov. 19(12), 825–826 (2020).
Chen, J. W. & Dhahbi, J. Lung adenocarcinoma and lung squamous cell carcinoma cancer classification, biomarker identification, and gene expression analysis using overlapping feature selection methods. Sci. Rep. 11(1), 13323 (2021).
Chen, Z. et al. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat. Rev. Cancer 14(8), 535–546 (2014).
Shukla, S., et al., Development of a RNA-Seq Based Prognostic Signature in Lung Adenocarcinoma. J. Natl. Cancer Inst., 109(1). (2017).
Travis, W. D. et al. International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society: international multidisciplinary classification of lung adenocarcinoma: executive summary. Proc. Am. Thorac. Soc. 8(5), 381–385 (2011).
Consortium, E.P., An integrated encyclopedia of DNA elements in the human genome. Nature. (2012).
Mattick, J. S. Non-coding RNAs: the architects of eukaryotic complexity. EMBO Rep. 2(11), 986–991 (2001).
Wang, Y., et al., Enhancer RNA (eRNA) in Human Diseases. Int. J. Mol. Sci., 2022. 23(19).
Sartorelli, V. & Lauberth, S. M. Enhancer RNAs are an important regulatory layer of the epigenome. Nat. Struct. Mol. Biol. 27(6), 521–528 (2020).
Shen, Y. et al. Insights into Enhancer RNAs: Biogenesis and Emerging Role in Brain Diseases. Neuroscientist 29(2), 166–176 (2023).
Raisner, R. et al. Enhancer Activity Requires CBP/P300 Bromodomain-Dependent Histone H3K27 Acetylation. Cell Rep. 24(7), 1722–1729 (2018).
Hsieh, C. L. et al. Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc. Natl. Acad. Sci. U S A 111(20), 7319–7324 (2014).
Zhang, Z. et al. Transcriptional landscape and clinical utility of enhancer RNAs for eRNA-targeted therapy in cancer. Nat. Commun. 10(1), 4562 (2019).
Theodorou, V. et al. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 23(1), 12–22 (2013).
Bernardo, G. M. et al. FOXA1 represses the molecular phenotype of basal breast cancer cells. Oncogene 32(5), 554–563 (2013).
Ji, L. et al. ADRB2 expression predicts the clinical outcomes and is associated with immune cells infiltration in lung adenocarcinoma. Sci. Rep. 12(1), 15994 (2022).
Zhang, Y., et al., eRNA-IDO: A One-stop Platform for Identification, Interactome Discovery, and Functional Annotation of Enhancer RNAs. Genom. Proteom. Bioinform., (2024).
Rusan, M. et al. Suppression of Adaptive Responses to Targeted Cancer Therapy by Transcriptional Repression. Cancer Discov. 8(1), 59–73 (2018).
Frankish, A. et al. GENCODE: reference annotation for the human and mouse genomes in 2023. Nucl. Acids Res. 51(D1), D942–D949 (2023).
Martin, M., CUTADAPT removes adapter sequences from high-throughput sequencing reads. EMBnet. J. (2011).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9(9), R137 (2008).
Ramirez, F., et al., deepTools: a flexible platform for exploring deep-sequencing data. Nucl. Acids Res., (2014).
Chen, W. et al. GCEN: An Easy-to-Use Toolkit for Gene Co-Expression Network Analysis and lncRNAs Annotation. Curr. Issues Mol. Biol. 44(4), 1479–1487 (2022).
Zheng, R. et al. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucl. Acids Res. 47(D1), D729–D735 (2019).
Zeng, W. et al. HiChIPdb: a comprehensive database of HiChIP regulatory interactions. Nucl. Acids Res. 51(D1), D159–D166 (2023).
Szklarczyk, D. et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucl. Acids Res. 51(D1), D638–D646 (2023).
Jiang, P. & Singh, M. SPICi: a fast clustering algorithm for large biological networks. Bioinformatics 26(8), 1105–1111 (2010).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genom. Res. 13(11), 2498–2504 (2003).
Kanehisa, M. et al. KEGG: biological systems database as a model of the real world. Nucl. Acids Res. 53(D1), D672–D677 (2025).
