
Abstract
Calcific aortic valve stenosis (CAVS) is steadily rising worldwide with no effective pharmacological agents available. Observational studies implicated dyslipidaemia as a risk factor for CAVS. Whether dyslipidaemia is causative for CAVS and the therapeutic potential of different lipid-modifying drug targets for CAVS treatment remains unclear. We appraised the relationship of genetically-proxied lipid traits and 12 lipid-modifying drug targets with CAVS risk using Mendelian randomization (MR). Genetic variants associated with lipid traits and variants in genes encoding lipid-modifying drug targets were retrieved from GLGC. Summary-level data for CAVS were obtained from the TARGET consortium and FinnGen. Validation analyses were performed using genetic instruments retrieved from liver-derived gene expression and circulation plasma levels of targets. Colocalisation and mediation analyses were performed to evaluate the robustness of our findings and explore potential mediators (i.e., lipoprotein a (Lp(a)), body mass index, apolipoprotein B (ApoB)). The MR analyses supported that total cholesterol and LDL-cholesterol level were independent causal risk factors. The drug-target MR analysis suggested that genetic mimicry of PCSK9 inhibition should reduce CAVS risk (OR = 0.63, 95% CI = 0.56–0.70), which was corroborated by colocalisation analysis. Secondary analyses supported a genetically proxied effect of liver-specific PCSK9 expression (OR = 0.94 per SD reduction in PCSK9 expression, 95% CI = 0.88–1.00) and circulating plasma levels of PCSK9 (OR = 0.86 per SD reduction in PCSK9 protein, 95% CI = 0.83–0.88) on CAVS risk. ApoB and Lp(a) mediated 55.9% and 4.5%, respectively, of the total effect of PCSK9 on CAVS risk. Multiple sensitivity analyses supported this observation. Our study supports total cholesterol, LDL-cholesterol as a causal factor for CAVS, and genetically proxied inhibition of PCSK9 may reduced its risk.
Similar content being viewed by others

Introduction
Calcific aortic valve stenosis (CAVS) due to progressive hemodynamic obstruction is the most prevalent valve heart disease, and will affect 12.6 million individuals worldwide, including 2%−3% of adults aged > 65 years1. Given the absence of pharmacological therapies for CAVS, there is considerable interest in identifying new agents or repurposing existing drugs to delay the progression or reverse course of the disease2,3.
Dyslipidaemia, as an important shared risk factor of different types of cardiovascular disease, have been intensively investigated for CAVS3,4,5, but findings were inconsistent. Whilst observational studies supported that elevated low-density lipoprotein cholesterol (LDL-C) as a risk factor for CAVS, randomized controlled trials (RCTs) failed to demonstrate any benefit of statin therapy on retard disease progression6,7,8. Recently, results from general population study suggested that triglyceride (TG) may affect progression of CAVS4. However, these findings get challenged where subsequent study indicated the association with triglycerides was attenuated after adjustment of LDL-C5. Additionally, observational and genetic studies have reported that elevated lipoprotein(a) [Lp(a)] concentration is associated with higher risk of CAVS9,10, but its association with hemodynamic progression is controversial11. Taken together, these findings suggest that the observed association between dyslipidaemia and CAVS risk may be mediated through mechanisms other than the difference in plasma lipids levels. It is biologically plausible that pathways regulating the biosynthesis and metabolism of lipids may be involved in the development of CAVS. However, the effects of other lipid-modifying targets on CAVS remain unknown except for statin. Therefore, it’s necessary to establish a comprehensive picture depicting the relationship between serum lipids, lipid-modifying targets and CAVS.
Mendelian randomization (MR) uses germline genetic proxies as instruments to evaluate the causality for lifelong modifiable exposures with disease, and thus overcome some of the limitations of traditional observational studies12. As an extension of MR, drug target MR has emerged as efficient way to estimate therapeutic potential by using genetic variants in genes encoding drug targets (referred to as cis variants) to mimic the actions of drugs. This approach has also been widely applied to predict likely beneficial effects of drug targets on cardiovascular diseases13, neurological diseases14 and cancers15.
In this study, we conducted MR analyses to assess the causal effects of lipids on CAVS and to explore the potential effects of 16 lipid-modifying targets on CAVS. Moreover, MR analyses using genetic variants of plasma protein quantitative trait loci (pQTL) and liver tissue-derived gene expression quantitative trait loci (eQTL) for potential targets were performed as sensitivity analyses. Finally, we evaluated the effect of genetically proxied lipid-lowering therapies on CAVS risk via several traits previously implicated in the disease (body mass index (BMI), lipoprotein A (Lp(a)), and apolipoprotein B (APOB))16,17.
Methods
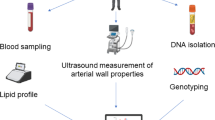
This study followed the Strengthening the Reporting of Observational Studies in Epidemiology-Mendelian Randomisation reporting guidelines (Supplementary table S1)18. An outline of the study can be found in Fig. 1.

Study overview. # The genetic variants were oriented to be in the lipid increasing direction, i.e., effect estimates in Mendelian randomisation analysis was scaled to per SD increase in serum lipids.
Data sources
Summary genetic associations with serum lipids (LDL-C, TG, high-density lipoprotein cholesterol (HDL-C) and total cholesterol (TC)) were obtained from the largest Genome-Wide Association Study (GWAS) performed by the Global Lipids Genetics Consortium (GLGC) with over 1 million Europeans, adjusting for sex, age, age squared, and population stratification19. The GWAS statistics without UK Biobank (UKB) participants was used to avoid the potential bias due to overlapped samples.
