
Abstract
Head and neck squamous cell carcinoma (HNSC) is a highly aggressive malignancy with poor prognosis, necessitating the identification of novel biomarkers for improved diagnosis and treatment. The exosome complex (EXOSC) family plays a crucial role in RNA metabolism, but its significance in HNSC remains poorly understood. We performed a comprehensive multi-omics analysis integrating data from TCGA, GEO, CPTAC, and the Human Protein Atlas to investigate the expression, prognostic value, and immune relevance of EXOSC genes in HNSC. We conducted differential expression analysis, survival analysis (OS, DSS, PFI), ROC curve evaluation, and clinicopathological correlation studies. Genetic alterations were examined using cBioPortal. Gene co-expression and enrichment analyses were used to elucidate potential molecular functions, and a protein–protein interaction (PPI) network was constructed via GeneMANIA. Immune infiltration, immune checkpoint correlations, and RNA modification associations were assessed using ssGSEA, Spearman correlation, and RNA modification databases. Experimental validation was performed by qRT-PCR in HNSC and normal cell lines. All EXOSC family members were significantly upregulated in HNSC tissues and cell lines. ROC analysis demonstrated favorable diagnostic potential, particularly for EXOSC2 (AUC = 0.910). Elevated expression of EXOSC2, EXOSC3, EXOSC8, and EXOSC9 was significantly associated with poor OS, DSS, and PFI. High expression of EXOSC2, EXOSC4, EXOSC5, and EXOSC9 correlated with advanced clinical stage, lymphovascular invasion, and poor therapeutic outcomes. cBioPortal analysis revealed EXOSC4 had the highest genetic alteration frequency (8%), primarily due to amplification. Immune infiltration analysis showed EXOSC gene expression was significantly correlated with various immune cell populations and immune checkpoint molecules, especially EXOSC3, EXOSC9, and EXOSC10. Functional enrichment and PPI network analyses indicated that EXOSC family genes participate in RNA metabolism, exoribonuclease activity, and immune-related pathways. A prognostic risk model based on EXOSC co-expressed genes demonstrated strong predictive performance for patient survival. Our study reveals that EXOSC family genes are significantly dysregulated in HNSC and are associated with tumor progression, prognosis, immune microenvironment modulation, and RNA modification. These findings highlight the potential of EXOSC members as novel diagnostic and prognostic biomarkers and suggest their relevance as therapeutic targets in HNSC.
Similar content being viewed by others

Introduction
Head and neck squamous cell carcinoma (HNSC) is one of the most common malignant tumors globally, originating primarily from the mucosal epithelium of the oral cavity, pharynx, and larynx1. Its high incidence and poor prognosis make it a significant public health concern2. It is a highly heterogeneous malignancy, ranking among the top causes of cancer-related mortality globally. Despite advancements in surgical interventions, radiotherapy, and systemic therapies, the overall survival rate of HNSC patients remains unsatisfactory, largely due to late-stage diagnosis, therapeutic resistance, and tumor recurrence3. The molecular heterogeneity of HNSC underscores the need for a deeper understanding of its genetic and epigenetic landscape to identify novel biomarkers and therapeutic targets. In this context, bioinformatics-driven analyses of cancer-associated gene families have emerged as a powerful approach to uncovering potential oncogenes or tumor suppressors that contribute to HNSC pathogenesis.
Emerging evidence suggests that RNA degradation is a critical mechanism of post-transcriptional gene regulation, influencing cancer progression by modulating the stability and turnover of oncogenic and tumor-suppressive transcripts4. Among the key players in RNA metabolism, the exosome complex (EXOSC) is central to RNA degradation and processing, functioning in both the nucleus and cytoplasm5. The EXOSC family, consisting of ten highly conserved subunits (EXOSC1-10), forms a multi-protein machinery responsible for the degradation of diverse RNA species, including aberrant and regulatory RNAs6. Beyond its canonical RNA degradation function, accumulating evidence suggests that EXOSC components contribute to oncogenesis. Several EXOSC subunits have been implicated in tumor development and progression across various malignancies7. For instance, EXOSC1 has been shown to modulate DNA stability and enhance the sensitivity of renal carcinoma cells to poly(ADP-ribose) polymerase (PARP) inhibitors8. Similarly, EXOSC2 plays a crucial role in breast cancer metastasis9, while EXOSC3 and EXOSC4 have been associated with colorectal tumorigenesis10,11. Additional studies indicate that EXOSC512, EXOSC813, EXOSC914, and EXOSC1015 participate in cancer progression, but their specific contributions to HNSC remain largely unexplored. Given the functional significance of EXOSC genes in multiple cancer types, elucidating their role in HNSC may provide novel insights into tumor biology and potential therapeutic targets.
In this study, we conducted a comprehensive bioinformatics analysis to investigate the expression profiles, prognostic value, and potential molecular functions of EXOSC family genes in HNSC. Using publicly available datasets, we explored the correlation between EXOSC expression and patient survival, tumor immune microenvironment characteristics, and key oncogenic pathways. Our findings provide new insights into the role of EXOSC genes in HNSC progression and highlight their potential as prognostic biomarkers and therapeutic targets for precision oncology.
Methods
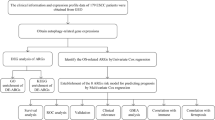
Expression profile analysis, survival analysis, and clinicopathological features of EXOSC family genes in HNSC
Expression data for EXOSC1[ENSG00000171311.13], EXOSC2[ENSG00000130713.16], EXOSC3[ENSG00000107371.14], EXOSC4[ENSG00000178896.9], EXOSC5[ENSG00000077348.9], EXOSC6[ENSG00000223496.3], EXOSC7[ENSG00000075914.13], EXOSC8[ENSG00000120699.13], EXOSC9[ENSG00000123737.13] and EXOSC10[ENSG00000171824.14] were retrieved from the TCGA-HNSC (head and neck squamous cell carcinoma) dataset in the TCGA database (https://portal.gdc.cancer.gov). The RNA-seq data, processed using the STAR pipeline, were extracted in TPM (transcripts per million) format and subsequently transformed using log2(value + 1) for normalization. The visualization of EXOSC gene expression data was carried out using the “ggplot2” package in R. Additionally, the correlation between the mRNA expression levels of EXOSC family genes and clinicopathological characteristics in HNSC was examined through R’s “ggplot2” package. A Kaplan-Meier survival analysis was conducted to establish a Cox proportional hazards regression model, which was used to assess overall survival (OS), disease-specific survival (DSS), and progression-free interval (PFI) based on EXOSC gene expression levels16. The “Survival” package in R software was used for this purpose, with hazard ratios (HR) and p-values calculated and documented. Results were visualized through forest plots, which were created using ggplot2. To evaluate the effectiveness of binary classification models, receiver operating characteristic (ROC) curve analysis was performed17. The ROC curve is a graphical representation that compares sensitivity and specificity, while the area under the curve (AUC) serves as a measure of classification accuracy, with values approaching 1 indicating higher predictive power. The “pROC” package in R was utilized to conduct the ROC curve analysis. Protein expression data were obtained from Clinical Proteomic Tumor Analysis Consortium (CPTAC) database, and subsequently analyzed using the UALCAN platform. And, representative immunohistochemistry (IHC) images of EXOSC family protein distribution in human HNSC tumor specimens and matched normal tissues were obtained from the publicly available Human Protein Atlas database (HPA).
EXOSC family genes expressions vlidation by GEO database
To further confirm the expression pattern of EXOSC family genes in HNSC, we employed datasets GSE142083 and GSE178537 obtained from the GEO database (https://www.ncbi.nlm.nih.gov/geo). The dataset encompass genes expression profiles derived from both normal and HNSC tissues.
Cell culture
The human oral keratinocyte (HOK) cell line and HN4 cell line were procured from the Shanghai Institute of Life Science Cell Bank Center (Shanghai, China). Both cell lines were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (100 U/mL penicillin and 100 µg/mL streptomycin) at 37 °C in a humidified atmosphere containing 5% CO₂. Cells in the exponential growth phase were harvested for subsequent experiments.
Quantitative real-time polymerase chain reaction (qRT-PCR)
The primer mix was validated on an ABI 7900HT system using the following protocol: initial enzyme activation at 50 °C for 2 min, followed by polymerase activation at 95 °C for 10 min. Amplification was carried out over 40 cycles, each consisting of denaturation at 95 °C for 15 s and annealing at 60 °C for 1 min. Finally, a melting curve analysis was performed with sequential steps at 95 °C for 15 s, 60 °C for 15 s, and 95 °C for 15 s to verify reaction specificity. EXOSC1-10 expression were quantified using the 2−ΔΔCt method. Primer sequences were: EXOSC1 forward: 5’-CACGGCTACATCTTTTCGTCGC-3’, reverse: 5’-CTCCCACATCTGGCAGTAACTG-3’; EXOSC2 forward: 5’-CTCGCTGGTAACTCAGAGGATG-3’, reverse: 5’-CTCCTGTTCCAAAAGCCTCTGG-3’; EXOSC3 forward: 5’-GGTGTCATTGGACAGGATGGTC-3’, reverse: 5’-TCTCCAGTGGATAGAGTTTTCCC-3’; EXOSC4 forward: 5’-GGACCGTAAGTCCTGTGAGATG-3’, reverse: 5’-AGGTCCCACCATCTGCCTGTAG-3’; EXOSC5 forward: 5’-GAACGGAAGCTGCTGATGTCCA-3’, reverse: 5’-GGTAGAAACGGAAGACGTGTTGC-3’; EXOSC6 forward: 5’-AGGGCTGCCAGCGCCTCTAC-3’, reverse: 5’-CTTGCGTCCGTAGTTGCTCAGG-3’; EXOSC7 forward: 5’-GTGAGGACTACCGATGTGTCGA-3’, reverse: 5’-CTGCTTTCACTCCCACCAAGAT-3’; EXOSC8 forward: 5’-GAACTGCCGTCCTGATGGAAGA-3’, reverse: 5’-GGCATCTGTTGATGGTGCTGCA-3’; EXOSC9 forward: 5’-CTGCCAGCATTGCTGCAATCGT-3’, reverse: 5’-GGTACAGGATCACGCTCTTCAG-3’; EXOSC10 forward: 5’-CTCTTTGGACCTCACGACTGCT-3’, reverse: 5’-AAGAGGCTCGCCTGCTTCTGAA-3’.
cBioPortal analysis of EXOSC family genes in HNSC
Genetic variations in EXOSC family genes within the TCGA dataset were investigated using cBioPortal (http://www.cbioportal.org/)18. This analysis incorporated all EXOSC family members (EXOSC1-EXOSC10) from the TCGA pan-cancer atlas cohort for HNSC. By entering the names of EXOSC genes into the platform’s search tool, mutation data were retrieved from both the “OncoPrint” and “Cancer Types Summary” modules within cBioPortal.
