
Abstract
Cervical high-grade squamous intraepithelial lesion (HSIL) is a well-recognized precursor to cervical squamous cell carcinoma (CSCC). This study aims to explore molecular mechanisms underlying this transition. The datasets of HSIL and CSCC were downloaded from gene expression omnibus database (GEO). HSIL and CSCC coexpression modules were analyzed by Weighted gene coexpression network analysis (WGCNA). Enrichment analysis were analyzed by Gene Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG). The immune microenvironment (TIME) was analyzed by CIBERSORT. Receiver operating characteristic (ROC) curve, and survival analysis were performed. The binding of FEN1 and its upstream regulator PCNA was determined by protein interaction, correlation analysis and rigid docking. Immune infiltration, immune checkpoint and docking with small molecule inhibitors was also analyzed. In vitro experiments were conducted to investigate the function and underlying mechanisms of FEN1 in HSIL and CSCC. A total of 30 sequentially expressed shared genes were identified as potential drug targets. Immune infiltration revealed an imbalance of M1/M2 macrophages, T γδ cells and T cells CD4 memory resting were negatively associated with tumor progression, forming a pro-cancer effect at the HSIL stage and progressing toward CSCC. The key gene FEN1 was over-expressed in HSIL and CSCC, and correlated with the prognosis of CSCC (P < 0.05). FEN1 interacts with PCNA to regulate macrophage-mediated immune infiltration, increase the risk of immune escape, and decrease the sensitivity to immune checkpoints. Small molecule inhibitor binds to FEN1 docking and inhibits its regulation. FEN1 expression is upregulated in both HSIL and CSCC, with a more pronounced increase in CSCC. Knockdown of FEN1 significantly inhibits the proliferation of cervical cancer cells. This study identifies FEN1 as a biomarker and therapeutic target to truncate or reverse CSCC progression at the HSIL stage.
Similar content being viewed by others


Introduction
This study focuses on the molecular mechanisms underlying the progression from high-grade squamous intraepithelial lesions (HSIL, Table 1) to invasive cervical squamous cell carcinoma (CSCC) driven by persistent high-risk human papillomavirus (HPV) infection. As the fourth most common malignancy in women worldwide, cervical cancer is projected to cause 13,960 new cases and 4310 deaths in the United States in 2023, with nearly 95% of cases directly arising from HSIL progression1. The objective of this research is to transform the current paradigm of cervical cancer prevention and treatment by identifying novel biomarkers capable of specifically blocking HSIL carcinogenesis. Current clinical practice faces three critical challenges: (1) The CIN grading system—the gold standard for HSIL diagnosis—has significant limitations, with interobserver variability in CIN2 diagnosis reaching 40%, often leading to confusion with CIN1/CIN3 lesions2,3; (2) Although p16INK4a has been widely adopted as an ancillary marker, its diagnostic accuracy strictly depends on subjective histopathological assessment4; (3) More alarmingly, even with standard treatment, CIN3 patients still face a 15–23% risk of progression to invasive carcinoma within 13 years, with lifetime risk soaring to 40%5. These clinical dilemmas underscore fundamental flaws in the current biomarker system.
The groundbreaking significance of this study lies in its systematic identification of key driver genes during HSIL-to-CSCC transition from the perspective of epigenetic regulatory networks. Departing from previous isolated investigations of individual biomarkers, we have innovatively developed a multi-omics integration platform, specifically targeting: (1) Molecular signatures capable of objectively distinguishing the true malignant potential of CIN2; (2) Early-warning indicators predictive of treatment-resistant HSIL; and (3) A quantitative diagnostic model independent of morphological interpretation. These innovations will directly address two core issues in current clinical guidelines—“histopathological diagnostic uncertainty” and “prognostic prediction blind spots”—providing novel tools for precision medicine in cervical cancer management.
Materials and methods
Data acquisition and preparation
We obtained the transcriptional profiles of high-grade squamous intraepithelial lesions and squamous-cell carcinoma squamous intraepithelial lesions from the gene expression synthesis (GEO) database (http:/www.ncbi.nlm.nih.gov/geo/). And, these datasets included more than 10 samples, and the subjects had not undergone preoperative chemoradiation. The datasets were analyzed using R 4.2.1 for background correction by using the ComBat function in the SVA package, quantile normalization, and summary. Findings were validated with an CSCC cohort from the Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/).
Evaluation of disparities in gene expression
The Limma algorithm, a linear model for microarray data, is widely used in high-throughput gene expression analysis, especially with microarrays. It incorporates a linear modeling framework that effectively synthesizes variance information, making it robust and adaptable to complex experimental designs and covariates. However, its assumption of homogeneity in gene variance may not be true for heterogeneous datasets. By integrating linear modeling with empirical Bayesian statistics, Limma provides a powerful tool for analyzing high-throughput gene expression data, allowing researchers to extract biologically significant insights related to differential gene expression under various experimental conditions. In this study, Limma was applied to analyze differential gene expression between HSIL and normal tissues, as well as between CSCC and normal tissues within the GSE dataset. A significance threshold of | log2 fold change (FC) |≥ 1 and a P-value cutoff of 0.05 were employed6,7 . The results were visualized using Volcano plots and heatmaps created with the ggplot2 package in R, with blue colors indicating low expression levels and red colors indicating high expression levels.
Construction of weighted gene coexpression network
WGCNA, a systematic biology approach, elucidates patterns of gene coexpression across diverse samples and aids in the identification of synergistic gene sets. This methodology has been instrumental in pinpointing candidate biomarker genes or therapeutic targets by leveraging the interconnectivity of gene sets and their relationship with phenotypes8. In this study, we implemented the R package “WGCNA” to construct a gene coexpression network for normal, HSIL, and CSCC samples. Initially, adjacency matrices were generated using a soft threshold B (b = 7) and gene–gene correlation matrices to quantify the connectivity between nodes. These adjacency matrices were then transformed into topological overlap matrices (TOMs). Subsequently, hierarchical clustering dendrograms were employed to delineate coexpression modules. Finally, the most relevant gene module was selected based on the module eigengene.
Gene enrichment analysis and central gene crossover analysis
To pinpoint potential therapeutic targets, we leveraged PandaOmics (https://pandaomics.com/)9,10,11 , an AI-driven computational platform dedicated to drug discovery and development, which yielded a set of 6,225 candidate cervical cancer-related drug targets12 . These potential targets were then cross-validated against the gene set identified through differential expression analysis (DEGs) and WGCNA using the jvenn online comparison tool.
To validate gene candidacy in HSIL-CC transition, 30 genes were analyzed for protein–protein interactions via STRING and visualized using Cytoscape’s “MCODE” algorithm. STRING integrates various types of evidence (such as literature, experimental data, database records, etc.) and provides comprehensive scores for gene and protein interactions13,14 . Gene Ontology (GO) and KEGG pathway enrichment analyses were conducted through Metascape (http://www.metascape.org/)15 , a comprehensive bioinformatics platform for gene enrichment analysis and functional annotation16, setting P < 0.05 as the significance threshold. Results were visualized using micro-health online network mapping platforms.