For CAVS outcomes, summary data were obtained from the Therapeutic targets for AoRtic stenosis using GEneTics (TARGET) consortium (13,765 cases)16 and the FinnGen cohort (R11 release with 11,047 cases) (https://www.finngen.fi/en)20. We also used various datasets for additional analysis in this paper and all individuals involved in these analyses are of European ancestry. A summary of these datasets is in Supplementary table S2.
Two-sample MR (TSMR)
We used univariable two-sample MR to investigate the associations of genetically predicted lipids on the risk of CAVS. The application of MR relies on 3 fundamental assumptions: (1) the genetic instrumental variable are strongly associated with an modifiable exposure (‘relevance’); (2) they do not share a common cause with the outcome (‘independence’); and (3) they affect the outcome exclusively through the exposure (‘exclusion restriction’). Genetic instruments were constructed by obtaining independent genetic variants (linkage disequilibrium[LD] clumping threshold of r2 < 0.001 within 10,000 kb window) associated with high-density lipoprotein cholesterol (HDL-C), LDL-C, total cholesterol (TC), and TG at genome-wide significance (P < 5 × 10−8) using the latest summary statistics from GLGC.
Multivariable MR (MVMR)
To find independent causal role of specific lipids traits on CAVS, multivariable MR was then used for positive findings from previous TSMR. Since TC and LDL-C are known to share considerable overlap, we therefore excluded TC from this analysis. Given that associations between genetically elevated levels of body mass index (BMI), lipoprotein-a [Lp(a)] and CAVS have been reported16, we performed multivariable MR additionally adjusted for BMI, and Lp(a) to further check the possible effect of LDL-C, and TG on CAVS. Genetic summary for Lp(a), and BMI have been obtained from the UK Biobank resource.
Drug-target MR (DTMR)
We retrieved recent guidelines for dyslipidaemia management and relevant reviews on lipid metabolism to compile a list of lipid-lowering drugs (e.g., statins, ezetimibe, PCSK9 inhibitors, fibrates, angiopoietin-like 3 (ANGPTL3) inhibitors and antisense oligonucleotide targeting apolipoprotein C-III (APOC3) mRNA)21,22,23 and subsequently used the DrugBank database (https://go.drugbank.com/) to identify target genes of these drugs (Supplementary table S3). These target genes were then classified as LDL-C-lowering targets (HMGCR, NPC1L1, PCSK9, ACLY, CETP, APOB, ABCG5 and ABCG8) and TG-lowering targets (PPARA, ANGPTL3, LPL and APOC3) based on their pharmacological effect. In the primary drug-target MR analysis, single-nucleotide polymorphisms (SNPs) were identified within ± 100 kb windows from 12 target gene and were robustly associated with lipids (e.g., LDL-C, and TG) levels at genome-wide significance. SNPs used as instruments in DTMR were permitted to be in weak linkage disequilibrium (r2 < 0.10) with each other to increase the proportion of variance in each respective drug target explained by the genetic instrument and maximize instrument strength24.
As a secondary analysis, two-sample MR were performed using cis-pQTL (QTLs that reside within ± 1 Mb region of the gene) and cis-eQTL to further examine results identified in the primary analyses. Total cholesterol was excluded from this analysis due to its observable overlapping with LDL-C. Given that most drug targets are proteins, we firstly estimated the association between genetically proxied plasma levels of potential drug targets (instrumented using cis-pQTLs) and lipids using genetic instruments obtained from three plasma proteomics studies (deCODE database with 4719 proteins measured in 35,559 individuals25, Fenland database with 4979 proteins measured in 10,708 individuals26 and UKB-PPP database with 2923 proteins measured in 54,219 individuals27. Then, we examined the associations between genetically proxied plasma level of the drug target (instrumented using cis-pQTLs) and CAVS. Finally, the cis-eQTL MR analysis was repeated using genetic instruments for relevant tissues derived from the Genotype-Tissue Expression project (GTEx-V8)28. We also calculated the LD correlation r2 between eQTLs and pQTLs using LDmatrix Tool (https://ldlink.nci.nih.gov/).
Colocalisation analysis
To evaluate the robustness of positive findings to confounding by linkage disequilibrium, we conducted Bayesian colocalisation with default parameters. Coloc provides the posterior probability for five hypotheses: no causal variant for neither trait (PP.H0), a causal variant only for the first trait (PP.H1), a causal variant only for the second trait (PP.H2), different causal variants underlying each trait (PP.H3), and a shared causal variant associated with both traits (PP.H4)29. Co-localisation analysis was performed by generating ± 100 kb windows around the gene region for drug target, and a posterior probability for the fifth hypothesis > 80% was considered as evidence of colocalization.
Statistical analysis
To test the validity of the instrumental variable for genetically proxied drug targets, positive control analysis were performed with coronary artery disease (CAD) as outcome. In estimating the causal effects of serum lipids on CAVS using TSMR design, the inverse-variance weighted (IVW) random-effects models was used as the primary analysis method. For drug-target MR, causal estimates were generated using random-effects IVW method after adjustment of LD structure between genetic variants. The Wald ratio method was used to estimate causal effect with a single instrumental variable available. To examine the robustness of the results and identify potential horizontal pleiotropy, we conducted sensitivity analyses which each make different assumptions using the weighted median, MR-Egger, weighted mode and MR pleiotropy Residual Sum and Outlier (MR-PRESSO)30. We repeated the analyses only using SNPs associated with no other lipid traits as genetic instruments for each lipids. The heterogeneity and pleiotropy between SNPs were evaluated by Cochran’s Q test and the MR-Egger intercept test, respectively. The strength of genetic instruments was assessed with F-statistics and an F-statistic > 10 indicates minimal weak instrument bias31. The MR Steiger test was carried out to establish the directionality. For a drug target associated with the risk of CAVS, colocalisation analysis was then performed. We repeated drug-target MR for significant associations using genetic variants with more stringent LD clumping threshold (r2 < 0.01 and r2 < 0.001, respectively) to test the robustness of our results. The effect size was scaled to per standard deviation (SD) changes in exposures.