Protein–Protein interaction (PPI) network construction of EXOSC family genes in HNSC
To explore the functional interactions of EXOSC family proteins, we utilized the GeneMANIA platform (http://genemania.org), a comprehensive bioinformatics tool that integrates diverse datasets covering 166,691 genes and approximately 660 million documented interactions across nine model organisms. GeneMANIA facilitates the prediction of gene function based on co-expression, co-localization, physical interactions, shared protein domains, and other association data. In this study, we used GeneMANIA to predict potential protein interaction partners of the EXOSC family in Homo sapiens and constructed a PPI network to elucidate their biological associations.
Immune-related analysis of EXOSC family genes in HNSC
To assess immune cell infiltration across 24 immune cell types in HNSC tissue samples, the ssGSEA algorithm was applied19. Correlation analyses were performed to examine associations between EXOSC gene expression levels, immune cells19, and immune checkpoint genes20 using Spearman correlation analysis. The relationships were illustrated using the “ggplot2” and “pheatmap” packages. Additionally, to compare immune cell infiltration in glioma samples with varying levels of EXOSC family genes expression, the Wilcoxon rank-sum test was employed.
RNA modification analysis of EXOSC family genes in HNSC
Expression data for EXOSC family genes and 44 marker genes associated with three major RNA modifications—m1A, m5C, and m6A—were retrieved from UCSC XENA21,22. Spearman correlation analysis was used to identify potential associations between EXOSC family genes expression and RNA modification-related genes using R (v4.0) and the “corrplot” package (v0.90). Correlation matrices and heatmaps were generated using “ggplot2” for visual interpretation.
Gene set enrichment analysis (GSEA) of EXOSC family genes in HNSC
GSEA was conducted using R software (version 3.6.3) to explore signaling pathways potentially associated with the expression of EXOSC family genes in HNSC. Patients were stratified into high and low expression groups based on the median expression value. The expression levels of EXOSC family genes were used as the phenotype label. Each analysis involved 1,000 permutations. The “c2.cp.v7.2.symbols.gmt” gene set from the Molecular Signatures Database (MSigDB) served as the reference. Pathways were considered significantly enriched if they met the criteria of false discovery rate (FDR) < 0.25, P-value < 0.05, and a normalized enrichment score (NES) > 1. Pathways with the highest NES values were prioritized for downstream interpretation.
Functional and pathway analysis
HNSC patient gene expression data in TPM format were extracted from the TCGA database. Co-expressed genes were identified using Spearman correlation coefficients (P < 0.01, |R| > 0.3). A Venn diagram was generated to depict the overlapping relationships among EXOSC1-EXOSC10. To investigate the biological pathways and functional roles associated with EXOSC family genes, the “clusterProfiler” package was utilized for Kyoto Encyclopedia of Genes and Genomes (KEGG)23 and Gene Ontology (GO)24 enrichment analyses. GO analysis was further divided into three categories: biological process (BP), molecular function (MF), and cellular component (CC).
Identification of prognostic genes
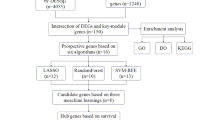
To systematically assess the prognostic relevance of genes co-expressed with EXOSC family members in head and neck squamous cell carcinoma (HNSC), we performed univariate Cox proportional hazards regression analysis using the “survival” R package, with a significance threshold set at P < 0.05. To account for potential multicollinearity among variables and improve model interpretability, we employed the least absolute shrinkage and selection operator (LASSO)-penalized Cox regression using the “glmnet” (version 4.1.7) R package. Subsequently, a nomogram was constructed with the “rms” R package to predict individual survival probabilities.
Construction of a prognostic model based on EXOSC family co-expression genes
A prognostic risk score model was established using centralized and standardized mRNA expression data from the HNSC training cohort. The risk score was defined as:
Risk score = Risk score=\(\:{\sum\:}_{i}^{n}xiyi\), where X represents the LASSO-derived coefficient for each gene and Y denotes the expression level of the corresponding EXOSC family co-expressed gene. Patients were stratified into high-risk and low-risk groups based on the median risk score. Overall survival (OS) between the two groups was compared to evaluate prognostic stratification. The model’s predictive performance was assessed using time-dependent receiver operating characteristic (ROC) curves generated with the “timeROC” R package. Clinical covariates included in the analysis comprised age, sex, histological grade, clinical stage, and TNM classification.
Statistical analysis
For data following a normal distribution, results were represented as means ± standard deviation. Comparisons of EXOSC family genes expression levels between normal and HNSC tissues were conducted using Wilcoxon rank-sum and Wilcoxon signed-rank tests in SPSS 23.0. Associations between EXOSC family genes expression and clinicopathological features were examined using the chi-square test. Statistical significance was defined as *p < 0.05, **p < 0.01, ***p < 0.001.
Results
Expressions and ROC of EXOSC family genes in HNSC
Analysis of data from the UCSC XENA database revealed differential expression patterns of EXOSC family members in head and neck squamous cell carcinoma (HNSC) based on 44 normal and 504 tumor samples (sample sizes detailed in Supplementary Table 1). As shown in Fig. 1A, all EXOSC family genes (EXOSC1-10) were significantly upregulated in HNSC tissues compared to normal tissues (P < 0.001). Figure 1B shows that EXOSC1-10 expression levels were significantly higher in 43 HNSC tumors than in paired normal samples (TCGA data). To evaluate their prognostic potential, receiver operating characteristic (ROC) curve analysis was conducted (sample sizes detailed in Supplementary Table 2). The results indicated high predictive accuracy for HNSC, with area under the curve (AUC) values of 0.862 for EXOSC1, 0.910 for EXOSC2, 0.873 for EXOSC3, 0.882 for EXOSC4, 0.798 for EXOSC5, 0.762 for EXOSC6, 0.695 for EXOSC7, 0.809 for EXOSC8, 0.870 for EXOSC9, and 0.794 for EXOSC10 (Fig. 1C). As shown in Fig. 1D-E, to further validate our findings, we analyzed the GSE142083 and GSE178537 datasets from the GEO database, which revealed that the expression levels of EXOSC1-10 were significantly elevated in HNSC tissues compared to normal controls (all P < 0.05). As shown in Fig. 1F, we used qRT-PCR to explore the gene levels of EXOSC family, we found that the expression levels of EXOSC1-10 were significantly higher in HNSC cell (HN4) than in normal cell (HOK) (all P < 0.05). Using data from the CPTAC, we compared the protein expression levels of EXOSC1-10 between normal tissues and primary tumor tissues in HNSC. Our analysis revealed significantly elevated total protein levels of EXOSC3-10 in primary HNSC tissues compared to normal controls (P < 0.001). In contrast, the expression levels of EXOSC1 and EXOSC2 showed no statistically significant differences between the two groups (Fig. 1G). Furthermore, immunohistochemistry (IHC) data obtained from HPA database revealed the expression profiles of the EXOSC family genes in clinical samples, as illustrated in Fig. 2A-I. Notably, the protein levels of EXOSC1, EXOSC2, EXOSC3, EXOSC4, EXOSC5, EXOSC7, EXOSC8, EXOSC9, and EXOSC10 were significantly upregulated in HNSC tissues compared to normal tissues.

Expressions and receiver operating characteristic (ROC) curve analysis of EXOSC family genes in Head and Neck Squamous Cell Carcinoma (HNSC). (*p < 0.05, **p < 0.01, ***p < 0.001). (A) Differential mRNA expression levels of EXOSC1-10 between HNSC tumors and normal tissues. (B) EXOSC1-10 expression levels between 43 HNSC tumors and paired normal samples. (C) Receiver operating characteristic (ROC) curves assessing the diagnostic performance of EXOSC family genes in distinguishing HNSC from normal tissue. (D) Validation of elevated EXOSC family gene expression in HNSC tissues compared to normal controls using the GSE142083 dataset. (E) Validation of elevated EXOSC family gene expression in HNSC tissues using the GSE178537 dataset. (F) Validation of elevated EXOSC family gene expression in HOK and HN4 cells by qRT-PCR. (G) Using data from the CPTAC, we compared the protein expression levels of EXOSC1-10 between normal tissues and primary tumor tissues in HNSC.

Validation of EXOSC family protein expressions. Immunohistochemical (IHC) analysis of EXOSC family expressions in HNSC and normal tissues was performed using data from the HPA data. (A) Representative IHC images from the HPA demonstrate EXOSC1 expression in HNSC primary tumor and matched normal tissue. (Antibody HPA038370, and 10X). (B) Representative IHC images from the HPA demonstrate EXOSC2 expression in HNSC primary tumor and matched normal tissue. (Antibody HPA021790, and 10X). (C) Representative IHC images from the HPA demonstrate EXOSC3 expression in HNSC primary tumor and matched normal tissue. (Antibody HPA020485, and 10X). (D) Representative IHC images from the HPA demonstrate EXOSC4 expression in HNSC primary tumor and matched normal tissue. (Antibody HPA024792, and 10X). (E) Representative IHC images from the HPA demonstrate EXOSC5 expression in HNSC primary tumor and matched normal tissue. (Antibody HPA053150, and 10X). (F) Representative IHC images from the HPA demonstrate EXOSC7 expression in HNSC primary tumor and matched normal tissue. (Antibody HPA057980, and 10X). (G) Representative IHC images from the HPA demonstrate EXOSC8 expression in HNSC primary tumor and matched normal tissue. (Antibody CAB034240, and 10X). (H) Representative IHC images from the HPA demonstrate EXOSC9 expression in HNSC primary tumor and matched normal tissue. (Antibody HPA048257, and 10X). (I) Representative IHC images from the HPA demonstrate EXOSC10 expression in HNSC primary tumor and matched normal tissue. (Antibody HPA028470, and 10X).