Zhang, Y., et al., ncFANs v2.0: an integrative platform for functional annotation of non-coding RNAs. Nucl. Acids Res., (2021).
Jeong, M. & Goodell, M. A. Noncoding Regulatory RNAs in Hematopoiesis. Curr. Top. Dev. Biol. 118, 245–270 (2016).
Otto, T. & Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 17(2), 93–115 (2017).
Liu, L. et al. The cell cycle in stem cell proliferation, pluripotency and differentiation. Nat. Cell Biol. 21(9), 1060–1067 (2019).
Chen, S. et al. The prognostic value and immunological role of CD44 in pan-cancer study. Sci. Rep. 13(1), 7011 (2023).
Chen, C. et al. The biology and role of CD44 in cancer progression: therapeutic implications. J. Hematol. Oncol. 11(1), 64 (2018).
Zhang, C. et al. CD44, a marker of cancer stem cells, is positively correlated with PD-L1 expression and immune cells infiltration in lung adenocarcinoma. Cancer Cell Int. 20(1), 583 (2020).
Kong, T. et al. CD44 Promotes PD-L1 Expression and Its Tumor-Intrinsic Function in Breast and Lung Cancers. Cancer Res. 80(3), 444–457 (2020).
Khan, I. & Steeg, P. S. Endocytosis: a pivotal pathway for regulating metastasis. Br. J. Cancer 124(1), 66–75 (2021).
Liu, J. et al. Wnt/beta-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target Ther. 7(1), 3 (2022).
Zhou, H. M. & Zhao, L. M. Wnt signaling pathway-derived score for predicting therapeutic resistance and tumor microenvironment in lung adenocarcinoma. Front Pharmacol. 13, 1091018 (2022).
Koni, M., V. Pinnaro, and M.F. Brizzi, The Wnt Signalling Pathway: A Tailored Target in Cancer. Int. J. Mol. Sci., (2020).
Wang, B. et al. PCAT19 Regulates the Proliferation and Apoptosis of Lung Cancer Cells by Inhibiting miR-25-3p via Targeting the MAP2K4 Signal Axis. Dis. Markers 2022, 2442094 (2022).
Braicu, C., et al., A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers (Basel), 2019.
Zheng, Q., S. Min, and Q. Zhou, Identification of potential diagnostic and prognostic biomarkers for LUAD based on TCGA and GEO databases. Biosci Rep, 2021.
Wang, Z. et al. Decreased HLF Expression Predicts Poor Survival in Lung Adenocarcinoma. Med. Sci. Monit. 27, e929333 (2021).
Lee, J. H., Xiong, F. & Li, W. Enhancer RNAs in cancer: regulation, mechanisms and therapeutic potential. RNA Biol. 17(11), 1550–1559 (2020).
Andersson, R. et al. An atlas of active enhancers across human cell types and tissues. Nature 507(7493), 455–461 (2014).
Hu, X., et al., The integrated landscape of eRNA in gastric cancer reveals distinct immune subtypes with prognostic and therapeutic relevance. iScience, (2022).
Luo, L. & Chen, X. Exploring the potential of eRNAs in cancer immunotherapy. Mol. Ther. Oncolytics 27, 197–199 (2022).
Li, W. et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498(7455), 516–520 (2013).
Pezone, A. et al. RNA Stabilizes Transcription-Dependent Chromatin Loops Induced By Nuclear Hormones. Sci. Rep. 9(1), 3925 (2019).
Acknowledgements
We are very grateful to the Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, for providing us with the cell lines, which provided us with great help in completing in vitro validation experiments.
Funding
This research was supported by Chengde Science and Technology Bureau under grant no.202204A071.
Author information
Authors and Affiliations
Contributions
AL completed the main analysis and writing of the first draft of the article; LZ, XD and YG completed some analyses; YD and JZ completed the validation experiments; XF helped to organize the data; PL designed the project and revised the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, A., Zhang, L., Du, X. et al. Deciphering functional landscape and clinical implications of enhancer RNAs in lung adenocarcinoma. Sci Rep 15, 29574 (2025). https://doi.org/10.1038/s41598-025-15485-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-15485-9