To examine whether the observed associations between genetically proxied inhibition of drug targets and CAVS risk could be attributed to a potential mediatory pathway (i.e. BMI, Lp(a), or ApoB)16,17, we performed two-sample MR analysis using the same variant selection criteria (P < 5 × 10−8 and r2 < 0.001). The effect of the mediator on the outcome was assessed with the product of coefficients method. Standard errors for the effects of genetically proxied lipid-modifying targets on CAVS risk via each potential mediator were derived with the delta method32. The mediated proportions were calculated by dividing the effect of the mediator by the total effect estimate of drug target on CAVS.
To detect potential reverse causality, reverse MR used CAVS summary data from the TARGET consortium as exposure and the plasma LDL-C and PCSK9 proteins as outcome data were utilized.
Finally, iterative leave-one-out analysis was conducted for PCSK9 by removing 1 SNP at a time from instruments to identify the presence of single influential variants.
Bonferroni-corrected significance levels of p-value < 0.0125 (0.05/4) and p-value < 4.16 × 10−3 (0.05/12, [12 drug targets tested]) were used to account for multiple testing. When the estimates were generated from multiple datasets of exposure and outcome, the combined effects were obtained using meta approach. All analyses were two-sided and were conducted using the ‘TwoSampleMR’ (V.0.5.7), ‘MendelianRandomization’ (V.0.9.0), ‘meta’ (V.6.5-0), and ‘Coloc’ (V.5.2.0) packages in R (version 4.3.0).
Results
Genetic variant selection
After clumping, we identified 374 SNPs for TC, 415 for HDL-C, 313 for LDL-C and 370 for TG (Supplementary table S4). Genetically proxied levels of lipids were significantly associated with CAD33,34 (Supplementary table S5). In drug-target MR, we evaluate 12 lipid-modifying targets’ effects on CAVS, namely HMGCR, NPC1L1, PCSK9, CETP, APOB, APOC3, ACLY, ANGPTL3, LPL, ABCG5, ABCG8, and PPARA. Genetic instruments for each of lipid-modifying targets are presented in Supplementary table S6. The positive control analyses identified associations in the expected direction between genetically proxied drug targets and CAD, ensuring the validity of the genetic instruments (Supplementary table S7). Characteristics of genetic variants used to instrument the cis-pQTL for plasma levels of PCSK9 protein and liver-derived cis-eQTL for PCSK9 gene are presented in Supplementary table S8. The F-statistics of all selected genetic instruments for lipids levels, and drug targets including the eQTL and pQTLs, ranged from 26.4 to 15850.2, indicating low risk of weak instrument bias.
Genetically causal associations of serum lipids with CAVS
There was limited evidence of an effect of genetically proxied HDL-C on CAVS after multiple testing correction (P = 0.14). We found genetically proxied TG levels were significantly associated with CAVS risk (IVW MR odds ratio (OR) = 1.27; 95% confidence interval (95% CI) = 1.18–1.36, Fig. 2). Significant associations between genetically higher serum TC and LDL-C and increased risk of CAVS were discovered in both TARGET and FinnGen datasets (Supplementary table S9). As estimated by the IVW method, the pooled odds of CAVS was 1.65 (95% CI = 1.53–1.77) for 1-SD increase in TC, and 1.53 (95% CI = 1.42–1.65) for 1-SD increase in LDL-C (Fig. 2). Given the collinearity between lipids traits, there is overlap between instruments for the four lipids traits (12%). We therefore repeated the analyses only using unique instrumental variables (241 SNPs for TC, 191 SNPs for LDL-C, 333 SNPs for TG and 376 for HDL-C) associated with no other lipid traits and all association remained (Supplementary table S10).

Causal effects of genetically proxied lipids on calcific aortic valve stenosis.
Moreover, results from multivariable MR analysis suggested that LDL-C (OR = 1.41, 95% CI = 1.31 to 1.51) were independent risk factors of CAVS after adjusting for known risk factors, including LP(a) and BMI (Fig. 3); whereas the association with triglycerides was attenuated (OR = 1.09, 95% CI = 1.02 to 1.21).

Results of multivariable Mendelian randomisation analysis of serum lipids and calcific aortic valve stenosis. (A) CAVS–LDL-C + Lp(a) + BMI. (B) CAVS–TG + Lp(a) + BMI.
Mendelian randomisation analysis of lipid-modifying targets and CAVS
For lipids traits associated with CAVS after multiple testing correction in univariable MR, we applied drug-target MR to investigate the association of genetically proxied lipid-modifying drug targets with CAVS risk from two independent datasets (Supplementary table S11). For drug targets that reached significance after multiple testing correction in both datasets, we then performed colocalization analyses to test the exclusion restriction assumption. Genetic mimicry of LPL and APOC3 enhancement equivalent to a 1-SD reduction in TG was significantly associated with lower CAVS risk. As shown in Supplementary table S12, the second hypothesis was the most likely (87%) in colocalisation analysis of ABCG5, suggesting that colocalisation finding may be underpowered. Besides, lowering of LDL-C caused by targeting ABCG5 should reduce the risk of CAVS (Table 1). However, there was weak evidence from colocalization analyses to support these associations (Supplementary table S12).