Survival analysis of EXOSC family genes in HNSC
Kaplan-Meier survival analysis using data from The Cancer Genome Atlas (TCGA) was performed to assess the association between EXOSC family gene expression and patient survival outcomes. The study evaluated overall survival (OS), disease-specific survival (DSS), and progression-free interval (PFI) in HNSC patients (Fig. 3). As shown in Fig. 3A, high expression of EXOSC2 (HR = 1.33, P = 0.036) and EXOSC3 (HR = 1.32, P = 0.040) was significantly associated with increased mortality risk, suggesting that elevated levels of these genes may predict poorer OS. Although other EXOSC family members (EXOSC1, EXOSC4–EXOSC10) exhibited a trend toward increased mortality risk, the associations were not statistically significant (P > 0.05). In the DSS analysis (Fig. 3B), high expression of EXOSC2 (HR = 1.42, P = 0.049), EXOSC3 (HR = 1.56, P = 0.014), EXOSC8 (HR = 1.75, P = 0.002), and EXOSC9 (HR = 1.49, P = 0.025) was significantly correlated with increased disease-specific mortality, indicating their potential as prognostic markers for poorer DSS. Other EXOSC genes (EXOSC1, EXOSC4–EXOSC7, and EXOSC10) showed a similar trend, but the associations were not statistically significant (P > 0.05). For PFI (Fig. 3C), high expression of EXOSC2 (HR = 1.41, P = 0.019), EXOSC4 (HR = 1.41, P = 0.017), EXOSC8 (HR = 1.71, P < 0.001), and EXOSC9 (HR = 1.41, P = 0.018) was significantly associated with an increased risk of disease progression, suggesting that elevated expression of these genes may predict shorter PFI. While other EXOSC family members (EXOSC1, EXOSC3, EXOSC5–EXOSC7, and EXOSC10) also exhibited a trend toward increased progression risk, these associations were not statistically significant (P > 0.05).

Prognostic significance of EXOSC family genes in HNSC. Survival analyses for EXOSC family genes in HNSC patients. Forest plots (A-C) showing overall survival (OS), disease specific survival (DSS) and progress free interval (PFI) stratified by high and low gene expression. (*p < 0.05, **p < 0.01, ***p < 0.001).
Correlation between expressions of EXOSC family and clinicopathological parameters of HNSC
The relationship between EXOSC family gene expression and clinicopathological factors in HNSC was systematically analyzed in a cohort of 504 patients, with 252 individuals in both the high- and low-expression groups for each gene (Supplementary Table 9). EXOSC2 expression was significantly associated with multiple pathological and clinical parameters. A higher proportion of patients in the high-expression group presented with advanced pathological T stage (T4; 23.4% vs. 15%, P = 0.00056), pathological stage IV (33.3% vs. 26.8%, P = 0.0054), and clinical N stage (N2/N3; 19.7% vs. 13.9%, P = 0.012). Additionally, its expression correlated with clinical stage IV (30.6% vs. 25.1%, P = 0.0469), male gender (39.7% vs. 33.7%, P = 0.0025), higher histologic grade (G3/G4; 14.5% vs. 10.5%, P = 0.000148), and lymphovascular invasion (21.3% vs. 14.3%, P = 0.00675). Similarly, EXOSC5 was significantly linked to pathological stage IV (31.7% vs. 28.4%, P = 0.014), clinical stage IV (30.8% vs. 24.9%, P = 0.0186), advanced clinical N stage (N2/N3; 19.5% vs. 14.1%, P = 0.011), male gender (40.5% vs. 32.9%, P = 0.00013), higher histologic grade (17.4% vs. 7.6%, P = 5.998e-07), lymphovascular invasion (21.1% vs. 14.6%, P = 0.0202), and prior radiation therapy (35.7% vs. 29.2%, P = 0.0248). EXOSC4 and EXOSC9 were associated with primary therapy outcomes, where high-expression groups exhibited a significantly greater proportion of patients with stable disease, partial response, or progressive disease (EXOSC4: 8.8% vs. 3.8%, P = 0.0024; EXOSC9: 8.6% vs. 4.1%, P = 0.0162). Additionally, EXOSC4 was linked to advanced pathological N stage (N2/N3; 24.8% vs. 17.5%, P = 0.0045) and lymph node dissection (43.9% vs. 38.1%, P = 0.00201). Other notable associations included EXOSC1, EXOSC7, and EXOSC8, which showed significant correlations with male gender (EXOSC1: 38.9% vs. 34.5%, P = 0.0265; EXOSC7: 39.1% vs. 34.3%, P = 0.0155; EXOSC8: 19.7% vs. 13.9%, P = 0.0285), histologic grade (EXOSC1: 16.3% vs. 8.7%, P = 2.11e-05; EXOSC7: 15.1% vs. 9.9%, P = 0.00127; EXOSC8: 15.5% vs. 9.5%, P = 0.00366), and lymphovascular invasion (EXOSC1: 20.2% vs. 15.5%, P = 0.0491; EXOSC7: 22.2% vs. 13.5%, P = 0.00218). Furthermore, EXOSC7 was associated with prior radiation therapy (35.7% vs. 29.2%, P = 0.0123).Conversely, no significant correlations were observed between EXOSC family gene expression and clinical T stage, M stage, race, alcohol history, smoking status, or age.
Mutation and correlation analysis of EXOSC family genes in HNSC
Epigenetic modifications play a critical role in the early stages of malignancy. Using the cBioPortal online tool, we analyzed genetic alterations in EXOSC family genes in HNSC, including mutations and copy number variations. The specific genetic alterations and their respective frequencies are illustrated in Fig. 4A. Among the EXOSC family members, EXOSC4 exhibited the highest alteration frequency (8%), with amplifications being the predominant alteration, suggesting that this gene is the most frequently altered in the analyzed samples. This was followed by EXOSC3 (3%), which displayed amplifications, deep deletions, and missense mutations. Other genes exhibited lower alteration frequencies: EXOSC2 (1.2%), EXOSC10 (1%), EXOSC5 (0.8%), EXOSC9 (0.8%), EXOSC1 (0.6%), EXOSC7 (0.4%), and EXOSC8 (0.4%), while EXOSC6 had the lowest alteration frequency (0.2%). These findings suggest that alterations in EXOSC4 may have a more significant impact on tumor initiation and progression. The broad distribution of EXOSC4 alterations further indicates their prevalence across multiple samples. In contrast, other genes, such as EXOSC1, EXOSC2, and EXOSC3, exhibited sparse alteration distributions, suggesting that these changes occur in only a small subset of cases. Overall, genetic alterations in EXOSC family genes were observed in 15.32% of the 496 HNSC patients, including structural variants, amplifications, and deep deletions. These alterations may significantly influence tumor initiation and progression, particularly as amplifications and deep deletions can lead to oncogene overexpression and loss of tumor suppressor gene function, respectively (Fig. 4B). To further investigate the relationships among EXOSC family members in HNSC, Pearson correlation analysis was performed. As shown in Fig. 4C, significant positive correlations were observed among all EXOSC family members (P < 0.001), except for EXOSC4 and EXOSC10 and EXOSC7 and EXOSC10, which did not show statistically significant associations.

Genetic alterations and expression correlations of EXOSC family genes in HNSC. (A-B) Genomic alteration profiles of EXOSC family genes in HNSC. (C) Co-expression analysis of EXOSC family genes in HNSC tissues. (*p < 0.05, **p < 0.01, ***p < 0.001).
Protein–protein interaction (PPI) network of EXOSC family genes in HNSC
To systematically identify potential functional partners of the EXOSC family in humans, we constructed a PPI network using GeneMANIA. The analysis identified 20 significant interacting proteins, including MPHOSPH6, DIS3, C1D, DIS3L, PNPT1, XRN1, KHSRP, ZFP36L1, ZFP36, SUPT6H, MTREX, PARN, AURKAIP1, TNPO1, HBS1L, DCP1A, DCP2, EXD3, EXD1, and DDX47. Among these, MPHOSPH6 and DIS3 exhibited the highest interaction scores (Fig. 5). Functional enrichment analysis revealed that the EXOSC family proteins and their interactors are primarily involved in biological processes and molecular functions such as the exoribonuclease complex, exosome (RNase complex), exonuclease activity, ncRNA catabolic process, regulation of mRNA catabolic process, RNA stability, and nuclease activity. These findings suggest that EXOSC family members may play a critical role in RNA degradation and post-transcriptional regulation within the tumor microenvironment.

Protein-protein interaction (PPI) network of the EXOSC family genes by GeneMANIA.
Correlation between EXOSC family genes expressions and immune cell infiltration in HNSC
As shown in Fig. 6, the relationship between the relative abundance of 24 immune cell types and EXOSC family gene expression in HNSC was assessed using the ssGSEA algorithm (sample sizes provided in Supplementary Table 3.1–3.4). EXOSC1 expression exhibited a significant positive correlation with NK CD56bright cells, NK cells, Th2 cells, and Tgd, while showing a negative correlation with TFH, Tcm, DC, iDC, B cells, neutrophils, Th17 cells, and mast cells (Fig. 6A). EXOSC2 expression was positively correlated with Th2 cells and NK CD56bright cells but negatively correlated with NK cells, CD8 + T cells, cytotoxic cells, NK CD56dim cells, Th1 cells, pDC, T cells, eosinophils, macrophages, B cells, Treg, TFH, DC, neutrophils, Th17 cells, iDC, and mast cells (Fig. 6B). EXOSC3 expression showed a significant positive correlation with aDC, Th2 cells, T helper cells, and cytotoxic cells, whereas it was negatively correlated with B cells, Th17 cells, Tem, pDC, NK cells, macrophages, eosinophils, DC, neutrophils, iDC, and mast cells (Fig. 6C).

Associations between EXOSC family genes expressions and tumor immune infiltration in HNSC. Bar plots showing correlations between EXOSC1-10 (A-J) expression and the infiltration of 24 immune cell types in HNSC. (*p < 0.05, **p < 0.01, ***p < 0.001).
EXOSC4 expression was positively correlated with NK CD56bright cells but negatively correlated with NK CD56dim cells, Th1 cells, cytotoxic cells, CD8 + T cells, aDC, iDC, DC, Th2 cells, TFH, neutrophils, eosinophils, T cells, Tem, Treg, Th17 cells, B cells, mast cells, T helper cells, and Tcm (Fig. 6D). EXOSC5 expression was positively associated with NK CD56bright cells and cytotoxic cells but negatively correlated with macrophages, Th17 cells, Th1 cells, Tcm, DC, Tgd, mast cells, and neutrophils (Fig. 6E). EXOSC6 expression was positively correlated with NK CD56bright cells but negatively associated with T helper cells, Th1 cells, iDC, mast cells, neutrophils, CD8 + T cells, pDC, TFH, DC, aDC, NK CD56dim cells, Treg, cytotoxic cells, B cells, Th17 cells, and T cells (Fig. 6F). EXOSC7 expression showed a significant positive correlation with NK CD56bright cells but was negatively correlated with T helper cells, cytotoxic cells, NK cells, Th2 cells, Tem, macrophages, B cells, T cells, aDC, NK CD56dim cells, Th1 cells, Tcm, TFH, Treg, DC, Th17 cells, eosinophils, iDC, neutrophils, and mast cells (Fig. 6G). EXOSC8 expression was positively associated with Th2 cells and T helper cells but negatively correlated with NK cells, pDC, macrophages, Tem, B cells, TFH, eosinophils, Th17 cells, DC, iDC, neutrophils, and mast cells (Fig. 6H). EXOSC9 expression was significantly positively correlated with Th2 cells, T helper cells, NK CD56bright cells, and CD8 + T cells, while showing a negative correlation with B cells, macrophages, TFH, eosinophils, DC, Th17 cells, neutrophils, iDC, and mast cells (Fig. 6I). EXOSC10 expression exhibited a significant positive correlation with Th2 cells, T helper cells, Tcm, aDC, Th1 cells, Treg, NK cells, Tem, and Tgd, while showing a negative correlation with B cells, NK CD56bright cells, DC, mast cells, and pDC (Fig. 6J).