To determine the infiltration of immune cells in HSIL and CSCC
CIBERSORTX represents a computational method for quantifying cell population abundance in complex mixtures using bulk RNA-seq data. It references a dataset of cell-specific gene expression profiles, like scRNA-seq, to deduce cell type proportions17. Useful in tumor immune microenvironment studies, it offers a wider range of cell types for quantification, including immune cells. In this study, CIBERSORTX was applied to GSE batch mRNA sequencing data to quantify immune cell subtype abundance.
To quantify and analyze the infiltration of immune cells within the tumor microenvironment, the CIBERSORT was employed to estimate the abundance of tumor-infiltrating immune cells from RNA sequencing data17,18. By employing the principle of linear support vector regression, CIBERSORT deconvolutes the expression matrix of immune cell subtypes to estimate the relative abundance of immune cells. This approach provides data on the expression of 13 common immune infiltrating cells. The relationship between DGEs and immune cell infiltration was explored using Spearman correlation analysis.
Analysis of hub genes
The integrated datasets encompassed GSE6315419, GSE2767820, and GSE780321, including 48 normal, 90 HSIL, and 77 cervical cancer tissues. We have performed rigorous internal validation to ensure the robustness of our findings. The TCGA-CSCC dataset was also utilized for external validation, further supporting the reliability and reproducibility of our findings. Differences in the expression of key genes were identified by Kruskal–wallis Test and Dunn’s Test. Subsequently, the diagnostic potential of these genes was evaluated by calculating the area under the Receiver Operating Characteristic (ROC) curve (AUC). Prognostic significance was verified through SURVIVAL analysis in the TCGA-CSCC dataset using the R SURVIVAL package and log-rank tests. Protein immunohistochemical staining results from the Human Protein Atlas (HPA, http://www.proteinatlas.org) validated gene expression in normal and cancerous tissues22.
GSEA
Gene Set Enrichment Analysis (GSEA) is a statistical method to assess if a predefined set of genes shows coherent and significant differences between two biological states, addressing limitations in traditional gene analysis, especially for high-throughput technologies like single-cell RNA sequencing23. By adopting a pathway-centric approach, GSEA aids in understanding the broader biological context of differential gene expression23,24. In this study, GSEA was performed using the clusterProfiler package (version 4.4.4) to identify pathways and mechanisms potentially involved in HSIL and CSCC. The analysis, performed using sangerbox 3.0, categorized samples into high and low expression groups based on hub gene expression to delineate pathways and molecular mechanisms associated with these conditions.
Docking technology
The three-dimensional structures of FEN1 and PCNA were retrieved from the α-fold protein structure database. Then, the docking tool HDOCK was used to initiate the protein–protein docking simulation25. Following an exploration of the Drugbank database (www.drugbank.ca) to identify small molecule inhibitors that interact with FEN1, the structural files were adapted, and a range of potential conformations between the small ligands and FEN1 were generated using the semi-flexible docking approach implemented in PyRX26. Visualization of the docking outcomes was ultimately performed by discovery studio.
Correlation analysis between diagnostic markers and infiltrating immune cells
The “ggstatsplot” package was employed to conduct Spearman’s correlation analysis between diagnostic markers and infiltrating immune cells27,28. Subsequently, the “ggplot2” package was utilized for the visualization of the obtained results.
. Association analysis of hub genes and immune checkpoint genes
We analyzed the expression of over 30 immune checkpoint genes and correlated them with FEN1 levels, considering P < 0.05 and r > 0.20 as significant positive correlation. A heatmap using Pearson coefficients visually represented the interaction between FEN1 and immune checkpoint genes in CSCC.
Quantitative polymerase chain reaction (qPCR)
Cervical samples were collected from 8 patients who underwent hysterectomy. Samples including cancerous tissues, precancerous lesion tissues, and adjacent healthy tissues were respectively collected. The expression level of FEN1 mRNA was verified by qPCR. Trizol reagent was added to extract the total RNA of the tissues, and the concentration of the RNA samples was determined using an ultraviolet spectrophotometer. According to the instruction manual of the reverse transcription kit (AG, Changsha, China), the RNA was reverse-transcribed into cDNA. Quantitative PCR was performed using the SYBR Pro Taq HS qPCR kit (AG 11701, AG). The primer sequences used in this study were as follows: the upstream primer sequence of FEN1 was 5′-ATCCCTTATCTTGATGCACCC-3′, the downstream primer sequence of FEN1 was 5′-AGTCAGGTGTCGCATTAGC-3′. GAPDH was used as the internal reference gene, with the upstream primer sequence of GAPDH being 5′-AGAAGGCTGGGGCTCATTTG-3′ and the downstream primer sequence of GAPDH being 5′-AGGGGCCATCCACAGTCTTC-3′. The relative mRNA expression level was analyzed using the 2-△△Ct method and normalized with GAPDH to analyze the changes in the FEN1 level.
Protein extraction and western blot
The FEN1 antibody was purchased from ABclonal Biotechnology (China), and the GAPDH antibody was purchased from proteintech (China). To extract proteins, the tissue samples were lysed in RIPA lysis buffer (cwbio, China). The protein concentration was determined using the BCA Protein Assay Kit (Thermo Fisher Scientific, USA). Proteins were separated by SDS–polyacrylamide gel electrophoresis and transferred onto a polyvinylidene fluoride (PVDF) membrane (Millipore). The membrane was blocked with 5% skimmed milk at 4 °C for 2–4 h, and then incubated with the specific anti-FEN1 antibody (1:1000) at 4 °C overnight. On the next day, the membrane was washed three times with TBST, and then incubated with an appropriate amount of horseradish peroxidase (HRP)-conjugated secondary antibody (BOSTER, 1:6000) at 4 °C for 2 h. Finally, the protein bands were visualized using an ECL kit (Biosharp, China).
Immunohistochemical assay
For immunohistochemistry, the sections of cervical tissue specimens fixed with 4% paraformaldehyde and embedded in paraffin were from the same subset of cases used for QPCR measurement. The sections were dewaxed by heating at 65 °C for 60 min and treating with an environmentally friendly dewaxing solution, and then hydrated in graded ethanol. Subsequently, they were heated in a microwave oven for 15 min in 0.01 M citrate buffer (pH 6.0) for antigen retrieval. After cooling for 30 min under running water at room temperature, the sections were then subjected to membrane permeabilization treatment with 0.3% Triton X-100. Non-specific binding sites were blocked with 3% H2O2 and 5% bovine serum albumin at room temperature. Then, the sections were incubated with primary antibodies (FEN1, 1:200, ABclonal Biotechnology catalog number A8999; Ki67, 1:200, proteintech catalog number: 66555-6-Ig) overnight at 4 °C. The sections were incubated with secondary antibodies, stained with DAB solution, and then counterstained with hematoxylin. After the sections were sealed with neutral gum and dried, images were captured using a digital pathology slide scanner (KFBIO, Ningbo, China).