Genetically proxied inhibition of PCSK9 was strongly associated with lower risk of CAVS in both TARGET and FinnGen datasets with pooled OR of 0.63 (95% CI = 0.56–0.70, P = 1.13 × 10−16, per SD reduction in LDL-C). Colocalisation analyses suggested that the PCSK9 region was shared by LDL-C and CAVS (PP.H4 = 99.3%) (Table 1), strengthening the observed causal effects of targeting PCSK9 on CAVS (Fig. 4). Additional analysis for the LD threshold with more stringent thresholds (r2 < 0.01 and r2 < 0.001) provided consistent evidence of an association between genetically proxied PCSK9 inhibition and decreased CAVS risk (Supplementary table S13). A total of 16 independent SNPs were used as instrumental variables for genetic predicted CAVS (Supplementary table S14), reverse MR shown that neither plasma LDL-C nor the PCSK9 proteins appeared to be causally influenced by genetically predicted CAVS (Supplementary table S15).

Genetic colocalization between PCSK9 and calcific aortic valve stenosis risk. (A) Colocalisation between LDL-C and calcific aortic valve stenosis in the PCSK9 region. (B) Colocalization analyses of cis-pQTL from deCODE for PCSK9. (C) Colocalization analyses of cis-pQTL from Fenland for PCSK9. (D) Colocalization analyses of cis-pQTL from UKB-PPP for PCSK9.
We found limited evidence of an genetic mimicries of other drug targets (HMGCR, ANGPTL3, NPC1L1, CETP, APOB, APOC3, ACLY, LPL, PPARA, ABCG5, and ABCG8) on CAVS (Supplementary table S11).
Triangulation of evidence using pQTL and eQTL data
Leveraging proteomic GWAS summary data from the deCODE, the Fenland cohorts and the UKB-PPP study, we performed two-sample MR analyses using cis-acting pQTLs to instrument inhibition of circulating PCSK9 protein levels to further examine the association between PCSK9 and CAVS. These cis-pQTLs are strongly associated with LDL-C levels (Supplementary table S16). MR results provided consistent evidence of an protective effect of lower levels of circulating PCSK9 protein on CAVS with a pooled OR of 0.86 (95% CI = 0.83–0.88, P = 3.38 × 10−21, Fig. 5), which in line with findings from our initial analyses. The results of the alternative MR methods were generally consistent (Supplementary table S17, Supplementary figure S1). Neither heterogeneity nor horizontal pleiotropy was detected (Supplementary table S18). Besides, results from leave-one-out analyses suggested that the overall estimate was not driven by a single influential variant, supporting the robustness of our findings (Supplementary figure S2). The funnel plot showed symmetric variation in effect size around the point estimate (Supplementary figure S3). Repeating the MR analysis using cis-pQTLs with more stringent thresholds (r2 < 0.01 and r2 < 0.001) on CAVS risk provided a very similar magnitude of effect (Supplementary table S13). We then assess genetic colocalization of PCSK9 protein with CAVS. PCSK9 protein showed strong evidence of colocalization in deCODE (PP.H4 = 99.4%), Fenland (PP.H4 = 99.4%) and UKB-PPP (PP.H4 = 99.4%) with the same causal variant (rs11591147) (Fig. 4, Supplementary table S12).

External validation of the causal relationship between PCSK9 and calcific aortic valve stenosis using data from (A) cis-eQTL and (B) cis-pQTLs.
Because PCSK9 is strongly expressed in the liver, which is the organ where LDL-C removal occurs, we further examine genetically proxied PCSK9 expression (instrumented using cis-eQTL) and CAVS. As none cis-eQTL in liver tissue reached genome wide significance based on the Genotype-Tissue Expression (GTEx), only one independent (r2 < 0.1) SNP in the PCSK9 gene with sub-genome-wide significance (P = 6.02 × 10−7) was available as an instrumental variable (rs553741), which is in strong LD with one of the SNPs used as cis-pQTLs of PCSK9 protein (rs472495, r2 = 0.86, Supplementary table S19). MR analyses using the cis-eQTL estimated an consistent association between lower level of liver PCSK9 expression and lower risk of CAVS with a pooled OR of 0.94 (95% CI = 0.88–1.00, P = 0.05, Fig. 5).
Mediation analysis
We hypothesised that the association between genetically proxied PCSK9 inhibition and CAVS may be mediated through established risk factors for CAVS, such as BMI, Lp(a), and ApoB. Therefore, we performed two-step MR analysis to investigate the mediating pathway from PCSK9 inhibition to CAVS.
MR analyses provided little evidence for genetically proxied inhibition of PCSK9 and BMI (Beta = −0.02, 95% CI = −0.042 to 0.002, P = 0.07, SD change in BMI per SD reduction in LDL-C, Supplementary table S20). However, genetically proxied inhibition of PCSK9 contributed significant alterations in Lp(a) (Beta = −0.08, 95% CI = −0.11 to −0.05, P = 2.97 × 10−7, SD change in Lp(a) per SD reduction in LDL-C) and a much greater extent in ApoB (Beta = −0.73, 95% CI = −0.78 to −0.67, P = 2.60 × 10−138, SD change in ApoB per SD reduction in LDL-C). Replication using PCSK9 pQTLs as genetic instruments on Lp(a) and ApoB provided similar estimates (Supplementary table S21-S22).