Association between EXOSC family genes expressions and immune checkpoint genes in HNSC
To further investigate the association between EXOSC family genes and immune regulation in HNSC, we conducted a comprehensive correlation analysis between EXOSC gene expression levels and immune checkpoint genes. The results of this analysis are presented in Supplementary Table 4. As shown in Fig. 7, EXOSC1 expression was significantly positively correlated with CTLA4, ENTPD1, HAVCR2, IDO1, LAG3, LGALS9, TGFB1, TNFRSF18, TNFRSF4, and TNFRSF14, while it was negatively correlated with CD244 and KDR. EXOSC2 exhibited a significant positive correlation with ADORA2A, LGALS9, TGFB1, TNFRSF18, and TNFRSF14, whereas it showed a negative correlation with CD244. EXOSC3 demonstrated a consistent positive correlation with 14 immune checkpoint genes, including CD274, CD96, CTLA4, HAVCR2, ICOS, IDO1, KIR2DL1, KIR2DL3, LAG3, LGALS9, PDCD1, TGFBR1, TIGIT, and TNFRSF14, while showing a negative correlation with KDR and VSIR. EXOSC4 exhibited a significant negative correlation with 21 checkpoint genes, including ADORA2A, BTLA, CD160, CD244, CD27, CD274, CD96, CSF1R, ENTPD1, HAVCR2, ICOS, IDO1, IL10, KDR, KIR2DL3, PDCD1, SIGLEC15, TGFBR1, TIGIT, TNFRSF9, and VSIR. In contrast, it showed a positive correlation with TGFB1 and TNFRSF18. EXOSC5 was positively correlated with 13 checkpoint genes, including ADORA2A, CD96, CTLA4, ENTPD1, HAVCR2, IDO1, KIR2DL1, LAG3, LGALS9, PDCD1, TNFRSF18, TNFRSF4, and TNFRSF14, while showing a negative correlation with CD274, IL10, KDR, TGFB1, and TGFBR1. EXOSC6 displayed a negative correlation with 16 checkpoint genes, including BTLA, CD244, CD27, CD274, CD96, CSF1R, CTLA4, ICOS, IDO1, KIR2DL1, KIR2DL3, LAG3, PDCD1, TIGIT, TNFRSF9, and VSIR, while exhibiting a positive correlation with SIGLEC15, TGFB1, TGFBR1, and TNFRSF18. EXOSC7 was negatively correlated with 13 checkpoint genes, including BTLA, CD160, CD244, CD27, CSF1R, ICOS, IL10, KDR, TGFB1, TGFBR1, TIGIT, TNFRSF9, and VSIR, whereas it showed a positive correlation with LGALS9, TNFRSF18, and TNFRSF14. EXOSC8 exhibited a positive correlation with 10 checkpoint genes, including CD160, CD274, CTLA4, HAVCR2, IDO1, LAG3, LGALS9, PDCD1, TGFBR1, and TNFRSF14, while also showing a positive correlation with KDR. EXOSC9 was positively correlated with 20 checkpoint genes, including ADORA2A, CD160, CD274, CD96, CTLA4, HAVCR2, ENTPD1, ICOS, IDO1, KIR2DL1, KIR2DL3, LAG3, LGALS9, PDCD1, TIGIT, TNFRSF18, TNFRSF4, TNFRSF9, and TNFRSF14. EXOSC10 exhibited a strong positive correlation with 25 immune checkpoint genes, including ADORA2A, BTLA, CD160, CD27, CD274, CD96, CSF1R, CTLA4, HAVCR2, ENTPD1, ICOS, IDO1, IL10, KDR, KIR2DL3, LAG3, LGALS9, PDCD1, SIGLEC15, TGFB1, TGFBR1, TIGIT, TNFRSF9, TNFRSF14, and VSIR.

Correlations between EXOSC family genes expressions and immune checkpoint molecules in HNSC. (*p < 0.05, **p < 0.01, ***p < 0.001).
Association between EXOSC family genes expression and RNA modification in HNSC
RNA modifications play a crucial role in cancer progression, including angiogenesis. In thymoma, the EXOSC family genes exhibits significant associations with RNA modification-related pathways. As detailed in Supplementary Table 5.1–5.3, correlation coefficients and p-values indicate strong positive associations between EXOSC family genes and key regulators of m1A, m5C, and m6A modifications. As shown in Fig. 8A, EXOSC family genes exhibit significant positive correlations with most m1A modification-related genes, including TRMT61A, TRMT10C, TRMT6, YTHDF1, ALKBH1 and ALKBH3. However, EXOSC4 and EXOSC7 are negatively associated with YTHDC1 and YTHDF3, respectively. Similarly, in m5C modification pathways (Fig. 8B), EXOSC family genes show significant positive correlations with NSUN5, NSUN2, and ALYREF, while EXOSC1, EXOSC4, and EXOSC7 are negatively associated with TET2, EXOSC4 is negatively correlated with NSUN7, DNMT1, DNMT3A and NSUN3, and EXOSC5 is negatively related to DNMT3B. Additionally, as depicted in Fig. 8C, EXOSC family genes demonstrate strong positive correlations with most m6A modification-related genes. while EXOSC3 is negatively associated with FTO, EXOSC4 is negatively correlated with ZC3H13, CBLL1, FTO, YTHDC2 and FMR1, and EXOSC7 is negatively related to FTO and YTHDF3. These findings suggest that RNA modifications may directly or indirectly regulate EXOSC family genes expression, highlighting a potential mechanistic link between EXOSC family genes and RNA modification pathways in HNSC.

Associations between EXOSC family genes expressions and RNA modification regulators in HNSC. Correlation analyses for RNA modification patterns: m1A (A), m5C (B), and m6A (C). (*p < 0.05, **p < 0.01, ***p < 0.001).
GSEA of EXOSC family genes in HNSC
To explore the functional roles of the EXOSC family genes in HNSC, we conducted a comprehensive GSEA. Based on the median expression levels, the HNSC cohort was stratified into high and low expression groups for each EXOSC family gene. Enrichment analysis revealed that high expression of EXOSC family genes was significantly associated with biological pathways such as keratinization, formation of the cornified envelope, and developmental biology, suggesting their potential involvement in epithelial differentiation and tumor development (Fig. 9A-J). These pathways were considered significantly enriched based on the thresholds of false discovery rate (FDR) < 0.25, P-value < 0.05, and normalized enrichment score (NES) > 1. Notably, pathways with the highest NES values were prioritized for downstream interpretation, providing insights into the biological relevance of EXOSC gene expression in HNSC.

Gene set enrichment analysis (GSEA) of the EXOSC family genes in HNSC. Significantly enriched pathways were identified using the following thresholds: false discovery rate (FDR) < 0.25, P-value < 0.05, and normalized enrichment score (NES) > 1. Reactome pathway enrichment analysis for EXOSC1-10 (A-J).
Genes co-expressed with EXOSC family genes in HNSC and enrichment analysis of EXOSC family co-expression genes in HNSC
Analysis of TCGA transcriptome data revealed that the number of genes positively correlated with EXOSC1, EXOSC2, EXOSC3, EXOSC4, EXOSC5, EXOSC6, EXOSC7, EXOSC8, EXOSC9, and EXOSC10 was 8,902; 11,397; 6,755; 3,928; 10,182; 11,163; 8,841; 8,796; 11,678; and 11,657, respectively. In contrast, the number of negatively correlated genes for these EXOSC family members was 1,276; 801; 1,447; 5,496; 2,072; 625; 2,307; 1,099; 856; and 471 (sample sizes provided in Supplementary Table 6.1–6.10). The top 10 positively and negatively correlated genes for each EXOSC family member are depicted in Fig. 10A–J (sample sizes provided in Supplementary Table 7.1–7.10). Additionally, a Venn diagram (Fig. 11A) highlights 40 intersecting genes co-expressed with the EXOSC family (|R| > 0.3, P < 0.001) (sample sizes provided in Supplementary Table 8). To further explore the biological roles of EXOSC family genes in HNSC, we performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses on their co-expressed genes (Fig. 11B). GO analysis categorized these genes into three functional domains: molecular functions (MF), cellular components (CC), and biological processes (BP). The most enriched BPs included ribonucleoprotein complex biogenesis, ribosome biogenesis, and ribonucleoprotein complex assembly. The primary CCs associated with EXOSC family co-expressed genes included the mitochondrial inner membrane, mitochondrial ribosome, and organellar ribosome. Key MFs influenced by these genes involved the structural constituent of ribosome, rRNA binding, and ATP-dependent activity acting on DNA. KEGG pathway analysis further indicated that EXOSC family co-expressed genes were primarily involved in the ribosome, spliceosome, and mRNA surveillance pathways.

Top 10 positively and top 10 negatively correlated genes with EXOSC family members in HNSC. Correlation networks for EXOSC1-10 (A-J). (*p < 0.05, **p < 0.01, ***p < 0.001)

Integrative analysis of co-expression networks and functional enrichment of EXOSC family genes in HNSC. (*p < 0.05, **p < 0.01, ***p < 0.001). (A) Venn diagram identifying 40 consensus co-expressed genes associated with EXOSC1-10 (|R| > 0.3, P < 0.001). (B) Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses of the 40 intersecting genes.