Cell culture and plasmid transfection
The cervical cancer cell lines, Hela and SiHa cells (Guangzhou Saikuk Biotechnology Co., Ltd., China), were cultured in Dulbecco’s Modified Eagle Medium (high glucose) supplemented with 10% fetal bovine serum (FBS; Corning), penicillin (100 U/mL, BI, Israel), and streptomycin (100 μg/mL, BI, Israel). All cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2. The FEN1 1-shRNA plasmids (shFEN1 1 -1, shFEN1 1 -2, shFEN1 1 -3) and the negative control-shRNA plasmid (shNC) were purchased from GeneCopoeia (Guangzhou, China). For plasmid transfection, briefly, Hela cells (3 × 103 cells/well) and SiHa cells (3 × 103 cells/well) were seeded into 96-well plates and cultured for 24 h. Then, 100 ng of plasmids were transfected using Lipo 6000™ (Beyotime Institute of Biotechnology, China). After incubation for 6–8 h, the original medium was replaced with the standard medium.
Cell proliferation assay
The Cell Counting Kit-8 (Biosharp, Hefei, China) was used to detect the cell proliferation ability. At 24 h, 48 h, and 72 h post-transfection, add a mixture of CCK-8 (10 µl) and complete medium (90 µl) to each well of the 96-well plate, followed by incubation at 37 °C for 2 h. Then, the absorbance of the cells was measured at 450 nm using a microplate reader (Biotek, USA).
Ethical approval and consent
This study has been approved by the Ethics Committee of the Hospital of Integrated Traditional Chinese and Western Medicine of Southern Medical University. All aspects of the study are in line with the standards set by the Declaration of Helsinki. Human cervical samples were obtained from individuals who underwent total resection surgery for cervical cancer, and this was carried out in accordance with the approval of the Ethics Committee of the Hospital of Integrated Traditional Chinese and Western Medicine of Southern Medical University (Approval Number: NFZXYEC-2018-00). All sample donors or their legal guardians/agents have signedawritten informed consent form before participating in the study.
Statistical analysis
R programming language (version 4.2.1) facilitated statistical analyses, while Cytoscape (version 3.10.0) and GraphPad Prism (version 9.5.1) were used for graphical representations. The Wilcoxon rank-sum test determined FEN1 expression differences across tissues.
Results
GEO information
Figure 1 depicts the research strategy. In accordance with the established criteria for dataset inclusion, we identified four datasets for the exploratory phase of our study: GSE63514, GSE27678, GSE7803, and TCGA-CSCC29. A comprehensive overview of these datasets’ informational details is provided in Table 2. Preprocessing and normalization procedures were applied to the three GSE datasets, which were subsequently combined for the discovery phase, while the TCGA-CSCC dataset was allocated for validation purposes. This finding provides a basis for subsequent analyses, where we will focus on those genes whose expression is consistently increased in the normal-HSIL-CSCC lineage that may serve as potential biomarkers or therapeutic targets.

Study protocol and methodological framework.
Identify shared genes that are sequentially increasing in HSIL and CSCC
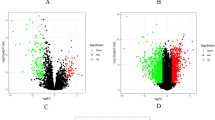
The LIMMA package was used to detect DEGs between HSIL and CSCC30. PCA showed strong concordance between HSIL, CSCC, and normal tissue components, suggesting that the transition from HSIL to CSCC is not primarily due to genetic alterations but rather to changes in oncogene expression, with tumor suppressor factors being suppressed (Fig. 2A). Given these observations, our subsequent analyses will focus on elucidating the specific gene expression changes that contribute to the progression from HSIL to CSCC (Fig. 2B). Elevated oncogenes were prioritized as potential biomarkers specific to HSIL or CSCC stages. Volcano plots and heatmaps revealed 534 DEGs in HSIL versus Normal (266 downregulated, 268 upregulated) (Fig. 2C,D). And there were 408 DEGs in CSCC versus Normal (216 downregulated, 192 upregulated) (Fig. 2E,F). DEGs associated with HSIL and CSCC are involved in the onset and progression of these diseases.

Analysis of Differentially Expressed Genes in HSIL versus CSCC. (A) PCA of normalized data, colored by disease stage, with 95% CI ellipses. (B) Box plots represent the distribution of Pearson correlation coefficients between each stage. Kruskal–Wallis test was used, then Wilcoxon rank sum test and Bonferroni correction test were used to compare the correlations. (C) Volcano plot for HSIL vs Normal, with upregulated (red) and downregulated (blue) genes. (D) Heatmap of Pearson correlations for HSIL vs Normal sample pairs. (E) Volcano plot for CSCC vs Normal, with gene expression changes. (F) Heatmap of Pearson correlations for CSCC vs Normal sample pairs.
We conducted WGCNA analysis, in addition to assessing differential expression between the two groups, to examine the association between disease progression and key genes. The optimal soft threshold was 7. When mean connectivity was tended to 0, the ordinate scale-free fit index, signed R2 approached the threshold value of 0.8 (red line) (Fig. 3A). A total of 10 modules were obtained by the dynamic tree cut algorithm. (Fig. 3B,C). Among these, the blue and purplish red modules demonstrated a progressively positive correlation with the transition from normal tissue to HSIL and then to CSCC (Fig. 3D). The correlation coefficients were 0.38 (P < 0.01) for the blue module and 0.28 (P < 0.01) for the purplish-red module. (Fig. 3E). The stepwise positive correlation of these modules with disease progression suggests that the genes within these modules play a significant role in the transition from normal tissue to HSIL and then to CSCC. Conversely, the brown module exhibited a negative correlation, with a coefficient of − 0.27 (P < 0.01). Our study aims to identify key genes that drive the progression from HSIL to CSCC. The blue and purplish-red modules, with their strong positive correlation and biological relevance, align well with this objective.

Weighted gene coexpression network analysis of HSIL and SCC. (A) Measurement of soft threshold power. (B) Genes significance across modules. (C) Cluster dendrogram of genes highly linked to HSIL and SCC in key modules. (D) Relationship between HSIL and SCC modules and traits. Each cell contains a correlation and a P value. (E) Scatter plot between gene salience (GS) and module members (MM) in blue and magenta.
Identification and functional enrichment analysis of shared genes
To investigate the concurrent pathogenesis of HSIL and CSCC and identify genes with potential therapeutic relevance, we intersected the genes predicted by DEGs, WGCNA, and PandaOmics, resulting in a set of 30 overlapping genes (Fig. 4A). Functional annotation and enrichment analysis of these genes revealed alterations potentially involved in the transition from HSIL to CSCC. KEGG pathway analysis showed enrichment in pathways related to DNA replication, cell cycle, and p53 signaling, crucial processes in cancer development (Fig. 4B). GO analysis confirmed enrichment in terms related to DNA replication and cell cycle processes, repair of double-strand breaks, and cellular components like telomere region and nucleus of chromosomes. Molecular function terms like ATP binding, DNA helicase activity, and DNA binding were enriched, indicating that regulation of DNA replication is a key factor in cancer cell development (Fig. 4C)31,32. Upregulation of these processes during tumorigenesis suggests they may play a pivotal role in the progression from HSIL to CSCC.