In addition, we examined the association between genetically proxied Lp(a) and ApoB levels on CAVS risk using independent genetic instruments across the genome (r2 < 0.001, P < 5 × 10−8). Following IVW MR results, there was a strong effect of genetically proxied Lp(a) (Beta = 0.26, 95% CI = 0.22–0.30, Supplementary table S23) and ApoB levels (Beta = 0.36, 95% CI = 0.24–0.48) on CAVS risk (Supplementary table S24).
We found that plasma Lp(a) levels partially mediated the total effect of genetically proxied inhibition of PCSK9 on CAVS (proportion mediated = 4.5%, P = 1.77 × 10−6), while lowering ApoB levels mainly mediated the association (mediation proportion: 55.9%, P = 9.56 × 10−9, Fig. 6).

Mediation analysis of the effect of genetically proxied PCSK9 inhibition on calcific aortic valve stenosis via potential mediators.
In replication analyses using PCSK9 cis-pQTLs as the genetic instruments, we evaluated whether Lp(a) or ApoB mediates the total effect of plasma level of PCSK9 protein on CAVS (Supplementary figure S4). We also found consistent results for plasma PCSK9 level and ApoB mainly mediated the total effect of PCSK9 levels on CAVS (proportion mediated = 69.1%, P = 1.86 × 10−7).
Discussion
In this MR analysis of 24,811 CAVS cases and 1,082,788 controls, we found strong evidence for an association of lifelong exposure to high LDL-cholesterol, and total cholesterol with a higher risk of CAVS. The drug-target MR analyses suggested that therapeutic inhibition of lipid-lowering drug target PCSK9 may reduce risk of CAVS, which was corroborated by colocalisation. Using cis-acting pQTLs measured in deCODE, Fenland, and UKB-PPP, we discovered that genetically predicted lower levels of circulating PCSK9 protein was associated with an decreased CAVS risk. Estimates based on PCSK9 expression data from liver further support this finding.
Previously, population-based cohort study and Mendelian randomization study found similar evidence regarding the association of triglycerides and aortic stenosis risk4,5. It should be noted that these previous analyses were harassed by the relatively small number of cases (< 2000 cases) and insufficient power due to limited numbers of independent genetic instruments available for triglycerides. Although the apparent causal association between triglycerides and aortic stenosis was detected in univariable MR, the results were attenuated after adjusting for other lipid markers which is in line with previous MR study by Nazarzadeh et al., implying triglycerides may not an independent risk factor.
Beyond the well-established function of PCSK9 in regulation of cholesterol metabolism, its role in CAVS has yet to be comprehensively evaluated and characterized. Circulating PCSK9 levels have previously been linked with bioprosthetic aortic valve degeneration35,36. Perrot and colleagues reported that PCSK9 is detectable only in close proximity to cell nuclei in healthy aortic valves, whereas PCSK9 is highly abundant in in stenotic ones37. Additionally, animal experiments indicated that PCSK9 knockout mice are less likely to develop aortic valve calcification compared to wild-type mice38. Furthermore, rare variants with stronger effects in PCSK9 were recently identified as a susceptibility locus for CAVS and aortic valve replacement39. These preclinical studies highlight the role of PCSK9 in the etiology of CAVS, and our study further unveiled that targeting PCSK9 should reduce the risk of CAVS, implicating that alirocumab might help to protect against it. Further investigations from clinical trials are needed to confirm this finding.
Additionally, we performed subsequent analyses to explore the potential mediatory role of BMI, Lp(a), and ApoB in the associations between PCSK9 and CAVS risk. Although observational studies40,41 and MR study42 identified evidence for an positive association between BMI and CAVS, our results found little evidence supporting effects of genetically proxied inhibition of PCSK9 on BMI. The evidence for associations of Lp(a) and ApoB with CAVS was replicated in our study. In mediation analysis, we found evidence that PCSK9 inhibition may lower levels of Lp(a). However, the estimated proportion mediated is at best modest (4.5%). This finding has also been supported by randomised control trials which shown that PCSK9 inhibition with evolocumab provides modest (16%) reduction in Lp(a) levels in patients with high Lp(a) levels43. Furthermore, genetically proxied PCSK9 exhibited stronger magnitude of association with ApoB (55.9%) than that of Lp(a), suggesting that PCSK9 inhibitors may exert their cardiovascular benefits mainly through reduction of apoB-containing lipoprotein particles rather than Lp(a) reduction. This is consistent with the notion that CAVS shares similar pathobiology with atherosclerotic vascular disease, involving intimal accumulation of apoB-containing particles, inflammation, and calcification17,44,45. Although our findings suggest that ApoB and Lp(a) may play a mediatory role for the protective effect of PCSK9 inhibition on CAVS, further functional research is needed to elucidate underlying mechanisms.
Recent studies together with ours using different IVs construction strategy have all identified the protective role of PCSK inhibitor on CAVS46,47,48, which further confirmed the validity of our finding. However, we systematically depict the relationship between serum lipids, lipid- modifying targets and CAVS in a robust way. In addition, we performed subsequent analyses to explore the potential mediatory role of BMI, Lp(a), and ApoB in the associations between potential drug targets and CAVS risk. Besides, we combined data from different outcome sources, which greatly increased the power of our study. Moreover, we integrated multi-omic data including transcriptomics and proteomic to confirm the causal links between the identified proteins and CAVS.