Development of a prognostic model and nomogram based on EXOSC family co-expression genes in HNSC
To identify prognostic biomarkers among EXOSC family co-expressed genes in HNSC, we first conducted univariate Cox regression analysis, which identified 12 candidate genes significantly associated with overall survival: AIMP1, BCCIP, CCT3, CCT4, ERAL1, EXOSC2, MRPL18, MTX1, PRPF19, RSL1D1, RUVBL1, and UTP6 (Fig. 12A). Subsequent LASSO Cox regression analysis further refined this list to five key prognostic genes: AIMP1, CCT3, CCT4, ERAL1, and RSL1D1 (Fig. 12B–C). Based on these five genes, we constructed a prognostic risk model with the following formula: Risk score = (AIMP1 × 0.2520) + (CCT3 × 0.0330) + (CCT4 × 0.0820) + (ERAL1 × 0.0131) + (RSL1D1 × 0.1855). To rigorously assess the predictive power of this EXOSC family co-expression signature, we applied the model to the TCGA HNSC cohort (n = 504). Patients were stratified into high-risk (n = 252) and low-risk (n = 252) groups based on the median risk score. The prognostic value of the model was supported by three main findings. First, clinical outcome analysis revealed that patients in the high-risk group exhibited significantly poorer overall survival and higher mortality rates compared to those in the low-risk group (Fig. 12D). Second, Kaplan–Meier survival analysis demonstrated a clear separation between the two groups, with high-risk patients showing significantly worse survival outcomes (P = 0.008; Fig. 12E). Third, time-dependent ROC curve analysis confirmed the model’s moderate predictive accuracy, with AUCs of 0.581, 0.633, and 0.618 for 1-, 3-, and 5-year overall survival, respectively (Fig. 12F). To further enhance the clinical utility of this model, we developed a nomogram that integrates both the risk score and relevant clinical variables (Fig. 13). Notably, the LASSO-derived risk score was among the most significant predictors of prognosis in multivariate analysis, underscoring its potential value in clinical decision-making.

Development of a prognostic risk model based on EXOSC family co-expression genes in HNSC using TCGA cohort data. (*p < 0.05, **p < 0.01, ***p < 0.001). (A) Univariate Cox regression analysis of the 12 prognostic EXOSC family co-expression genes, presented as forest plots of hazard ratios (HRs) with 95% confidence intervals. (B) Feature coefficients across varying lambda values in LASSO regression. (C) Partial likelihood deviance versus log(λ) in the LASSO Cox regression model. (D) Risk stratification analysis as follows: Top: Patient distribution into high-risk (red) and low-risk (blue) groups based on median risk score. Middle: Survival status (alive = blue; deceased = red). Bottom: Expression heatmap of the 5 prognostic genes across risk groups. (E) Kaplan-Meier analysis demonstrating significant survival difference between risk groups (log-rank test). (F) Time-dependent receiver operating characteristic (ROC) curves assessing model predictive performance.

Construction of a clinical nomogram for survival prediction in HNSC patients. Integrative prognostic nomogram incorporating molecular risk signatures and clinicopathological features for individualized survival prediction in HNSC.
Discussion
The findings presented in this study provide a comprehensive analysis of the EXOSC family’s role in HNSC, highlighting their differential expression, prognostic significance, association with clinicopathological parameters, genetic alterations, immune cell infiltration, and immune checkpoint regulation. These results not only underscore the potential of EXOSC family genes as biomarkers for HNSC but also shed light on their functional roles in tumor biology and immune modulation. Our study revealed significant upregulation of all EXOSC family genes (EXOSC1–EXOSC10) in HNSC tissues compared to normal tissues, suggesting their potential involvement in tumorigenesis. These findings are consistent with previous studies demonstrating the oncogenic roles of EXOSC family members in various malignancies, including breast and colorectal cancers7,9,11,25. Notably, the diagnostic potential of EXOSC family genes was evaluated using ROC curve analysis, where EXOSC2 exhibited the highest AUC value (0.910), indicating superior predictive accuracy for distinguishing HNSC from normal tissues. The robust diagnostic performance of the EXOSC family in our study suggests that these genes could serve as promising biomarkers for early HNSC detection, potential prognostic indicator for risk stratification, and novel therapeutic target. Survival analysis highlighted the prognostic significance of EXOSC2, EXOSC3, EXOSC8, and EXOSC9 in HNSC. Patients with high expression of these genes exhibited significantly worse OS, DSS, and PFI, emphasizing their role in tumor aggressiveness and progression. Importantly, the diagnostic value of the EXOSC family in HNSC is notably high, with several members demonstrating exceptional AUC values (e.g., EXOSC2 AUC = 0.910), reinforcing their potential as reliable biomarkers. This contrasts with findings in other cancers, where EXOSC gene upregulation may be present but often lacks comparable diagnostic discrimination. Furthermore, the strong association of EXOSC2, EXOSC3, EXOSC8, and EXOSC9 with unfavorable survival outcomes-OS, DSS, and PFI-highlights their clinical relevance in prognostication. These associations are markedly stronger in HNSC compared to prior reports in malignancies such as colorectal carcinoma and liver carcinoma, where similar survival correlations were either absent or limited to a subset of EXOSC genes (e.g., EXOSC4 or EXOSC10)11,15. Our study provides strong evidence that EXOSC family genes, particularly EXOSC2, EXOSC3, EXOSC8, and EXOSC9, are significantly associated with poor survival outcomes in HNSC patients. These findings highlight their potential as prognostic biomarkers and therapeutic targets, paving the way for further research into their roles in tumor progression, immune regulation, and treatment resistance.
The correlation between EXOSC family gene expression and advanced clinicopathological features, such as higher pathological T stage, clinical stage IV, and lymphovascular invasion, underscores their potential role in tumor aggressiveness. For instance, EXOSC2 and EXOSC5 were significantly associated with advanced disease stages and higher histologic grades, suggesting their involvement in promoting tumor invasion and metastasis. These findings are supported by studies showing that exosome complex components facilitate the expression of genes involved in epithelial-mesenchymal transition (EMT) and extracellular matrix remodeling26. The association of EXOSC4 and EXOSC9 with primary therapy outcomes, particularly in patients with stable or progressive disease, highlights their potential as predictors of treatment resistance. This is consistent with the role of exosome complex genes in modulating DNA damage response and repair pathways, which are critical for radiation and chemotherapy resistance27. EXOSC1, EXOSC7, and EXOSC8 showed significant associations with male gender, higher histologic grades, and lymphovascular invasion, suggesting a role in tumor differentiation and aggressive phenotypes. These findings indicate that certain EXOSC genes may contribute to disease progression and resistance to therapy, providing valuable insights for risk stratification and personalized treatment approaches in HNSC patients.
Malignant progression unfolds through well-defined histopathological stages, a process fundamentally driven by the sequential accumulation of genetic mutations and their consequent impact on transcriptional programs28. Our study revealed that genetic alterations in EXOSC family genes are relatively common in HNSC, with a total alteration frequency of 15.32% across 496 patients. The analysis of genetic alterations revealed that EXOSC4 exhibited the highest alteration frequency (8%), predominantly through amplifications. The high frequency of EXOSC4 amplifications suggests that this gene may function as an oncogenic driver in HNSC. Gene amplifications often lead to increased mRNA and protein expression, which can enhance cell proliferation, invasion, and survival. Previous studies have shown that amplifications of ribonucleoprotein complex genes can disrupt RNA processing, promote genomic instability, and facilitate tumor growth29. Our findings support this hypothesis, as EXOSC4 alterations were widespread and frequently involved copy number gains, reinforcing its potential role in tumor progression. This suggests that EXOSC4 may play a pivotal role in HNSC pathogenesis, potentially through the overexpression of oncogenic transcripts.
We also constructed a PPI network to investigate the potential molecular partners and functional associations of the EXOSC family genes in HNSC. The EXOSC family constitutes a core component of the RNA exosome complex, which is essential for RNA processing and degradation, particularly in the regulation of non-coding RNAs and mRNA surveillance pathways. Through GeneMANIA analysis, we identified 20 significant interacting proteins, including MPHOSPH6, DIS3, C1D, and PNPT1, with MPHOSPH6 and DIS3 showing the strongest interaction scores. Notably, DIS3 encodes a catalytic subunit of the exosome complex and possesses 3´ to 5´ exoribonuclease and endoribonuclease activities30. It plays a central role in RNA quality control and turnover, and mutations in DIS3 have been implicated in multiple cancers, including multiple myeloma30. Its interaction with multiple EXOSC components in our analysis underscores its central role in exosome-mediated RNA decay in HNSC. MPHOSPH6, another high-confidence interactor, is known to associate with the nuclear exosome targeting complex and assist in rRNA processing31. Dysregulation of rRNA metabolism is frequently observed in cancers, where it supports enhanced protein synthesis and tumor progression31. Therefore, the interaction between EXOSC family members and MPHOSPH6 may reflect an oncogenic role of altered ribonucleolytic activity in HNSC. Functional enrichment analysis revealed that EXOSC family proteins and their interactors are significantly involved in pathways related to exonuclease activity, RNA degradation, ncRNA catabolic processes, and regulation of RNA stability. These biological processes are consistent with the known functions of the exosome complex and reinforce the hypothesis that EXOSC dysregulation contributes to oncogenesis by disturbing RNA homeostasis. Recent studies have shown that aberrant RNA processing and degradation can promote tumorigenesis through the accumulation of non-functional or deleterious RNA species, altered splicing, and disrupted post-transcriptional gene regulation32. Moreover, the implication of regulation of mRNA catabolic process in our analysis suggests that EXOSC family genes may influence cancer-relevant transcripts, including those involved in immune modulation, proliferation, and apoptosis.
The tumor microenvironment (TME) plays a pivotal role in HNSC progression, influencing tumor immune evasion, treatment response, and overall prognosis33. Using ssGSEA analysis, we identified significant correlations between EXOSC family gene expression and the relative abundance of various immune cell types, highlighting their potential role in immune regulation within the TME. Our findings suggest that EXOSC family genes are widely involved in immune modulation, with some members exhibiting strong positive associations with immunosuppressive cell types (e.g., Th2 cells, Treg, NK CD56bright cells), while others show negative correlations with cytotoxic and antigen-presenting cells (e.g., CD8 + T cells, dendritic cells (DC), neutrophils, and NK cells). This suggests that alterations in EXOSC gene expression may shape the immunosuppressive landscape of HNSC tumors, contributing to immune evasion mechanisms. EXOSC1, EXOSC2, and EXOSC4 are Positively Correlated with Th2 Cells and NK CD56bright Cells. Th2 cells and NK CD56bright cells are known to be involved in pro-tumorigenic immune responses, suppressing cytotoxic T lymphocyte (CTL) activity and promoting an immunosuppressive TME34. EXOSC1, EXOSC2, and EXOSC4 expression positively correlated with Th2 cells and NK CD56bright cells, suggesting that these genes may contribute to a tumor-permissive immune environment. Notably, EXOSC2 exhibited a strong negative correlation with cytotoxic immune cells, including CD8 + T cells, NK CD56dim cells, and Th1 cells, reinforcing its potential role in immune evasion. These findings suggest that targeting EXOSC1, EXOSC2, or EXOSC4 could reverse immunosuppressive signaling and enhance anti-tumor immunity. T helper cells play a dual role in tumor immunity, depending on the balance between Th1 (anti-tumor) and Th2 (pro-tumor) responses35. EXOSC3, EXOSC8, and EXOSC10 expression positively correlated with T helper cells, but their impact on Th1 vs. Th2 differentiation needs further exploration. EXOSC10 exhibited a strong positive correlation with Treg and Th2 cells, which are known for their immunosuppressive roles. Tregs suppress anti-tumor immune responses, facilitating tumor immune escape and promoting a tolerogenic microenvironment36. Given these associations, EXOSC10 may be a key modulator of immunosuppression in HNSC and could serve as a potential therapeutic target for immunomodulation. CD8 + T cells and cytotoxic NK cells are crucial for tumor clearance, mediating direct tumor cell killing via perforin and granzyme pathways37. The negative correlation between EXOSC4, EXOSC5, and EXOSC6 expression and CD8 + T cells/cytotoxic cells suggests that these genes may be involved in suppressing immune-mediated tumor clearance, leading to a weaker anti-tumor response. This could explain why high expression of EXOSC4 is linked to poor therapy response, as tumors with lower CD8 + T cell infiltration tend to be more resistant to immunotherapy. DCs are key antigen-presenting cells that activate T cell responses against tumors. The observed negative correlation between EXOSC1, EXOSC3, EXOSC6, and EXOSC7 expression and DC infiltration suggests that these genes might contribute to immune escape by impairing antigen presentation. Similar trends have been reported in lung and pancreatic cancers, where decreased DC infiltration was linked to worse prognosis and immune evasion mechanisms38.