Analysis of shared gene function enrichment and immune infiltration in HSIL and SCC. (A) Shared DEG between HSIL and SCC. (B) The shared genes are represented by a sangki map, showing KEGG enrichment. (C) GO of biological processes (BP), cellular component (CC), and molecular functions (MF). (D) In Normal, HSIL and CSCC group, infiltrating immune cells were mapped into superimposed bar graph. (E) Box plot showed that the types of immune cells in Normal, HSIL and SCC groups were significantly different. (F) PPI shows 30 protein interactions. *P < 0.05, **P < 0.01, ***P < 0.001.
Immune infiltrates in HSIL and CSCC
The spatial decon (version 1.8.0) safeTME deconvolution method was employed to analyze the immune infiltrates within HSIL and CSCC samples, aiming to elucidate the influence on the immune microenvironment. This approach facilitates the quantification of immune cell types via spatial transcriptome data. A heat map depicting the abundance of immune cells across normal, HSIL, and CSCC tissues was generated. The proportion of immune cells across the three stages was investigated to uncover the dynamics of immune cell populations during CSCC development.
Findings indicate that the transition from HSIL to CSCC is marked by a predominant expression of resting CD4 T cells and gamma-delta T cells, which are key effectors of the innate immune response (Fig. 4D, E)33,34. The anti-tumor effects appear to wane as their expression diminishes in the context of CSCC progression. Among the three stages, the most pronounced difference was observed in macrophage expression, a cell type that significantly contributes to tumor advancement. Notably, M1 macrophages, which exert anti-cancer effects, were found to be less prevalent in CSCC compared to HSIL, whereas M0 macrophages, capable of differentiating into M1 and M2 phenotypes, showed a more significant decrease in CSCC than in HSIL. No significant difference was detected between HSIL and CSCC in terms of M2 macrophage presence. M1 macrophages secrete pro-inflammatory cytokines that enhance CD8 + T cell function, thereby inhibiting tumor growth. In contrast, M2-type macrophages release immunosuppressive factors that promote angiogenesis, tissue remodeling, and metastasis. For instance, M2 tumor-associated macrophages (TAMs) drive tumor cell invasion through the CCL18-CCR8 axis35,36. These observations lead to the hypothesis that an imbalance in M1/M2 macrophages during the HSIL stage, favoring a pro-cancer phenotype, may be a trigger for the progression to cervical cancer. Collectively, the results suggest the existence of an immunosuppressive microenvironment in the HSIL stage, characterized by a macrophage M1/M2 imbalance and a reduced proportion of resting memory CD4 T cells and gamma-delta T cells. This microenvironment could potentially attenuate the anti-tumor immune response and facilitate the acceleration of CSCC tumorigenesis.
The value of hub genes in HSIL and CSCC
A PPI network using 30 genes identified FEN1, CDK1, TOP2A, CHEK1, and RRM2 as key genes (Fig. 4F). We evaluated these genes as potential predictive and differential markers for HSIL and CSCC. The expression of these genes was elevated in both conditions (P < 0.01) (Fig. 5A). ROC curve analysis assessed their diagnostic value, with FEN1, CDK1, TOP2A, and RRM2 showing AUC > 0.7 for HSIL, while CHEK1 did not perform as a diagnostic marker (AUC = 0.541) (Fig. 5B). All five genes were specific for CSCC diagnosis (Fig. 5C). The HPA database confirmed high protein expression of Fen1, CDK1, TOP2A, and RRM2 in cancer tissues, excluding CHEK1 due to data absence (Fig. 6A). Prognostic analysis revealed FEN1 (HR = 0.18, P = 0.044), CDK1, TOP2A, CHEK1, and RRM2 as significantly associated with CSCC prognosis (P < 0.05), suggesting high FEN1 expression is linked to poorer outcomes (Fig. 5D).

Identification and Validation of Common Diagnostic Biomarkers in HSIL and CSCC. (A) Differential expression of 5 genes in Normal, HSIL and CSCC groups. (B) ROC curves of 5 pivotal genes in HSIL vs Normal group. (C) ROC curves of five pivotal genes in the CSCC versus Normal group. (D) Prognostic analysis of 5 pivotal genes in CSCC. *P < 0.05, **P < 0.01, ***P < 0.001. Kruskal–Wallis test with Dunn’s or ANOVA with Tukey’s multiple comparison post hoc. Data aremeans + SEM.

HPA database immunohistochemical staining of the protein expression levels of the hub genes. (A) Correlation between FEN1 and PCNA in the TCGA-CSCC data set (B).
Tracing FEN1 and PCNA routes from HSIL to CSCC.
To delve into the underlying mechanism of FEN1 upregulation, an exploration of the STRING database was conducted to identify genes that interact with or are co-expressed alongside FEN1. It was found that PCNA is implicated as a transcription factor influencing FEN1 expression (Fig. 7A). Examining a vast dataset from the TCGA-CSCC cohort revealed a strong correlation between the expression levels of FEN1 and PCNA (Fig. 6B)37. Insights from protein interaction studies suggest that FEN1 engages in direct competition for PCNA binding with its PIP (PCNA-interacting protein) sequence-derived residues (Fig. 7B)38. This interaction predominantly involves the hydrophobic pocket on the front face of PCNA, which is crucial for the assembly of PCNA-FEN1 complexes. The rigid docking of FEN1 and PCNA exhibited a binding energy of − 434.92 kcal mol with a confidence of 0.9967. The interaction surface possessed properties mainly through hydrogen bonding and Johannes Diderik van der Waals contact.

FEN1 and PCNA Functions in the Progression from HSIL to CSCC. (A) Genes that interact with FEN1 or co-express with FEN1. (B) Protein docking of FEN1 and PCNA. (C) GSEA analysis of PCNA (left) and FEN1(right) in HSIL group. (D) GSEA analysis of PCNA and FEN1 in SCC Group. GSEA, Gene set enrichment analysis. (E) The correlation analysis of PCNA (left) and FEN1(right) in HSIL group. (F) The correlation analysis of PCNA (left) and FEN1(right) in CSCC group.
Gene set enrichment analysis (GSEA) revealed that both FEN1 and PCNA were significantly enriched in HSIL and CSCC, as well as in pathways related to sister chromatid DNA replication and mitosis (Fig. 7C,D). These observations imply that FEN1, along with its upstream regulators, plays a pivotal role in DNA replication during cervical carcinogenesis, facilitating the accumulation of gene mutations or chromosomal abnormalities that contribute to the progression of cervical cancer. Consequently, targeting the PCNA-FEN1 pathway could effectively disrupt DNA replication and control the advancement of cervical cancer. Correlation analysis indicated that FEN1 expression is negatively associated with TCM, CD8 + T cells, NK cells, and B cells, while exhibiting a positive correlation with Treg cells and macrophages (Fig. 7E,F).