As for other lipid-modifying targets analyzed in our study, not significant associations were identified in either MR or colocalisation analyses. Seveal reasons may explain such pehnomen. It’s probability that the impact of these targets on cholesterol levels in serum, cholesterol in bile, and the absorption of cholesterol from the intestine would eventually yield null effects on cholesterol oversaturation49,50. In addtion, mechanism-specific effect of PCSK9 inhibition may also result in such result. The inhibition of mevalonate synthesis by PCSK9 inhibitor in the cholesterol biosynthesis pathway may lower levels apolipoprotein B-100-containing lipoproteins, including Lp(a), which has been found to be strongly correlated with the risk of CAVS in both observational and MR studies51,52. However, statins and Ezetimibe had little to mild effects on Lp(a) as reported by randomised control trials53. Furthermore, statistical power can be limited in colocalization analysis, which can result in low posterior probabilities for shared causal variants. Therefore, failure in the co-localisation finding of ABCG5 and CAVS may reflect limited power.
Among the strengths of our analysis is the strict instrument selection and validation process employed. By using cis-acting variants with F-statistics > 10, horizontal pleiotropy should be minimised and we showed the validity of our instrument from positive control study. Besides, the ineffectiveness of statin therapy on CAVS was successfully predicted in our study using genetic instruments mimicry of HMGCR inhibition, which provides additional confidence to our results. Additionally, our MR findings of the alternative MR methods were generally consistent and were supported by colo-calization, reverse MR, various proteomic GWAS, and transcriptome-wide association study in liver samples. Furthermore, our MR findings withstood multiple sensitivity analyses including leave-one-out analyses and validation analysis with more stringent LD thresholds. Moreover, we included replication analyses using datasets from different resources (i.e. CAVS, pQTL) where possible.
Despite the strengths of the study, some limitations should be considered. Firstly, we cannot definitively rule out the possibility of horizontal pleiotropy. Nevertheless, various sensitivity analyses were conducted to test the assumptions of MR analyses and colocalization analysis was also performed to exclude bias caused by linkage disequilibrium. Secondly, the genetic variants used as a proxy for the effect of PCSK9 inhibition predicted one standard deviation change, and thus our mendelian randomisation estimates may not be equivalent to those reported by randomised controlled trials. This is because drugs are often taken for a short period, whereas germline genetic variants represent the effect of lifelong changes in lipid levels. Thirdly, as there were no genome-wide significant genetic instrumental variables for gene expression around the cis-region of PCSK9, we may have limited power to detect the effect of PCSK9 expression on CAVS even though we combined outcome data from different sources. Additionally, the gene-environment or gene-gene interactions, off-target and nonlinearity effects of drug targets could not be assessed on the basis of the summary-level data. Lastly, GWAS data sets used in the present MR only included individuals of European ancestry, which limits the generalizability of these findings to non-European populations.
Conclusion
In summary, our study demonstrates that high LDL-cholesterol, and total cholesterol are at increased risk for CAVS and the use of PCSK9 inhibitors may protect against CAVS, potentially through a mechanism involving the lowering of ApoB levels. The underlying mechanisms should be elucidated in further research, and clinical evidence for the pharmacological effect of PCSK9 inhibitors on CAVS incidence and progression is also needed.
Data availability
Data are available in a public, open access repository. The latest GLGC GWAS summary statistics can be accessed at http://csg.sph.umich.edu/willer/public/glgc-lipids2021/. The summary statistics of FinnGen GWAS data can be accessed at https://www.finngen.fi/en. Gene expression eQTL data is available from the GTEx project via https://www.gtexportal.org/home/. The summary level statistics from TARGET GWAS can be accessed via https://doi.org/10.5281/zenodo.7505361. Plasma proteome from the deCODE study is available from https://www.decode.com/summarydata/. Summary statistics of plasma proteome from Fenland study can be obtained from www.omicscience.org/apps/pgwas. Proteo-genomic summary association of UKB-PPP study is available from http://ukb-ppp.gwas.eu. Several summary statistics of genome wide association studies (GWAS) used in this study are publicly available on the MRC Integrative Epidemiology Unit (IEU) Open GWAS project (https://gwas.mrcieu.ac.uk/).
Abbreviations
- HDL-C:
-
high-density lipoprotein cholesterol
- LDL-C:
-
low-density lipoprotein cholesterol
- TG:
-
triglyceride
- TC:
-
total cholesterol
- CAVS:
-
calcific aortic valve stenosis
- MR:
-
Mendelian randomisation
- MVMR:
-
multivariable MR
- TSMR:
-
two-sample MR
- DTMR:
-
drug-target MR
- BMI:
-
body mass index
- pQTL:
-
protein quantitative trait loci
- eQTL:
-
expression quantitative trait loci
- UKB:
-
UK Biobank
- GLGC:
-
Global Lipids Genetics Consortium
- TARGET:
-
Therapeutic targets for AoRtic stenosis using GEneTics
- HMGCR:
-
hydroxymethylglutaryl-CoA reductase
- NPC1L1:
-
Niemann-Pick C1-like protein 1
- LPL:
-
Lipoprotein Lipase
- PCSK9:
-
proprotein convertase subtilisin/kexin type 9
- CETP:
-
cholesteryl ester transfer protein, APOB: apolipoprotein B
- APOC3:
-
apolipoprotein C-III
- ACLY:
-
ATP citrate lyase
- ANGPTL3:
-
angiopoietin like 3
- ABCG5:
-
ATP Binding Cassette Subfamily G Member 5
- ABCG8:
-
ATP Binding Cassette Subfamily G Member 8
- PPARA:
-
peroxisome proliferator-activated receptor-alpha
- SD:
-
standard deviation
- OR:
-
odds ratio
- CI:
-
confidence interval
- SNP:
-
single-nucleotide polymorphisms
- IVW:
-
inverse variance weighted
- LD:
-
linkage disequilibrium.