Immune checkpoints play a critical role in regulating immune responses by preventing excessive immune activation39. However, tumors can exploit these pathways to evade immune surveillance, leading to immune escape and progression. The correlation analysis between EXOSC family gene expression and immune checkpoint genes provides new insights into how these genes might contribute to immune modulation in HNSC. Our findings reveal that specific EXOSC family genes are significantly correlated with key immune checkpoint molecules, suggesting a potential role in immune escape mechanisms. Notably, EXOSC1, EXOSC3, EXOSC5, EXOSC9, and EXOSC10 exhibited strong positive correlations with multiple immune checkpoint genes, including PDCD1 (PD-1), CD274 (PD-L1), CTLA4, HAVCR2 (TIM-3), IDO1, and LAG3, which are known mediators of T cell exhaustion and tumor immune evasion40. Conversely, EXOSC4, EXOSC6, and EXOSC7 showed significant negative correlations with several immune checkpoint genes, suggesting a potential role in maintaining an anti-tumor immune response. EXOSC1, EXOSC3, and EXOSC10 were positively correlated with multiple immune checkpoint genes (e.g., PDCD1, CTLA4, LAG3, IDO1, and HAVCR2). These immune checkpoints are key mediators of T cell exhaustion, suppressing T cell proliferation and cytotoxic activity41. The strong positive correlation between EXOSC3 and 14 immune checkpoints, including PDCD1, CTLA4, and LAG3, suggests that EXOSC3 may be involved in promoting an immunosuppressive microenvironment, potentially leading to reduced anti-tumor immunity and resistance to checkpoint blockade therapy. Similarly, EXOSC10 exhibited the most extensive positive correlations (25 immune checkpoints), including PDCD1, CD274, CTLA4, and TIGIT, further suggesting a role in immune suppression and tumor progression. Given these associations, targeting EXOSC3 or EXOSC10 may enhance immune checkpoint inhibitor (ICI) efficacy in HNSC patients by reducing immunosuppressive signaling. Unlike EXOSC1, EXOSC3, and EXOSC10, EXOSC4 and EXOSC6 showed significant negative correlations with multiple immune checkpoints, including PDCD1, CTLA4, CD274, and CSF1R. EXOSC4 was negatively correlated with 21 checkpoint genes, while EXOSC6 exhibited negative correlations with 16 checkpoint genes, suggesting a potential role in maintaining anti-tumor immune responses. This suggests that low EXOSC4 or EXOSC6 expression may be associated with better immune activation and improved response to immunotherapy. EXOSC5, EXOSC8, and EXOSC9 demonstrated diverse associations with immune checkpoint genes, showing both positive and negative correlations depending on the checkpoint molecule. For example, EXOSC5 was positively correlated with CTLA4 and PDCD1 but negatively correlated with IL10 and TGFB1, suggesting a dual role in immune regulation. EXOSC8 was positively associated with CD274 (PD-L1), IDO1, and CTLA4, supporting a role in immune suppression, while its positive correlation with KDR might indicate involvement in tumor angiogenesis. EXOSC9 had a strong positive correlation with 20 immune checkpoint genes, including PDCD1, CTLA4, LAG3, and TIGIT, suggesting that it may contribute to an immunosuppressive tumor microenvironment. This suggests their potential involvement in immune escape mechanisms and highlights their possible value as biomarkers for immunotherapeutic responsiveness. Notably, while previous studies have mainly linked EXOSC genes to RNA processing and tumor cell proliferation in other cancers, our findings uniquely emphasize their immune-related functions in HNSC. This suggests a context-dependent role of EXOSC genes, potentially shaped by the tissue-specific immune landscape. Among the EXOSC members, EXOSC10 showed strong positive correlations with a wide range of immune checkpoints, positioning it as a potential immunotherapeutic target, either alone or in combination with checkpoint inhibitors. Its distinct association with both effector and regulatory immune components suggests a dual role in modulating immune surveillance and immune suppression. Overall, our findings underscore the innovative potential of EXOSC genes as immune-related biomarkers and therapeutic targets in HNSC, expanding their known functional repertoire. Future experimental work is warranted to elucidate the mechanistic basis of these interactions and to explore whether modulation of EXOSC expression can enhance the efficacy of existing immunotherapies.
Emerging evidence indicates that RNA modifications such as m6A, m5C, and m1A play pivotal roles in post-transcriptional gene regulation and are closely implicated in cancer progression, including HNSC42,43,44. Our study provides novel insights into the regulatory interplay between the EXOSC family of RNA exosome complex components and RNA modification regulators in HNSC, suggesting a mechanistic framework through which RNA modifications may contribute to tumor biology via modulation of EXOSC gene expression. In the study, we observed widespread and significant correlations between EXOSC family genes expressions and core regulators of m1A, m5C, and m6A modifications. Notably, most EXOSC genes exhibited strong positive associations with m1A-related genes, including TRMT61A, TRMT10C, TRMT6, and YTHDF1, implicating a coordinated expression pattern that may reflect shared regulatory mechanisms or functional convergence in RNA metabolism. Prior studies have shown that m1A writers such as TRMT61A are involved in maintaining translation fidelity and cellular stress responses45. The strong correlation with YTHDF1, an m1A/m6A reader, further suggests that EXOSC genes may be embedded within RNA modification-dependent regulatory circuits that fine-tune mRNA stability and translation in HNSC cells. Interestingly, selective negative correlations were also observed; for instance, EXOSC4 and EXOSC7 displayed inverse associations with YTHDC1 and YTHDF3, respectively. These exceptions may indicate subunit-specific roles within the EXOSC complex or feedback mechanisms aimed at buffering aberrant modification-induced transcriptomic shifts. Similar heterogeneity has been reported in other cancers, where specific RNA-modifying enzymes exert context-dependent effects on gene networks46. Within the m5C regulatory network, EXOSC family genes demonstrated positive correlations with NSUN2, NSUN5, and the reader ALYREF. NSUN2, in particular, has been implicated in enhanced proliferation and metastasis in various cancers through m5C-dependent regulation of mRNA stability47. These findings suggest that m5C modifications may modulate EXOSC gene function or expression, contributing to oncogenic phenotypes. Conversely, EXOSC1, EXOSC4, and EXOSC7 were negatively associated with the DNA demethylase TET2, a finding of particular interest given the dual role of TET enzymes in both DNA and RNA methylation landscapes. The negative correlation between EXOSC4 and multiple m5C regulators, including NSUN3, NSUN7, DNMT1, and DNMT3A, hints at a possible suppressive interplay, potentially reflective of EXOSC4-mediated degradation of RNA species enriched in these modifications. The m6A landscape similarly revealed robust positive correlations between EXOSC genes and key m6A writers/readers, reinforcing the notion that RNA modification and degradation machineries are co-regulated in HNSC. For instance, positive correlations with METTL3, WTAP, and YTHDF1 were prevalent across the family, which may support enhanced RNA turnover or selectivity in target mRNA surveillance. Nevertheless, EXOSC4 and EXOSC7 consistently showed negative associations with the eraser FTO and reader YTHDF3, as well as additional components like ZC3H13 and CBLL1, indicating a potentially unique regulatory axis for these subunits. The functional implications of these inverse relationships merit further mechanistic investigation but may represent a compensatory balance between RNA stabilization and degradation pathways. Collectively, these results highlight the tight interconnection between RNA modification machinery and the RNA exosome complex, positioning EXOSC genes as potential integrators of epitranscriptomic regulation in HNSC. Previous studies have demonstrated that alterations in RNA modification pathways can reprogram cancer transcriptomes, affecting splicing, export, translation, and decay48. Our findings suggest that EXOSC genes may not only respond to these regulatory cues but also participate actively in shaping RNA homeostasis in an RNA modification-dependent manner. Given the emerging role of RNA modifications as therapeutic targets, particularly m6A and m5C regulators49, the EXOSC family may represent a novel class of co-targetable factors that modulate tumor cell plasticity and response to treatment.
To further elucidate the biological functions of EXOSC family genes in HNSC, we conducted GSEA, which revealed significant enrichment of pathways related to keratinization, formation of the cornified envelope, and developmental biology. These findings provide critical insights into how EXOSC family members may contribute to tumor progression through modulation of epithelial differentiation and cellular developmental programs. Keratinization, a hallmark of squamous epithelial cells, is a tightly regulated process involving the terminal differentiation of keratinocytes50. In the context of HNSC, aberrant keratinization has been widely associated with tumor differentiation status and clinical behavior. Poorly differentiated HNSC tumors often exhibit reduced keratinization, while well-differentiated tumors tend to retain keratin pearl formation51. Dysregulation of genes involved in keratinocyte differentiation pathways may facilitate a dedifferentiated, more aggressive tumor phenotype52. The association between EXOSC gene expression and keratinization-related pathways in our analysis suggests that EXOSC family members may play a role in maintaining epithelial identity or, conversely, promoting epithelial dedifferentiation when dysregulated. The enrichment of the Formation of the Cornified Envelope pathway further supports this hypothesis. The cornified envelope is an insoluble protein layer formed during the late stages of keratinocyte differentiation, contributing to the physical barrier of stratified squamous epithelium53. Its formation involves cross-linking of involucrin, loricrin, and small proline-rich proteins by transglutaminases. Recent studies have implicated disruption of cornified envelope components in carcinogenesis54. The involvement of EXOSC family genes in this pathway implies that they may regulate post-transcriptional processing of mRNAs encoding key structural proteins, thereby influencing epithelial barrier integrity and tumor invasiveness. Moreover, the enrichment of Developmental Biology pathways underscores the role of EXOSC family genes in regulating fundamental cellular programs often hijacked during tumorigenesis. Cancer is frequently described as a disease of altered development, where embryonic gene expression programs are reactivated to support proliferation, migration, and immune evasion55. In HNSC, activation of developmental pathways such as Wnt, Notch, and Hedgehog signaling has been linked to epithelial–mesenchymal transition (EMT), tumor heterogeneity, and therapy resistance56. Given that the RNA exosome complex is known to modulate the stability and maturation of diverse classes of RNAs, the EXOSC family may influence these developmental processes by regulating non-coding RNAs, pre-mRNAs, or transcription factor transcripts essential for oncogenic reprogramming. Collectively, these results highlight a novel connection between EXOSC family gene expression and key epithelial and developmental processes in HNSC. This suggests a dual role for EXOSC genes in both maintaining normal epithelial differentiation and facilitating malignant transformation when dysregulated. Our findings are consistent with prior research demonstrating the oncogenic potential of RNA exosome components through disruption of RNA surveillance and transcriptome stability.