FEN1’s role in immune evasion
In CSCC, immune rejection score and MDSC levels were elevated versus HSIL, indicating FEN1 high expression may enhance immune evasion ( \* MERGEFORMAT Fig. 8A,B). Examining FEN1’s influence on immunotherapy response, we found FEN1 negatively correlated with PDCD1 (PD-1), TIGIT, CTLA4, and HAVCR2, suggesting poor response to anti-PD-1, anti-CTLA4, and anti-TIGIT therapies with high FEN1 (Fig. 8C). However, FEN1 expression positively correlated with LGALS9 and TNFSF15. LGALS9, expressed by microglia and macrophages, and TNFSF15, expressed in monocytes and macrophages, are involved in immune regulation and tumor immune escape39,40. Targeting LGALS9 and TNFSF15 may be a potential therapeutic approach for CSCC with high FEN1 expression.

The high level of FEN1 indicates the adverse reaction of SCC to immunotherapy. (A) Immune rejection score. (B) MDSC level. (C) Correlation between FEN1 expression and immune checkpoint inhibitor expression. (D) FEN1 docks with small molecule inhibitors. *P < 0.05.
The molecular docking analysis revealed that FEN1 possesses a substantial binding affinity for all the inhibitors tested (Fig. 8D). The majority of the ligands were observed to be encapsulated within the active pocket of FEN1, which could potentially prevent the ligand from interacting with other receptors and consequently interfering with downstream signaling pathways. This finding suggests that FEN1 could be a promising drug target, with the potential to influence the progression of HSIL and CSCC.
By integrating the results of DEGs analysis, WGCNA module analysis, immune infiltration analysis, and functional enrichment analysis, we revealed key genes and pathways for the transition from HSIL to CSCC. In particular, the role of FEN1 and its related pathways in disease progression and immune escape provides a new direction for future therapeutic strategies.
The expression differences of FEN1 in HSIL and CSCC tissues
To validate the expression differences of FEN1 in HSIL and CSCC tissues, qPCR, Western Blot, and IHC staining were performed on normal tissues, high-grade squamous intraepithelial lesion (HSIL) tissues, and cervical squamous cell carcinoma (CSCC) tissues. QPCR results confirmed that FEN1 expression levels gradually increased with disease progression, showing an upward trend in normal tissues, HSIL, and CSCC tissues (Fig. 9A). Western Blot further demonstrated that the protein expression level of FEN1 exhibited a progressive increase during the pathological process from normal tissues to HSIL and then to CSCC (Fig. 9B,C). Subsequently, IHC results revealed that FEN1 protein was predominantly localized in the nucleus (Fig. 9F), with elevated expression observed in both the HSIL and CSCC groups (Fig. 9E), and a more significant increase in the CSCC group. Additionally, compared with the normal group, Ki67 levels were increased in both the HSIL and CSCC groups, with a more pronounced increase in the CSCC group (Fig. 9D). These results indicate that FEN1 expression gradually increases from normal tissues to HSIL and then to CSCC.

QPCR analysis showed that the mRNA levels of FEN1 gradually increased in the HSIL and CSCC groups compared with the control group. (A) Western blotting was used to detect FEN1 protein expression in normal tissues, HSIL tissues, and CSCC tissues (B). Quantitative results of Western blotting analysis (C). Quantitative analysis of Ki-67 expression detected by immunohistochemistry (D). Quantitative analysis of FEN1 expression detected by immunohistochemistry (E). Immunohistochemical detection of Ki-67 and FEN1 expression in tissues, with a scale bar of 100 μm shown in the figure. (F) Cell proliferation CCK-8 assay showed that the growth rate of Hela cells significantly decreased and cell proliferation was inhibited after FEN1 gene knockdown. (G) Cell proliferation CCK-8 assay showed that the growth rate of SiHa cells significantly decreased and cell proliferation was inhibited after FEN1 gene knockdown. (H) Data presentation (A–E): Values are expressed as mean ± SD. Statistical significance was determined by Student’s *t*-test or one-way ANOVA (P < 0.05, *P < 0.01, **P < 0.001).
To further investigate whether HSIL promotes tumor growth through FEN1, specific shRNA was used to knock down endogenous FEN1 in Hela and SiHa cells. As shown by CCK-8 assays, FEN1 gene knockdown significantly inhibited the proliferative activity of human cervical cancer cell lines Hela and SiHa compared with the control group (P < 0.05) (Fig. 9G,H). Quantitative analysis showed that the survival rates of Hela cells and SiHa cells in the FEN1 knockdown group decreased in a time-dependent manner at 24 h, 48 h and 72 h after transfection, indicating that FEN1 plays a critical role in maintaining the proliferative capacity of cervical cancer cells. Taken together, these data suggest that HSIL promotes uterine lesions and facilitates cancer development by increasing FEN1 expression.
Discussion
This meta-analysis, which builds upon previous research, comprehensively delineates the transcriptomic characteristics of high-grade intraepithelial neoplasia and pinpoints five pivotal genes (FEN1, CDK1, TOP2A, CHEK1, RRM2) linked to the progression from HSIL to CSCC. Our analysis meticulously assessed the diagnostic and prognostic efficacy of these genes in HSIL and CSCC. To the best of our knowledge, our study is the first to assess and corroborate the diagnostic and prognostic significance of FEN1 in cervical lesions.
The transcriptomic profiles of normal epithelial cells and LSIL have been noted to be uniform, with a subset of dysregulated genes present in CSCC also observed in a portion of HSIL, hinting at potential heterogeneity within HSIL41. Research on the transcriptomic landscape of hepatocarcinogenesis has revealed that dysplastic lesions and early-stage cancers are homogeneous, whereas advanced liver cancer is heterogeneous42. Therefore, this study concentrated on the heterogeneity between HSIL and CSCC.
We identified 30 differentially expressed genes (DEGs) that sequentially increased across the three GSE datasets. Cell cycle, DNA repair, and oncogenic p53 pathways are activated in HSIL and CSCC. A robust correlation between these pathways and HPV has been established. HR-HPV E6 and E7 oncoproteins disrupt p53 and PRB, causing cell cycle dysfunction and promoting the DNA damage response (DDR), key in cervical carcinogenesis43,44. CIBERSORT analysis indicated enrichment of common gene populations in immune cell activation pathways, suggesting diminished immune response in HSIL and CSCC patients. The tumor-promoting status in HSIL patients is attributed to alterations in M0 numbers, M1/M2 imbalance, and diminished recruitment of T cells γδ and T cells CD4 memory resting, establishing a tumor-promoting mechanism. As an unconventional subset of T lymphocytes, γδ T cells possess the ability to recognize antigens independently and are predominantly localized in peripheral mucosal tissues. Studies have demonstrated that γδ T cells activate anti-tumor immune responses by secreting pro-inflammatory cytokines such as interferon-γ (IFN-γ), interferon-α (IFN-α), and tumor necrosis factor-α (TNF-α). Furthermore, γδ T cells can significantly suppress oxidative phosphorylation, the Hedgehog signaling pathway, and angiogenesis-related pathways, thereby modulating the tumor microenvironment and inhibiting tumor progression45. Network analysis identified five hub genes. Following differential expression analysis, ROC diagnostic assessment, HPA website validation, and KM prognostic evaluation, we demonstrated that increased FEN1 protein expression is associated with adverse prognosis in CSCC patients, providing insights into HSIL to CSCC transformation.