References
Yadgir, S. et al. Global burden of disease study 2017 nonrheumatic valve disease collaborators. Global, regional, and National burden of calcific aortic valve and degenerative mitral valve diseases, 1990–2017. Circulation 141, 1670–1680 (2020).
Baumgartner, H. et al. 2017 ESC/EACTS guidelines for the management of valvular heart disease. Eur. Heart J. 38, 2739–2791 (2017).
Smith, J. G. et al. Cohorts for heart and aging research in genetic epidemiology (CHARGE) extracoronary calcium working group. Association of low-density lipoprotein cholesterol-related genetic variants with aortic valve calcium and incident aortic stenosis. JAMA 312 (17), 1764–1771 (2014).
Kaltoft, M., Langsted, A. & Nordestgaard, B. G. Triglycerides and remnant cholesterol associated with risk of aortic valve stenosis: Mendelian randomization in the Copenhagen general population study. Eur. Heart J. 41 (24), 2288–2299 (2020).
Nazarzadeh, M. et al. Plasma lipids and risk of aortic valve stenosis: a Mendelian randomization study. Eur. Heart J. 41 (40), 3913–3920 (2020).
Cowell, S. J. et al. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl. J. Med. 352, 2389–2397 (2005).
Rossebø, A. B. et al. Intensive lipid Lowering with Simvastatin and Ezetimibe in aortic stenosis. N Engl. J. Med. 359, 1343–1356 (2008).
Chan, K. L., Teo, K., Dumesnil, J. G., Ni, A. & Tam, J. ASTRONOMER investigators. Effect of lipid Lowering with Rosuvastatin on progression of aortic stenosis: results of the aortic stenosis progression observation: measuring effects of Rosuvastatin (ASTRONOMER) trial. Circulation 121, 306–314 (2010).
Moore, M. K., Jones, G. T., McCormick, S., Williams, M. J. A. & Coffey, S. Association between lipoprotein(a), LPA genetic risk score, aortic valve disease, and subsequent major adverse cardiovascular events. Eur. J. Prev. Cardiol. 31 (10), 1303–1311 (2024).
Satterfield, B. A. et al. Associations of genetically predicted Lp(a) (Lipoprotein [a]) levels with cardiovascular traits in individuals of European and African ancestry. Circ. Genom Precis Med. 14 (4), e003354 (2021).
Arsenault, B. J. et al. Lipoprotein(a) and calcific aortic valve stenosis progression: A systematic review and Meta-Analysis. JAMA Cardiol. 9 (9), 835–842 (2024).
Sanderson, E. et al. Mendelian randomization. Nat. Rev. Methods Primers. 2 (1), 6 (2022).
Ference, B. A. et al. Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. N Engl. J. Med. 375 (22), 2144–2153 (2016).
Williams, D. M., Finan, C., Schmidt, A. F., Burgess, S. & Hingorani, A. D. Lipid Lowering and alzheimer disease risk: a Mendelian randomization study. Ann. Neurol. 87 (1), 30–39 (2020).
Fang, S. et al. Richardson tg.association between genetically proxied PCSK9 Inhibition and prostate cancer risk: A Mendelian randomisation study. PLoS Med. 20 (1), e1003988 (2023).
Yu Chen, H. et al. Dyslipidemia, inflammation, calcification, and adiposity in aortic stenosis: a genome-wide study. Eur. Heart J. 44 (21), 1927–1939 (2023).
Small, A. M. et al. Multiancestry Genome-Wide association study of aortic stenosis identifies multiple novel loci in the million veteran program. Circulation 147 (12), 942–955 (2023).
Skrivankova, V. W. et al. Strengthening the reporting of observational studies in epidemiology using Mendelian randomization: the STROBE-MR statement. JAMA 326 (16), 1614–1621 (2021).
Graham, S. E. et al. The power of genetic diversity in genome-wide association studies of lipids. Nature 600, 675–679 (2021).
Kurki, M. I. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613 (7944), 508–518 (2023).
Mach, F. et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur. Heart J. 41 (1), 111–188 (2020).
Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 139 (25), e1082–e1143 (2019).
Borén, J., Taskinen, M. R., Björnson, E. & Packard, C. J. Metabolism of triglyceride-rich lipoproteins in health and dyslipidaemia. Nat. Rev. Cardiol. 19 (9), 577–592 (2022).
Woolf, B. et al. A drug target for erectile dysfunction to help improve fertility, sexual activity, and wellbeing: Mendelian randomisation study. BMJ 383, e076197 (2023).
Ferkingstad, E. et al. Large-scale integration of the plasma proteome with genetics and disease. Nat. Genet. 53, 1712–1721 (2021).
Pietzner, M. et al. Mapping the proteo-genomic convergence of human diseases. Science 374, eabj1541 (2021).
Sun, B. B. et al. Plasma proteomic associations with genetics and health in the UK biobank. Nature 622 (7982), 329–338 (2023).
The GTEx Consortium. The GTEx consortium atlas of genetic regulatory effects across human tissues. Science 369 (6509), 1318–1330 (2020).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10 (5), e1004383 (2014).
Morrison, J. et al. Mendelian randomization accounting for correlated and uncorrelated pleiotropic effects using genome-wide summary statistics. Nat. Genet. 52, 740–747 (2020).