The EXOSC family, which forms the core of the RNA exosome complex, plays a critical role in RNA processing, degradation, and surveillance6. Our TCGA transcriptome data analysis identified a substantial number of genes co-expressed with EXOSC family members, revealing both positively and negatively correlated genes. Notably, the number of positively correlated genes varied across EXOSC members, with EXOSC9 (11,678) and EXOSC10 (11,657) showing the highest co-expression, while EXOSC4 (3,928) exhibited the lowest number of positively correlated genes. These differences suggest distinct functional roles and regulatory mechanisms among EXOSC genes in HNSC pathogenesis. Interestingly, we identified 40 intersecting genes that were co-expressed with multiple EXOSC family members (|R| > 0.3, P < 0.001). This overlapping gene set may represent a core regulatory network influenced by EXOSC genes in HNSC, further emphasizing their potential role in tumor progression and RNA metabolism. Gene Ontology (GO) enrichment analysis revealed that co-expressed genes of the EXOSC family are highly enriched in ribonucleoprotein complex biogenesis, ribosome biogenesis, and ribonucleoprotein complex assembly. These findings are consistent with previous studies showing that dysregulated ribosome biogenesis is a hallmark of cancer, facilitating increased protein synthesis to sustain rapid tumor growth57. EXOSC1, EXOSC2, and EXOSC10 showed strong correlations with genes involved in ribosome biogenesis, reinforcing their pro-tumorigenic role through the enhancement of translational capacity. EXOSC4 had the fewest positively correlated genes, suggesting that its role in ribosome biogenesis may be more restricted, or potentially inhibitory. The involvement of EXOSC genes in ribonucleoprotein complex assembly further supports their function in mRNA processing and decay, processes that are critical for maintaining the stability and turnover of oncogenic transcripts in cancer cells. GO analysis of cellular component (CC) enrichment revealed that EXOSC family co-expressed genes are significantly associated with mitochondrial inner membranes, mitochondrial ribosomes, and organellar ribosomes. Given that mitochondrial function is closely linked to cancer metabolism and chemoresistance58, this suggests that the EXOSC family may contribute to mitochondrial biogenesis and energy metabolism in HNSC. EXOSC9 and EXOSC10 showed the strongest enrichment in mitochondrial components, suggesting a role in mitochondria-related RNA processing or translation. Since mitochondrial ribosomes regulate oxidative phosphorylation (OXPHOS), the correlation between EXOSC family genes and mitochondrial ribosome components raises the possibility that EXOSC genes modulate metabolic reprogramming in HNSC, potentially influencing tumor progression and therapy resistance. The molecular function (MF) enrichment analysis revealed that EXOSC family co-expressed genes are significantly involved in: Structural constituent of ribosomes, rRNA binding and ATP-dependent activity acting on DNA. These findings align with the known functions of EXOSC genes as key regulators of RNA degradation59. The strong enrichment in rRNA binding and ribosome structural functions suggests that EXOSC genes may contribute to ribosomal RNA processing and stability, thereby supporting cancer cell proliferation by enhancing protein translation. KEGG pathway analysis demonstrated that EXOSC family co-expressed genes are primarily involved in the ribosome, spliceosome, and mRNA surveillance pathways. These pathways play pivotal roles in post-transcriptional gene regulation, and their dysregulation is a common feature in cancer60. EXOSC1, EXOSC3, and EXOSC10 were enriched in ribosome-related pathways, reinforcing their role in enhancing protein synthesis, a key feature of aggressive tumors. Given that tumor cells exploit ribosomal activity to support unchecked proliferation, targeting ribosome biogenesis has emerged as a potential therapeutic strategy in HNSC61. Our findings suggest that dysregulated EXOSC family expression could serve as a potential target for therapies aimed at disrupting ribosome function in HNSC. The spliceosome pathway was significantly enriched in EXOSC family co-expressed genes, particularly in EXOSC2, EXOSC5, and EXOSC6. Aberrant alternative splicing is a known driver of cancer progression, leading to the production of oncogenic protein isoforms that promote survival, invasion, and therapy resistance62. These results suggest that EXOSC genes might influence splicing factor regulation, affecting the expression of tumor-promoting splice variants in HNSC. The enrichment of EXOSC-related genes in the mRNA surveillance pathway highlights their potential role in post-transcriptional regulation of tumor suppressor genes. The EXOSC complex is involved in the degradation of aberrant mRNAs, preventing the accumulation of defective transcripts. However, in the context of HNSC, it is possible that EXOSC overexpression enhances the degradation of tumor-suppressive mRNAs, leading to uncontrolled tumor growth.
We then systematically explored the prognostic potential of EXOSC family co-expression genes in HNSC and successfully constructed a robust risk model and nomogram to predict patient outcomes. Our analysis identified five key co-expression genes-AIMP1, CCT3, CCT4, ERAL1, and RSL1D1-which collectively demonstrated significant predictive value for overall survival. The EXOSC family genes are involved in RNA degradation and processing, critical functions in post-transcriptional regulation that are often dysregulated in cancer32. Our findings suggest that dysregulation of EXOSC family co-expression partners may reflect broader transcriptomic instability in HNSC. For instance, RSL1D1 has been shown to mediate p53-dependent cell cycle regulation, which may account for its prognostic impact in HNSC63. Furthermore, CCT3 and CCT4, members of the chaperonin-containing TCP-1 complex, are known to assist in the folding of key oncogenic proteins, including cyclins and p53. Overexpression of CCT family members have been reported in various malignancies and is associated with enhanced tumor growth, metastasis, and chemoresistance64. Similarly, ERAL1, a mitochondrial RNA chaperone, may reflect mitochondrial dysfunction and altered metabolic reprogramming, hallmarks of many aggressive tumors65. Our prognostic model demonstrated significant discriminative power, with high-risk patients exhibiting substantially poorer overall survival. The time-dependent ROC analysis further validated the model’s utility, yielding AUC values above 0.6 across 1-, 3-, and 5-year survival predictions. Although the AUCs were moderate, they were comparable to or better than those reported for other RNA-based prognostic signatures in HNSC. Importantly, we also developed a nomogram that incorporated both molecular (risk score) and clinical parameters, enhancing its translational potential. The LASSO-derived risk score emerged as one of the strongest independent predictors of survival, underscoring its robustness. Notably, the EXOSC family and its associated co-expressed genes may also offer insights into therapeutic vulnerabilities. Previous studies have shown that inhibition of RNA processing pathways can sensitize tumors to chemotherapy or immunotherapy32. Thus, components of the EXOSC-associated risk model may not only serve as prognostic biomarkers but also as potential therapeutic targets.
The findings from this study have significant clinical implications, particularly in the development of personalized treatment strategies for HNSC. The strong prognostic value of EXOSC2, EXOSC3, EXOSC8, and EXOSC9 suggests that they could serve as novel biomarkers for patient risk stratification. Additionally, given their roles in immune modulation, EXOSC family genes may represent potential targets for combination therapies involving immune checkpoint inhibitors. Future research should focus on functional validation studies to elucidate the precise molecular mechanisms underlying EXOSC gene-mediated tumor progression and immune regulation in HNSC. Additionally, prospective clinical trials assessing the utility of EXOSC-based biomarkers in guiding therapeutic decisions would be valuable.
In conclusion, this study provides a comprehensive bioinformatics analysis of EXOSC family genes in HNSC, revealing their significant upregulation in tumor tissues and strong prognostic implications. High expression levels of EXOSC2, EXOSC3, EXOSC4, EXOSC8, and EXOSC9 were associated with poorer survival outcomes, while genetic alterations, particularly in EXOSC4, emerged as critical events in tumor progression. Immune infiltration analysis further suggested that EXOSC genes may influence the tumor microenvironment by modulating immune cell populations. Functional enrichment analyses linked EXOSC genes to key oncogenic pathways, emphasizing their role in tumorigenesis. Collectively, these findings highlight the potential of EXOSC family members as prognostic biomarkers and therapeutic targets in HNSC, paving the way for further research into their mechanistic roles and clinical applications.
Data availability
All supporting data are provided in the supplementary files.
References
Johnson, D. E. et al. Head and neck squamous cell carcinoma [J]. Nat. Rev. Dis. Primers. 6 (1), 92 (2020).
Nguyen, E. S. et al. Prognostic factors and outcomes of de Novo sinonasal squamous cell carcinoma: A systematic review and Meta-analysis [J]. Otolaryngol. Head Neck Surg. 166 (3), 434–443 (2022).
Hartl, D. M. et al. Review of outcomes after salvage surgery for recurrent squamous cell carcinoma of the head and neck. Cancers (Basel), 15(19), (2023).
Kolapalli, S. P., Nielsen, T. M. & Frankel, L. B. Post-transcriptional dynamics and RNA homeostasis in autophagy and cancer [J]. Cell. Death Differ. 32 (1), 27–36 (2025).
Ogami, K. & Suzuki, H. I. Nuclear RNA exosome and pervasive transcription: dual sculptors of genome function [J]. Int. J. Mol. Sci., 22(24), (2021).
Fasken, M. B. et al. The RNA exosome and human disease [J]. Methods Mol. Biol. 2062, 3–33 (2020).
Zhang, Y. et al. Integrated bioinformatic investigation of exoscs in hepatocellular carcinoma followed by the preliminary validation of EXOSC5 in cell proliferation [J]. Int. J. Mol. Sci., 23(20), (2022).
Liu, Q. et al. Exosome component 1 cleaves single-stranded DNA and sensitizes human kidney renal clear cell carcinoma cells to poly(ADP-ribose) polymerase inhibitor. Elife 10, (2021).
Lv, C. G. et al. EXOSC2 mediates the Pro-tumor role of WTAP in breast cancer cells via activating the Wnt/β-Catenin signal [J]. Mol. Biotechnol. 66 (9), 2569–2582 (2024).