Flap endonuclease 1 (FEN1), a pivotal protein in the base excision repair (BER) pathway, is essential for Okazaki fragment maturation during DNA replication46. Cancer cells, characterized by uncontrolled DNA replication, require enhanced DNA repair mechanisms, making FEN1 mutations or dysregulation critical for genomic instability and cancer progression47. Clinical data link FEN1 expression to the invasiveness and prognosis of epithelial ovarian and breast cancers, suggesting its potential as a biomarker48,49. Aberrant FEN1 can promote malignant transformation and carcinogen susceptibility.
Cell cycle-dependent kinase 1 (CDK1), a serine/threonine kinase, and topoisomerase 2 alpha (TOP2A), governing DNA topology, are both associated with cancer progression and drug resistance50,51. Checkpoint kinase 1 (CHEK1) and ribonucleotide reductase subunit M2 (RRM2) play roles in genome stability, tumor resistance, and apoptosis52. In breast cancer, RRM2 upregulation contributes to resistance to GTI-2040, tamoxifen, doxorubicin, and cisplatin53,54. These proteins collectively highlight the complexity of cancer biology and the importance of targeting DNA repair and cell cycle pathways for therapy.
Protein–protein interactions are critical for directing FEN1 towards distinct biochemical metabolic pathways. FEN1 interacts with various proteins to form specific protein complexes, which are pivotal in DNA replication and repair. These interacting proteins include PCNA, replication protein A (RPA), apurinic/apyrimidinic endonuclease 1 (Ape1), PARP1, polymerase, DNA replication helicase/nuclease 2 (DNA2), and P30055. When FEN1 interacts with PCNA, it is recruited to replication-deficient regions to remove RNA primers and repair DNA base excision repair (BER) sites. Furthermore, PCNA can strongly enhance the endonuclease and exonuclease activities of FEN156.
We performed GSEA on target genes in HSIL and CSCC, revealing FEN1 and PCNA enrichment in DNA replication pathways. In HSIL, these genes were associated with chromosome organization and segregation, while in CSCC, they were linked to sister chromatid pathways. These findings suggest that HPV splicing integrates DNA replication metabolites, causing chromosome segregation and immune homeostasis imbalance, driving cervical cancer progression57. These genes appear to mediate the transition from HSIL to CSCC, highlighting their role in carcinogenesis.
We investigated the regulatory roles of PCNA and its downstream molecule FEN1 on immune cell populations in cervical cancer58. Our findings showed that FEN1 expression was negatively associated with the infiltration of immune-promoting cells (TCM, CD8 + T cells, NK cells, B cells) but positively with immunosuppressive cells (Treg cells, macrophages)59. Elevated FEN1 may enhance DNA damage repair capacity in cancer cells, enabling them to sustain proliferation under persistent genotoxic stress—such as double-strand breaks induced by HPV integration—and accelerate the transition from precancerous lesions to invasive carcinoma. During the S phase, FEN1 interacts with PCNA to participate in DNA replication. Its high expression may disrupt cell cycle checkpoint control, promote abnormal cell proliferation, and evade apoptosis60. This correlation may explain the poor prognosis linked to FEN1 in cervical cancer. The study also highlighted the importance of immunosuppressive genes (PD-1, PD-L1, CTLA-4) in tumor immune evasion, which is crucial for tumor growth and metastasis. FEN1’s significant positive correlation with immunosuppression and its higher expression in CSCC compared to HSIL suggest its pivotal role in immune evasion mechanisms in cervical cancer.
Consequently, FEN1 emerges as a promising drug target, and small molecule inhibitors targeting FEN1 have been developed61. These small molecule inhibitors are organic compounds with low molecular weight that can penetrate the cell membrane by targeting the active site of proteins, thereby reducing their activity or interfering with cellular reactions. Molecular docking results suggest that small-molecule inhibitors are predominantly encapsulated within the active pocket of FEN1. We believe that these inhibitors inhibit FEN1’s activity by interfering with the active site it uses to bind PCNA, which represents an essential concept for the development of FEN1 inhibitors.
During the experimental validation phase, we employed multi-dimensional detection methods to systematically evaluate the expression characteristics of FEN1: CCK-8 assays were used to detect the proliferative capacity of cervical cancer cells with FEN1 knockdown, real-time fluorescent quantitative PCR (qPCR) was applied to analyze mRNA expression levels, Western blotting (WB) was conducted to detect protein expression, and immunohistochemistry (IHC) was combined for localization and quantitative analysis of tissue samples. The results confirmed a significant positive correlation between FEN1 expression levels and the malignancy degree of cervical cancer.
From the perspective of molecular evolution, tumorigenesis and development fundamentally represent a malignant evolutionary process driven by cumulative genetic variations. This study revealed that abnormally high expression of FEN1 not only accelerates the accumulation rate of gene mutations but also significantly enhances the mutagenic potential of tumor cells by promoting genomic instability. Specific findings include: (1) Analysis of clinical samples showed that FEN1 expression levels exhibited a gradient increase with the progression of cervical lesions (from normal tissues, HSIL to CSCC); (2) In vitro functional experiments confirmed that FEN1 gene knockdown significantly inhibited the proliferative activity of cervical cancer cells (p < 0.01). These discoveries collectively reveal the critical role of FEN1 in the malignant progression of cervical cancer, spanning from clinical correlations to functional mechanisms.
Bioinformatics suggests that FEN1 is upregulated in the progression from HSIL to CSCC, and this has been verified in vitro experiments. It indicates that FEN1 is closely related to the progression of HSIL. Advanced techniques like single-cell sequencing could enhance our understanding of macrophage influence in this transition, potentially revealing new therapeutic targets. However, this study fails to establish causation between FEN1 overexpression and disease progression. Our lab is conducting experimental research on FEN1’s role in tumor virus-induced cancer cell transformation to address these limitations.
Conclusion
Our findings validate FEN1 as a promising biomarker for prognostication and immunotherapeutic strategies, especially effective in HSIL therapy and CSCC prevention. It elucidates FEN1’s role in immune dysregulation and evasion mechanisms.
Data availability
Data are contained within the article and Supplementary Materials.