Staiger, D. & Stock, J. H. Instrumental variables regression with weak instruments. Econometrica 65 (3), 557–586 (1997).
Sanderson, E. Multivariable Mendelian randomization and mediation. Cold Spring Harb Perspect. Med. 11 (2), a038984 (2021).
Richardson, T. G. et al. Characterising metabolomic signatures of lipid-modifying therapies through drug target Mendelian randomisation. PLoS Biol. 20 (2), e3001547 (2022).
Wang, Q. et al. Metabolic profiling of angiopoietin-like protein 3 and 4 inhibition: a drug-target Mendelian randomization analysis. Eur. Heart J. 42 (12), 1160–1169 (2021).
Salaun, E. et al. Hemodynamic deterioration of surgically implanted bioprosthetic aortic valves. J. Am. Coll. Cardiol. 72, 241–251 (2018).
Nsaibia, M. J. et al. Association between plasma lipoprotein levels and bioprosthetic valve structural degeneration. Heart 102, 1915 (2016).
Perrot, N. et al. Genetic and in vitro inhibition of PCSK9 and calcific aortic valve stenosis. JACC Basic. Transl Sci. 5 (7), 649–661 (2020).
Poggio, P. et al. PCSK9 involvement in aortic valve calcification. J. Am. Coll. Cardiol. 72, 3225–3227 (2018).
Rämö, J. et al. Rare genetic variants in ldlr, apob, and PCSK9 are associated with aortic stenosis. Circulation 150 (22), 1767–1780 (2024).
Kjeldsen, E. W. et al. Cardiovascular risk factors and aortic valve stenosis: towards 10-year absolute risk charts for primary prevention. Eur. J. Prev. Cardiol. https://doi.org/10.1093/eurjpc/zwae177 (2024).
Larsson, S. C., Wolk, A., Håkansson, N. & Bäck, M. Overall and abdominal obesity and incident aortic valve stenosis: two prospective cohort studies. Eur. Heart J. 38 (28), 2192–2197 (2017).
Kaltoft, M., Langsted, A. & Nordestgaard, B. G. Obesity as a causal risk factor for aortic valve stenosis. J. Am. Coll. Cardiol. 75, 163–176 (2020).
O’Donoghue, M. L. et al. Lipoprotein(a), PCSK9 Inhibition and cardiovascular risk: insights from the FOURIER trial. Circulation 139, 1483–1492 (2019).
O'Brien, KD et al. Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of ‘degenerative’ valvular aortic stenosis. Arterioscler. Thromb. Vasc Biol. 16 (4), 523–532 (1996).
Siudut, J. et al. Apolipoproteins and lipoprotein(a) as factors modulating fibrin clot properties in patients with severe aortic stenosis. Atherosclerosis 344, 49–56 (2022).
Hou, Y. et al. Genetic proxy of lipid-lowering drugs and calcific aortic valve stenosis: A Mendelian randomization study. Heliyon 10 (13), e34089 (2024).
Yang, L. et al. Causal relationships between Lipid-Lowering drug target and aortic disease and calcific aortic valve stenosis: A Two-Sample Mendelian randomization. Rev. Cardiovasc. Med. 25 (8), 292 (2024).
Xu, D. et al. Identifying novel drug targets for calcific aortic valve disease through Mendelian randomization. Atherosclerosis 402, 119110 (2025).
Temel, R. E. et al. Hepatic Niemann- pick C1- like 1 regulates biliary cholesterol concentration and is a target of Ezetimibe. J. Clin. Invest. 117, 1968–1978 (2007).
Sudhop, T. et al. Inhibition of intestinal cholesterol absorption by Ezetimibe in humans. Circulation 106, 1943–1948 (2002).
Arsenault, B. J. et al. Lipoprotein(a) levels, genotype, and incident aortic valve stenosis: a prospective Mendelian randomization study and replication in a case-control cohort. Circ. Cardiovasc. Genet. 7 (3), 304–310 (2014).
Bhatia, H. S. et al. Oxidized phospholipids and calcific aortic valvular disease. J. Am. Coll. Cardiol. 84 (25), 2430–2441 (2024).
Robinson, J. G. et al. Efficacy and safety of Alirocumab in reducing lipids and cardiovascular events. N Engl. J. Med. 372 (16), 1489–1499 (2015).
Acknowledgements
We are grateful to all participants and investigators from the Genetic Investigation of deCODE Consortium, Fenland Consortium, FinnGen cohort, UK Biobank, UKB-PPP consortium, Global Lipids Genetics Consortium, GTEx Consortium and the Therapeutic targets for AoRtic stenosis using GEneTics (TARGET) Consortium for sharing GWAS summary statistics. We would like to thank all the other investigators for making summary statistics openly available.
Author information
Authors and Affiliations
Contributions
CGW, QP, ZC, and LY designed the study and initial analysis plan. CGW, QP, ZC, and LY contributed to the statistical analysis, performed the data analysis, and wrote the draft of the manuscript. YLZ, JJ, YL, and YXH contributed to discussion and revision of the manuscript. CGW, QP, ZC, and LY critically reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and informed consent
All GWAS studies had been approved by its Ethical Review Committee and permits the use of summary data without restriction. Human subjects were not included in this study and participant consent was not required for this study because all summary-level genetic data were obtained from sources available to the public.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, C., Pu, Q., Zou, Y. et al. Genetic insights into causal effects of lipids and lipid-modifying targets on calcific aortic valve stenosis: a Mendelian randomized study. Sci Rep 15, 29475 (2025). https://doi.org/10.1038/s41598-025-15525-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-15525-4