Tsuda, M. et al. Aberrant expression of MYD88 via RNA-controlling CNOT4 and EXOSC3 in colonic mucosa impacts generation of colonic cancer [J]. Cancer Sci. 112 (12), 5100–5113 (2021).
Pan, Y. et al. EXOSC4 functions as a potential oncogene in development and progression of colorectal cancer [J]. Mol. Carcinog. 57 (12), 1780–1791 (2018).
Chen, X. et al. EXOSC5 promotes proliferation of gastric cancer through regulating AKT/STAT3 signaling pathways [J]. J. Cancer. 13 (5), 1456–1467 (2022).
Cui, K. et al. EXOSC8 promotes colorectal cancer tumorigenesis via regulating ribosome biogenesis-related processes [J]. Oncogene 41 (50), 5397–5410 (2022).
Yoshino, S. et al. EXOSC9 depletion attenuates P-body formation, stress resistance, and tumorigenicity of cancer cells [J]. Sci. Rep. 10 (1), 9275 (2020).
Meng, Z. Y. et al. EXOSC10 is a novel hepatocellular carcinoma prognostic biomarker: a comprehensive bioinformatics analysis and experiment verification [J]. PeerJ, 11e15860. (2023).
Liu, J. et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics [J]. Cell, 173(2), 400 – 16.e11 (2018).
De Hond, A. A. H., Steyerberg, E. W. & Van Calster, B. Interpreting area under the receiver operating characteristic curve [J]. Lancet Digit. Health. 4 (12), e853–e5 (2022).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data [J]. Cancer Discov. 2 (5), 401–404 (2012).
Bindea, G. et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer [J]. Immunity 39 (4), 782–795 (2013).
Hu, F. F. et al. Expression profile of immune checkpoint genes and their roles in predicting immunotherapy response. Brief. Bioinform, 22(3), (2021).
Haruehanroengra, P. et al. RNA modifications and cancer [J]. RNA Biol. 17 (11), 1560–1575 (2020).
Wang, S. et al. UCSCXenaShiny: an R/CRAN package for interactive analysis of UCSC Xena data [J]. Bioinformatics 38 (2), 527–529 (2022).
Kanehisa, M. et al. KEGG for taxonomy-based analysis of pathways and genomes [J]. Nucleic Acids Res. 51 (D1), D587–d92 (2023).
The Gene Ontology Resource. 20 years and still going strong [J]. Nucleic Acids Res. 47 (D1), D330–d8 (2019).
Huang, Y. H. et al. EXOSC5 maintains cancer stem cell activity in endometrial cancer by regulating the NTN4/integrin β1 signalling axis [J]. Int. J. Biol. Sci. 20 (1), 265–279 (2024).
Yang, E. et al. Exosome-mediated metabolic reprogramming: the emerging role in tumor microenvironment remodeling and its influence on cancer progression [J]. Signal. Transduct. Target. Ther. 5 (1), 242 (2020).
Meng, X., Yang, S. & Camp, V. J. A. The Interplay Between the DNA Damage Response, RNA Processing and Extracellular Vesicles. Front. Oncol. 9, 1538. (2019).
Garnis, C., Buys, T. P. & Lam, W. L. Genetic alteration and gene expression modulation during cancer progression. Mol Cancer, 3, 9. (2004).
Wickramasinghe, V. O. & Venkitaraman, A. R. RNA processing and genome stability: cause and consequence [J]. Mol. Cell. 61 (4), 496–505 (2016).
Ohguchi, Y. & Ohguchi, H. DIS3: the enigmatic gene in multiple myeloma. Int. J. Mol. Sci., 24(4), (2023).
Peng, R. R. et al. The 5.8S pre-rRNA maturation factor, M-phase phosphoprotein 6, is a female fertility factor required for oocyte quality and meiosis [J]. Cell. Prolif. 53 (3), e12769 (2020).
Barbieri, I. & Kouzarides, T. Role of RNA modifications in cancer [J]. Nat. Rev. Cancer. 20 (6), 303–322 (2020).
Elmusrati, A., Wang, J. & Wang, C. Y. Tumor microenvironment and immune evasion in head and neck squamous cell carcinoma [J]. Int. J. Oral Sci. 13 (1), 24 (2021).
Wang, Z. et al. T cells, particularly activated CD4(+) cells, maintain anti-CD20-mediated NK cell viability and antibody dependent cellular cytotoxicity [J]. Cancer Immunol. Immunother. 71 (2), 237–249 (2022).
Silva, R., Lopes, M. F. & Travassos, L. H. Distinct T helper cell-mediated antitumor immunity: T helper 2 cells in focus [J]. Cancer Pathog Ther. 1 (1), 76–86 (2023).
Li, C. et al. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects [J]. Mol. Cancer. 19 (1), 116 (2020).
Prager, I. & Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity [J]. J. Leukoc. Biol. 105 (6), 1319–1329 (2019).
Hegde, S. et al. Dendritic cell paucity leads to dysfunctional immune surveillance in pancreatic cancer [J]. Cancer Cell. 37 (3), 289–307e9 (2020).
Francis, D. M. et al. Blockade of immune checkpoints in lymph nodes through locoregional delivery augments cancer immunotherapy. Sci. Transl Med., 12(563), (2020).
Andrews, L. P. et al. LAG-3 and PD-1 synergize on CD8(+) T cells to drive T cell exhaustion and hinder autocrine IFN-γ-dependent anti-tumor immunity [J]. Cell 187 (16), 4355–72e22 (2024).
Baessler, A. & Vignali, D. A. A. T cell exhaustion [J]. Annu. Rev. Immunol. 42 (1), 179–206 (2024).
Deng, X. et al. The roles and implications of RNA m(6)A modification in cancer [J]. Nat. Rev. Clin. Oncol. 20 (8), 507–526 (2023).
Li, M. et al. 5-methylcytosine RNA methyltransferases and their potential roles in cancer [J]. J. Transl Med. 20 (1), 214 (2022).
Liu, Y. et al. Research progress of N1-methyladenosine RNA modification in cancer [J]. Cell. Commun. Signal. 22 (1), 79 (2024).
Monshaugen, I. et al. Depletion of the m1A writer TRMT6/TRMT61A reduces proliferation and resistance against cellular stress in bladder cancer. Front. Oncol. 13, 1334112. (2023).
Dalhat, M. H. et al. Dissecting the oncogenic properties of essential RNA-modifying enzymes: a focus on NAT10 [J]. Oncogene 43 (15), 1077–1086 (2024).
Yang, Y. et al. NOP2 facilitates EZH2-mediated epithelial-mesenchymal transition by enhancing EZH2 mRNA stability via m5C methylation in lung cancer progression [J]. Cell. Death Dis. 15 (7), 506 (2024).
Bártová, E. & Stixová, L. Svobodová Kovaříková A. N4-acetylcytidine and other RNA modifications in epitranscriptome: insight into DNA repair and cancer development [J]. Epigenomics 17 (6), 411–422 (2025).
Dai, Y. et al. RNA methylation and breast cancer: insights into m6A, m7G and m5C [J]. Mol. Biol. Rep. 52 (1), 27 (2024).
Deo, P. N. & Deshmukh, R. Pathophysiology of keratinization [J]. J. Oral Maxillofac. Pathol. 22 (1), 86–91 (2018).
Lin, C. et al. Innovative organ-on-a-chip platforms for exploring tumorigenesis and therapy in head and neck cancer [J]. J. Transl Med. 23 (1), 798 (2025).
Liu, S. et al. 5-Hydroxymethylation highlights the heterogeneity in keratinization and cell junctions in head and neck cancers [J]. Clin. Epigenetics. 12 (1), 175 (2020).
Elias, P. M. et al. Formation and functions of the corneocyte lipid envelope (CLE) [J]. Biochim. Biophys. Acta. 1841 (3), 314–318 (2014).
Candi, E., Schmidt, R. & Melino, G. The cornified envelope: a model of cell death in the skin [J]. Nat. Rev. Mol. Cell. Biol. 6 (4), 328–340 (2005).
Sell, S. et al. Cancer: A problem of developmental biology; scientific evidence for reprogramming and differentiation therapy [J]. Curr. Drug Targets. 17 (10), 1103–1110 (2016).
Nakaya, Y. & Sheng, G. EMT in developmental morphogenesis [J]. Cancer Lett. 341 (1), 9–15 (2013).
Elhamamsy, A. R. et al. Ribosome biogenesis: A central player in cancer metastasis and therapeutic resistance [J]. Cancer Res. 82 (13), 2344–2353 (2022).
Genovese, I. et al. Mitochondria: insights into crucial features to overcome cancer chemoresistance [J]. Int. J. Mol. Sci., 22(9), (2021).
Morton, D. J. et al. The RNA exosome and RNA exosome-linked disease [J]. Rna 24 (2), 127–142 (2018).
Bradley, R. K. & Anczuków, O. RNA splicing dysregulation and the hallmarks of cancer [J]. Nat. Rev. Cancer. 23 (3), 135–155 (2023).
Jiao, L. et al. Ribosome biogenesis in disease: new players and therapeutic targets [J]. Signal. Transduct. Target. Ther. 8 (1), 15 (2023).
Zhang, Y. et al. Alternative splicing and cancer: a systematic review [J]. Signal. Transduct. Target. Ther. 6 (1), 78 (2021).
Ding, L. et al. Mutations in DNA binding domain of p53 impede RSL1D1-p53 interaction to escape from degradation in human colorectal cancer cells [J]. Exp. Cell. Res. 417 (1), 113211 (2022).
Dong, Y. et al. CCTs as new biomarkers for the prognosis of head and neck squamous cancer [J]. Open. Med. (Wars). 15 (1), 672–688 (2020).
Britton, R. A. et al. Isolation and preliminary characterization of the human and mouse homologues of the bacterial cell cycle gene era [J]. Genomics 67 (1), 78–82 (2000).
Author information
Authors and Affiliations
Contributions
Xuezhong Zhang and Qingmei Jia proposed the study idea. Xuezhong Zhang, Mengmeng Zhao, Tingting Chu, Jiashawei and Qingmei Jia collected and analyzed the data. Xuezhong Zhang drafted the manuscript. Xuezhong Zhang and Qingmei Jia critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The datasets analyzed in this study were sourced from publicly available repositories.
Consent to participate
As this research utilized anonymized, publicly accessible data, obtaining individual participant consent was not required.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, X., Zhao, M., Chu, T. et al. Comprehensive bioinformatics analysis of EXOSC family genes in head and neck squamous cell carcinoma. Sci Rep 15, 30361 (2025). https://doi.org/10.1038/s41598-025-15758-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-15758-3