Abbreviations
- AUC:
-
Area under curve
- CSCC:
-
Cervical squamous cell carcinoma
- DEGs:
-
Differential expression genes
- GO:
-
Gene ontology
- GSEA:
-
Gene set enrichment analysis
- HPV:
-
High-risk human papillomavirus
- HSIL:
-
High-grade squamous intraepithelial lesion
- HPA:
-
Human protein atlas
- ROC:
-
Receiver operating characteristic
References
Perkins, R. B., Wentzensen, N., Guido, R. S. & Schiffman, M. Cervical cancer screening: A review. JAMA 330, 547–558. https://doi.org/10.1001/jama.2023.13174 (2023).
Zhao, Y. et al. Real-world effectiveness of primary screening with high-risk human papillomavirus testing in the cervical cancer screening programme in China: A nationwide, population-based study. BMC Med. 19, 164. https://doi.org/10.1186/s12916-021-02026-0 (2021).
Elfström, K. M. et al. Organized primary human papillomavirus-based cervical screening: A randomized healthcare policy trial. PLoS Med. 18, e1003748. https://doi.org/10.1371/journal.pmed.1003748 (2021).
Vink, F. J. et al. Classification of high-grade cervical intraepithelial neoplasia by p16(ink4a), Ki-67, HPV E4 and FAM19A4/miR124-2 methylation status demonstrates considerable heterogeneity with potential consequences for management. Int. J. Cancer 149, 707–716. https://doi.org/10.1002/ijc.33566 (2021).
Norman, I. et al. Atypical glandular cells and development of cervical cancer: Population-based cohort study. Int. J. Cancer 151, 2012–2019. https://doi.org/10.1002/ijc.34242 (2022).
SDA, et al. Identification of differentially expressed genes in cervical cancer patients by comparative transcriptome analysis. Biomed. Res. Int. 2021, 8810074. https://doi.org/10.1155/2021/8810074 (2021).
Kurmyshkina, O., Kovchur, P., Schegoleva, L. & Volkova, T. Markers of angiogenesis, lymphangiogenesis, and epithelial-mesenchymal transition (plasticity) in CIN and early invasive carcinoma of the cervix: Exploring putative molecular mechanisms involved in early tumor invasion. Int. J. Mol. Sci. 21, 6515 (2020).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinf. 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Le, M. H. N. et al. An in-depth review of AI-powered advancements in cancer drug discovery. Biochim. Biophys. Acta. Mol. Basis Dis. 1871, 167680. https://doi.org/10.1016/j.bbadis.2025.167680 (2025).
Mkrtchyan, G. V. et al. High-confidence cancer patient stratification through multiomics investigation of DNA repair disorders. Cell Death Dis. 13, 999 (2022).
Ren, F. et al. AlphaFold accelerates artificial intelligence powered drug discovery: efficient discovery of a novel CDK20 small molecule inhibitor. Chem. Sci. 14, 1443–1452 (2023).
Kamya, P. et al. PandaOmics: An AI-driven platform for therapeutic target and biomarker discovery. J. Chem. Inf. Model 64, 3961–3969. https://doi.org/10.1021/acs.jcim.3c01619 (2024).
Wagemann, O. et al. Exploratory analysis of the proteomic profile in plasma in adults with Down syndrome in the context of Alzheimer’s disease. Alzheimers Dement. 21, e70040 (2025).
Sasamoto, N. et al. Plasma proteins and persistent postsurgical pelvic pain among adolescents and young adults with endometriosis. Am. J. Obstet. Gynecol. 231(240), e241–e240 (2024).
Isali, I. et al. A systematic review and in silico study of potential genetic markers implicated in cases of overactive bladder. Am. J. Obstet. Gynecol. 228, 36–47 (2023).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523. https://doi.org/10.1038/s41467-019-09234-6 (2019).
Chen, B., Khodadoust, M. S., Liu, C. L., Newman, A. M. & Alizadeh, A. A. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol. Biol. 1711, 243–259 (2018).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015).
Liu, L. et al. Integrated bioinformatics combined with machine learning to analyze shared biomarkers and pathways in psoriasis and cervical squamous cell carcinoma. Front. Immunol. 15, 1351908 (2024).
Zhang, W. Y., Liu, Y. J., He, Y. & Chen, P. Suppression of long noncoding RNA NCK1-AS1 increases chemosensitivity to cisplatin in cervical cancer. J. Cell Physiol. 234, 4302–4313. https://doi.org/10.1002/jcp.27198 (2019).
van Dam, P. A. et al. Potential new biomarkers for squamous carcinoma of the uterine cervix. ESMO Open 3, e000352 (2018).
Pontén, F., Schwenk, J. M., Asplund, A. & Edqvist, P. H. The Human Protein Atlas as a proteomic resource for biomarker discovery. J. Intern. Med. 270, 428–446. https://doi.org/10.1111/j.1365-2796.2011.02427.x (2011).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A 102, 15545–15550 (2005).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Yan, Y., Tao, H., He, J. & Huang, S. Y. The HDOCK server for integrated protein-protein docking. Nat. Protoc. 15, 1829–1852. https://doi.org/10.1038/s41596-020-0312-x (2020).
Wishart, D. S. et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074-d1082. https://doi.org/10.1093/nar/gkx1037 (2018).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 14, 7 (2013).
Bindea, G. et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795. https://doi.org/10.1016/j.immuni.2013.10.003 (2013).
Tian, J., Cui, X., Sun, J. & Zhang, J. Retraction notice to Exosomal microRNA-16–5p from adipose mesenchymal stem cells promotes TLR4-mediated M2 macrophage polarization in septic lung injury. Int. Immunopharmacol. 111, 109078. https://doi.org/10.1016/j.intimp.2022.109078 (2022).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Macheret, M. & Halazonetis, T. D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 10, 425–448. https://doi.org/10.1146/annurev-pathol-012414-040424 (2015).
Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 12, 31–46. https://doi.org/10.1158/2159-8290.Cd-21-1059 (2022).
Saura-Esteller, J. et al. Gamma delta T-cell based cancer immunotherapy: Past-present-future. Front. Immunol. 13, 915837. https://doi.org/10.3389/fimmu.2022.915837 (2022).
Speiser, D. E., Chijioke, O., Schaeuble, K. & Münz, C. CD4(+) T cells in cancer. Nat. Cancer 4, 317–329. https://doi.org/10.1038/s43018-023-00521-2 (2023).
Qian, B. Z. & Pollard, J. W. Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51 (2010).
Guan, F. et al. Tissue macrophages: Origin, heterogenity, biological functions, diseases and therapeutic targets. Signal Transduct. Target Ther. 10, 93 (2025).
Querol-Audí, J. et al. Repair complexes of FEN1 endonuclease, DNA, and Rad9-Hus1-Rad1 are distinguished from their PCNA counterparts by functionally important stability. Proc. Natl. Acad. Sci. U S A 109, 8528–8533. https://doi.org/10.1073/pnas.1121116109 (2012).
Bruning, J. B. & Shamoo, Y. Structural and thermodynamic analysis of human PCNA with peptides derived from DNA polymerase-delta p66 subunit and flap endonuclease-1. Structure 12, 2209–2219. https://doi.org/10.1016/j.str.2004.09.018 (2004).
Anand, P. et al. Single-cell RNA-seq reveals developmental plasticity with coexisting oncogenic states and immune evasion programs in ETP-ALL. Blood 137, 2463–2480. https://doi.org/10.1182/blood.2019004547 (2021).
Xu, W. D., Li, R. & Huang, A. F. Role of TL1A in inflammatory autoimmune diseases: A comprehensive review. Front Immunol 13, 891328. https://doi.org/10.3389/fimmu.2022.891328 (2022).
Li, C. & Hua, K. Single-cell transcriptomics provides insights into the origin and immune microenvironment of cervical precancerous lesions. Cancer Commun. 43, 1055–1058. https://doi.org/10.1002/cac2.12451 (2023).
Desjonqueres, E., Campani, C., Marra, F., Zucman-Rossi, J. & Nault, J. C. Preneoplastic lesions in the liver: Molecular insights and relevance for clinical practice. Liver Int. 42, 492–506. https://doi.org/10.1111/liv.15152 (2022).
Squarzanti, D. F. et al. Human papillomavirus type 16 E6 and E7 oncoproteins interact with the nuclear p53-binding protein 1 in an in vitro reconstructed 3D epithelium: New insights for the virus-induced DNA damage response. Virol. J. 15, 176. https://doi.org/10.1186/s12985-018-1086-4 (2018).
Templeton, C. W. & Laimins, L. A. p53-dependent R-loop formation and HPV pathogenesis. Proc. Natl. Acad. Sci. U S A 120, e2305907120. https://doi.org/10.1073/pnas.2305907120 (2023).
Li, J. et al. Multiomics profiling reveals the benefits of gamma-delta (γδ) T lymphocytes for improving the tumor microenvironment, immunotherapy efficacy and prognosis in cervical cancer. J. Immunother. Cancer 12, e008355 (2024).
Sobhy, M. A. et al. Implementing fluorescence enhancement, quenching, and FRET for investigating flap endonuclease 1 enzymatic reaction at the single-molecule level. Comput. Struct. Biotechnol. J. 19, 4456–4471. https://doi.org/10.1016/j.csbj.2021.07.029 (2021).
Wang, C. et al. A DNA nano firework for imaging and inhibitor screening of flap endonuclease 1 in living cells. Anal. Chem. 95, 8621–8631. https://doi.org/10.1021/acs.analchem.3c00930 (2023).
Berfelde, J., Hildebrand, L. S., Kuhlmann, L., Fietkau, R. & Distel, L. V. FEN1 inhibition as a potential novel targeted therapy against breast cancer and the prognostic relevance of FEN1. Int. J. Mol. Sci. https://doi.org/10.3390/ijms25042110 (2024).
Mesquita, K. A. et al. FEN1 blockade for platinum chemo-sensitization and synthetic lethality in epithelial ovarian cancers. Cancers https://doi.org/10.3390/cancers13081866 (2021).
Wang, Q., Bode, A. M. & Zhang, T. Targeting CDK1 in cancer: Mechanisms and implications. NPJ. Precis. Oncol. 7, 58. https://doi.org/10.1038/s41698-023-00407-7 (2023).
Uusküla-Reimand, L. & Wilson, M. D. Untangling the roles of TOP2A and TOP2B in transcription and cancer. Sci. Adv. 8, eadd4920. https://doi.org/10.1126/sciadv.add4920 (2022).
Mu, R. et al. Genetic variants of CHEK1, PRIM2 and CDK6 in the mitotic phase-related pathway are associated with nonsmall cell lung cancer survival. Int. J. Cancer 149, 1302–1312. https://doi.org/10.1002/ijc.33702 (2021).
Putluri, N. et al. Pathway-centric integrative analysis identifies RRM2 as a prognostic marker in breast cancer associated with poor survival and tamoxifen resistance. Neoplasia 16, 390–402. https://doi.org/10.1016/j.neo.2014.05.007 (2014).
Jiao, Y. et al. RRM2 alleviates doxorubicin-induced cardiotoxicity through the AKT/mTOR signaling pathway. Biomolecules https://doi.org/10.3390/biom12020299 (2022).
Henry, R. A., Balakrishnan, L., Ying-Lin, S. T., Campbell, J. L. & Bambara, R. A. Components of the secondary pathway stimulate the primary pathway of eukaryotic Okazaki fragment processing. J. Biol. Chem. 285, 28496–28505. https://doi.org/10.1074/jbc.M110.131870 (2010).
Blair, K. et al. Mechanism of human Lig1 regulation by PCNA in Okazaki fragment sealing. Nat. Commun. 13, 7833. https://doi.org/10.1038/s41467-022-35475-z (2022).
Gusho, E. & Laimins, L. Human papillomaviruses target the DNA damage repair and innate immune response pathways to allow for persistent infection. Viruses https://doi.org/10.3390/v13071390 (2021).
Wang, S. et al. Down-regulation of DNA key protein-FEN1 inhibits OSCC growth by affecting immunosuppressive phenotypes via IFN-γ/JAK/STAT-1. Int. J. Oral Sci. 15, 17. https://doi.org/10.1038/s41368-023-00221-8 (2023).
Wang, X., Xu, S., Fu, T., Wu, Y. & Sun, W. Combination of downregulating FEN1 and PD-1 blockade enhances antitumor activity of CD8+ T cells against HNSCC cells in vitro. J. Oral Pathol. Med. 52, 834–842. https://doi.org/10.1111/jop.13485 (2023).
Zheng, L. et al. Functional regulation of FEN1 nuclease and its link to cancer. Nucleic Acids Res. 39, 781–794 (2011).
Yang, F., Hu, Z. & Guo, Z. Small-molecule inhibitors targeting FEN1 for cancer therapy. Biomolecules https://doi.org/10.3390/biom12071007 (2022).
Acknowledgements
Not applicable.
Funding
The work was supported by the National Science Foundation of China [grant numbers 82074289, 81873205, 82104910], Project of Guangdong Bureau of Traditional Chinese Medicine [grant number 20231004].
Author information
Authors and Affiliations
Contributions
FX Zhang contributed to the design of experimental methods, the analysis of experimental data, and the review and revision. XM Chen, CY Chu, ML Chen, YH Yu and YY Liu contributed to literature retrieval, study conceptualization, manuscript writing and critical revision, prepared Fig. 1–7. Y Yang, YT You, J Zhou PP Jiang and N Song were responsible for data extraction, analysis and interpretation. The authors agreed to the final version for submission.
Corresponding authors
Ethics declarations
Competing interests
Our authors declared no competing interest related to this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.

Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, F., Chen, X., Chu, C. et al. FEN1 plays a key role in the transition from HSIL to CSCC. Sci Rep 15, 30122 (2025). https://doi.org/10.1038/s41598-025-15642-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-15642-0

{kind=link}